
A Customized Human Mitochondrial DNA Database (hMITO DB v1.0) for Rapid Sequence Analysis, Haplotyping and Geo-Mapping


Abstract
:
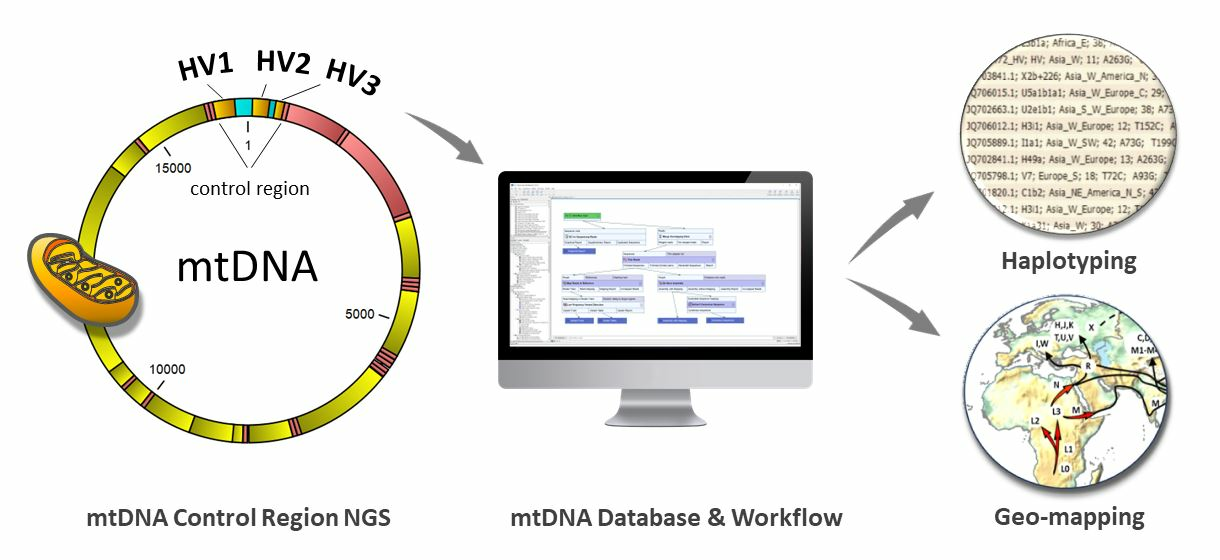{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. hMITO DB Haplogroup Distribution and Geographic Origins
Phylogenetic Tree of Representative Human Mitogenomes
2.2. Utility of hMITO DB for Sequence Analysis, Haplotyping, and Geo-Mapping of an mtDNA NGS Dataset from Cervical Cytology Samples and Cancer Cell Lines
2.2.1. Read Mapping and Visualization of Mapped Tracks
2.2.2. Low Frequency Variant Track and Table
2.3. mtDNA Haplotyping and Comparison to Self-Reported Race/Ethnicity
2.4. FASTQ File Sizes and Workflow Runtimes
3. Discussion
4. Materials and Methods
4.1. Construction and Content of Customized Reference Database

4.2. mtDNA Sequence Analysis and BLAST Workflows
4.3. Cell Samples and mtDNA Control Region Sequencing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Sun, N.; Finkel, T. Cardiac mitochondria: A surprise about size. J. Mol. Cell. Cardiol. 2015, 82, 213–215. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, B. From bioblasts to mitochondria: Ever expanding roles of mitochondria in cell physiology. Front. Physiol. 2010, 1, 7. [Google Scholar] [CrossRef]
- Herrera-Cruz, M.S.; Simmen, T. Over Six Decades of Discovery and Characterization of the Architecture at Mitochondria-Associated Membranes (MAMs). Adv. Exp. Med. Biol. 2017, 997, 13–31. [Google Scholar] [CrossRef]
- Moon, D.O. Calcium’s Role in Orchestrating Cancer Apoptosis: Mitochondrial-Centric Perspective. Int. J. Mol. Sci. 2023, 24, 8982. [Google Scholar] [CrossRef] [PubMed]
- Nass, M.M.; Nass, S. Intramitochondrial fibers with DNA characteristics I. Fixation and electron staining reactions. J. Cell Biol. 1963, 19, 593–611. [Google Scholar] [CrossRef]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef]
- Bandelt, H.J.; Kloss-Brandstätter, A.; Richards, M.B.; Yao, Y.G.; Logan, I. The case for the continuing use of the revised Cambridge Reference Sequence (rCRS) and the standardization of notation in human mitochondrial DNA studies. J. Hum. Genet. 2014, 59, 66–77. [Google Scholar] [CrossRef]
- DiMauro, S. A Brief History of Mitochondrial Pathologies. Int. J. Mol. Sci. 2019, 22, 5643. [Google Scholar] [CrossRef]
- Wallace, D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef]
- Beadnell, T.C.; Scheid, A.D.; Vivian, C.J.; Welch, D.R. Roles of the mitochondrial genetics in cancer metastasis: Not to be ignored any longer. Cancer Metastasis Rev. 2018, 37, 615–632. [Google Scholar] [CrossRef]
- Kim, M.; Mahmood, M.; Reznik, E.; Gammage, P.A. Mitochondrial DNA is a major source of driver mutations in cancer. Trends Cancer 2022, 8, 1046–1059. [Google Scholar] [CrossRef] [PubMed]
