
SAM/SAH Mediates Parental Folate Deficiency-Induced Neural Cell Apoptosis in Neonatal Rat Offspring: The Expression of Bcl-2, Bax, and Caspase-3
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
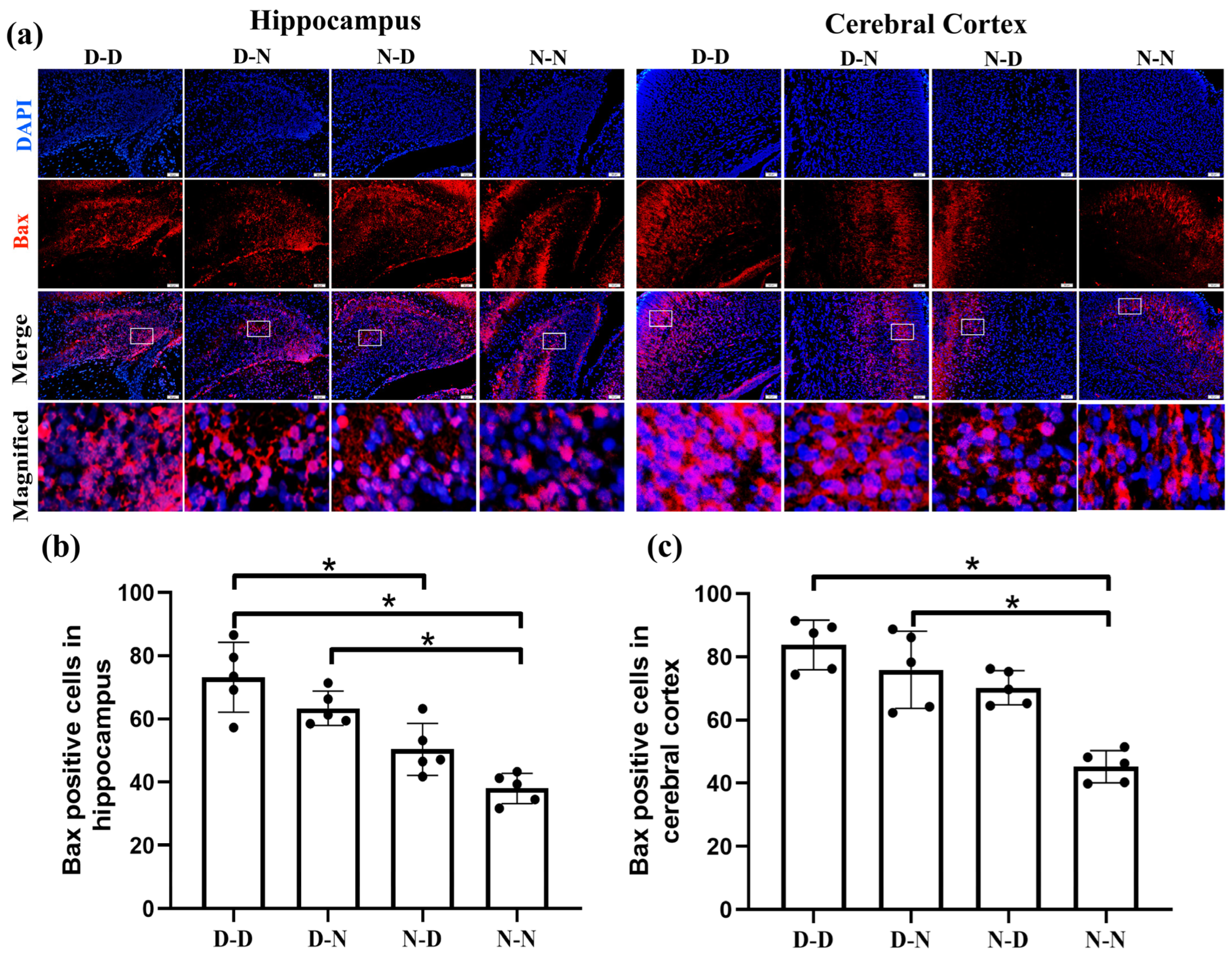
2.1. Parental Folate Deficiency Decreased the Expression of Bcl-2 and Increased the Expression of Bax of Neural Cells in the Brain of Neonatal Offspring Rats