- Scheid, A.D.; Beadnell, T.C.; Welch, D.R. The second genome: Effects of the mitochondrial genome on cancer progression. Adv. Cancer Res. 2019, 142, 63–105. [Google Scholar] [CrossRef] [PubMed]
- Smith, D. The Past, Present and Future Of Mitochondrial Genomics: Have We Sequenced Enough Mtdnas? Brief. Funct. Genom. 2015, 15, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Embley, T.; Martin, W. Eukaryotic Evolution, Changes and Challenges. Nature 2006, 440, 623–630. [Google Scholar] [CrossRef]
- Degli Esposti, M.; Chouaia, B.; Comandatore, F.; Crotti, E.; Sassera, D.; Lievens, P.M.; Daffonchio, D.; Bandi, C. Evolution of mitochondria reconstructed from the energy metabolism of living bacteria. PLoS ONE 2014, 5, e96566. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef]
- Akbari, M.; Nilsen, H.L.; Montaldo, N.P. Dynamic features of human mitochondrial DNA maintenance and transcription. Front. Cell Dev. Biol. 2022, 10, 984245. [Google Scholar] [CrossRef]
- Lee, E.Y.; Lee, H.Y.; Oh, S.Y.; Jung, S.E.; Yang, I.S.; Lee, Y.H.; Yang, W.I.; Shin, K.J. Massively parallel sequencing of the entire control region and targeted coding region SNPs of degraded mtDNA using a simplified library preparation method. Forensic Sci. Int. Genet. 2016, 22, 37–43. [Google Scholar] [CrossRef]
- Vinueza-Espinosa, D.C.; Cuesta-Aguirre, D.R.; Malgosa, A.; Santos, C. Mitochondrial DNA control region typing from highly degraded skeletal remains by single-multiplex next-generation sequencing. Electrophoresis 2023. [Google Scholar] [CrossRef]
- Bodner, M.; Perego, U.A.; Gomez, J.E.; Cerda-Flores, R.M.; Rambaldi Migliore, N.; Woodward, S.R.; Parson, W.; Achilli, A. The Mitochondrial DNA Landscape of Modern Mexico. Genes 2021, 12, 1453. [Google Scholar] [CrossRef]
- Cann, R.L.; Stoneking, M.; Wilson, A.C. Mitochondrial DNA and human evolution. Nature 1987, 325, 31–36. [Google Scholar] [CrossRef]
- Torroni, A.; Schurr, T.G.; Yang, C.C.; Szathmary, E.J.; Williams, R.C.; Schanfield, M.S.; Troup, G.A.; Knowler, W.C.; Lawrence, D.N.; Weiss, K.M. Native American mitochondrial DNA analysis indicates that the Amerind and the Nadene populations were founded by two independent migrations. Genetics 1992, 130, 153–162. [Google Scholar] [CrossRef] [PubMed]
- García-Olivares, V.; Muñoz-Barrera, A.; Lorenzo-Salazar, J.M.; Zaragoza-Trello, C.; Rubio-Rodríguez, L.A.; Díaz-de Usera, A.; Jáspez, D.; Iñigo-Campos, A.; González-Montelongo, R.; Flores, C. A benchmarking of human mitochondrial DNA haplogroup classifiers from whole-genome and whole-exome sequence data. Sci. Rep. 2021, 11, 20510. [Google Scholar] [CrossRef] [PubMed]
- van Oven, M.; Kayser, M. Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum. Mutat. 2009, 30, E386–E394. [Google Scholar] [CrossRef]
- Qiagen Digital Insights. Available online: https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-clc-microbial-genomics-module/ (accessed on 12 July 2023).
- Lott, M.T.; Leipzig, J.N.; Derbeneva, O.; Xie, H.M.; Chalkia, D.; Sarmady, M.; Procaccio, V.; Wallace, D.C. mtDNA Variation and Analysis Using Mitomap and Mitomaster. Curr. Protoc. Bioinform. 2013, 44, 1.23.1–1.23.26. [Google Scholar] [CrossRef] [PubMed]
- MITOMASTER. Available online: https://www.mitomap.org/foswiki/bin/view/MITOMASTER/WebHome (accessed on 12 July 2023).
- Schönherr, S.; Weissensteiner, H.; Kronenberg, F.; Forer, L. Haplogrep 3—An interactive haplogroup classification and analysis platform. Nucleic Acids Res. 2023, 51, W263–W268. [Google Scholar] [CrossRef] [PubMed]
- Halogrep 3. Available online: https://haplogrep.i-med.ac.at/ (accessed on 12 July 2023).
- Behar, D.M.; van Oven, M.; Rosset, S.; Metspalu, M.; Loogväli, E.L.; Silva, N.M.; Kivisild, T.; Torroni, A.; Villems, R. A “Copernican” reassessment of the human mitochondrial DNA tree from its root. Am. J. Hum. Genet. 2012, 90, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Ingman, M.; Gyllensten, U. Mitochondrial genome variation and evolutionary history of Australian and New Guinean aborigines. Genome Res. 2003, 13, 1600–1606. [Google Scholar] [CrossRef] [PubMed]
- Nagle, N.; van Oven, M.; Wilcox, S.; van Holst Pellekaan, S.; Tyler-Smith, C.; Xue, Y.; Ballantyne, K.N.; Wilcox, L.; Papac, L.; Cooke, K.; et al. Aboriginal Australian mitochondrial genome variation—An increased understanding of population antiquity and diversity. Sci. Rep. 2017, 7, 43041. [Google Scholar] [CrossRef]
- Dryomov, S.V.; Nazhmidenova, A.M.; Starikovskaya, E.B.; Shalaurova, S.A.; Rohland, N.; Mallick, S.; Bernardos, R.; Derevianko, A.P.; Reich, D.; Sukernik, R.I. Mitochondrial genome diversity on the Central Siberian Plateau with particular reference to the prehistory of northernmost Eurasia. PLoS ONE 2021, 16, e0244228. [Google Scholar] [CrossRef]
- Wielgus, K.; Danielewski, M.; Walkowiak, J. Svante Pääbo, reader of the Neanderthal genome. Acta Physiol. 2023, 237, e13902. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Robles, A. Dental evolutionary rates and its implications for the Neanderthal-modern human divergence. Sci. Adv. 2019, 5, eaaw1268. [Google Scholar] [CrossRef]
- Cabrera, V.M.; Marrero, P.; Abu-Amero, K.K.; Larruga, J.M. Carriers of mitochondrial DNA macrohaplogroup L3 basal lineages migrated back to Africa from Asia around 70,000 years ago. BMC Evol. Biol. 2018, 18, 98. [Google Scholar] [CrossRef] [PubMed]
- Rito, T.; Vieira, D.; Silva, M.; Conde-Sousa, E.; Pereira, L.; Mellars, P.; Richards, M.B.; Soares, P. A dispersal of Homo sapiens from southern to eastern Africa immediately preceded the out-of-Africa migration. Sci. Rep. 2019, 9, 4728. [Google Scholar] [CrossRef] [PubMed]
- Bentley, D.R.; Balasubramanian, S.; Swerdlow, H.P.; Smith, G.P.; Milton, J.; Brown, C.G.; Hall, K.P.; Evers, D.J.; Barnes, C.L.; Bignell, H.R.; et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008, 456, 53–59. [Google Scholar] [CrossRef]
- Head, S.R.; Komori, H.K.; LaMere, S.A.; Whisenant, T.; Van Nieuwerburgh, F.; Salomon, D.R.; Ordoukhanian, P. Library construction for next-generation sequencing: Overviews and challenges. BioTechniques 2014, 56, 61–77. [Google Scholar] [CrossRef]
- Bairoch, A. The Cellosaurus, a Cell-Line Knowledge Resource. J. Biomol. Tech. JBT 2018, 29, 25–38. [Google Scholar] [CrossRef]
- Cellosaurus. Available online: https://www.cellosaurus.org (accessed on 12 July 2023).
- Wen, B.; Xie, X.; Gao, S.; Li, H.; Shi, H.; Song, X.; Qian, T.; Xiao, C.; Jin, J.; Su, B.; et al. Analyses of genetic structure of Tibeto-Burman populations reveals sex-biased admixture in southern Tibeto-Burmans. Am. J. Hum. Genet. 2004, 74, 856–865. [Google Scholar] [CrossRef]
- Johnson, J.A.; Moore, B.; Hwang, E.K.; Hickner, A.; Yeo, H. The accuracy of race & ethnicity data in US based healthcare databases: A systematic review. Am. J. Surg. 2023, in press. [CrossRef]
- Hsieh, M.C.; Pareti, L.A.; Chen, V.W. Using NAPIIA to improve the accuracy of Asian race codes in registry data. J. Regist. Manag. 2011, 38, 190–195. [Google Scholar]
- Cardena, M.M.; Ribeiro-Dos-Santos, A.; Santos, S.; Mansur, A.J.; Pereira, A.C.; Fridman, C. Assessment of the relationship between self-declared ethnicity, mitochondrial haplogroups and genomic ancestry in Brazilian individuals. PLoS ONE 2013, 8, e62005. [Google Scholar] [CrossRef] [PubMed]
- Cook, L.A.; Sachs, J.; Weiskopf, N.G. The quality of social determinants data in the electronic health record: A systematic review. J. Am. Med. Inform. Assoc. JAMIA 2021, 29, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.W.; Ball, A.L.; Chadwick, A.E.; Alfirevic, A. The Role of Mitochondrial DNA Variation in Drug Response: A Systematic Review. Front. Genet. 2021, 12, 698825. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Shi, J.; Stein, R.; Liu, X.; Baldassano, R.N.; Forrest, C.B.; Chen, Y.; Huang, J. Missing data matter: An empirical evaluation of the impacts of missing EHR data in comparative effectiveness research. J. Am. Med. Inform. Assoc. JAMIA 2023, 30, 1246–1256. [Google Scholar] [CrossRef]
- WorldAtlas Continents. Available online: https://www.worldatlas.com/continents (accessed on 12 July 2023).
- United Nations Statistical Division-Geographic Regions. Available online: https://unstats.un.org/unsd/methodology/m49/ (accessed on 12 July 2023).
- Wallace, D.C.; Chalkia, D. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a021220. [Google Scholar] [CrossRef]
- Vilar, M.G.; Chan, C.W.; Santos, D.R.; Lynch, D.; Spathis, R.; Garruto, R.M.; Lum, J.K. The origins and genetic distinctiveness of the Chamorros of the Marianas Islands: An mtDNA perspective. Am. J. Hum. Biol. Off. J. Hum. Biol. Counc. 2013, 25, 116–122. [Google Scholar] [CrossRef]
- Qiagen CLC Genomics Workbench System Requirements. Available online: https://digitalinsights.qiagen.com/technical-support/system-requirements/ (accessed on 12 July 2023).
- Shen-Gunther, J.; Xia, Q.; Stacey, W.; Asusta, H.B. Molecular Pap Smear: Validation of HPV Genotype and Host Methylation Profiles of ADCY8, CDH8, and ZNF582 as a Predictor of Cervical Cytopathology. Front. Microbiol. 2020, 11, 595902. [Google Scholar] [CrossRef]
- How to BLAST Guide. National Center for Biotechnology Information. Available online: https://ftp.ncbi.nlm.nih.gov/pub/factsheets/HowTo_BLASTGuide.pdf (accessed on 21 August 2023).









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen-Gunther, J.; Gunther, R.S.; Cai, H.; Wang, Y. A Customized Human Mitochondrial DNA Database (hMITO DB v1.0) for Rapid Sequence Analysis, Haplotyping and Geo-Mapping. Int. J. Mol. Sci. 2023, 24, 13505. https://doi.org/10.3390/ijms241713505
Shen-Gunther J, Gunther RS, Cai H, Wang Y. A Customized Human Mitochondrial DNA Database (hMITO DB v1.0) for Rapid Sequence Analysis, Haplotyping and Geo-Mapping. International Journal of Molecular Sciences. 2023; 24(17):13505. https://doi.org/10.3390/ijms241713505
Chicago/Turabian StyleShen-Gunther, Jane, Rutger S. Gunther, Hong Cai, and Yufeng Wang. 2023. "A Customized Human Mitochondrial DNA Database (hMITO DB v1.0) for Rapid Sequence Analysis, Haplotyping and Geo-Mapping" International Journal of Molecular Sciences 24, no. 17: 13505. https://doi.org/10.3390/ijms241713505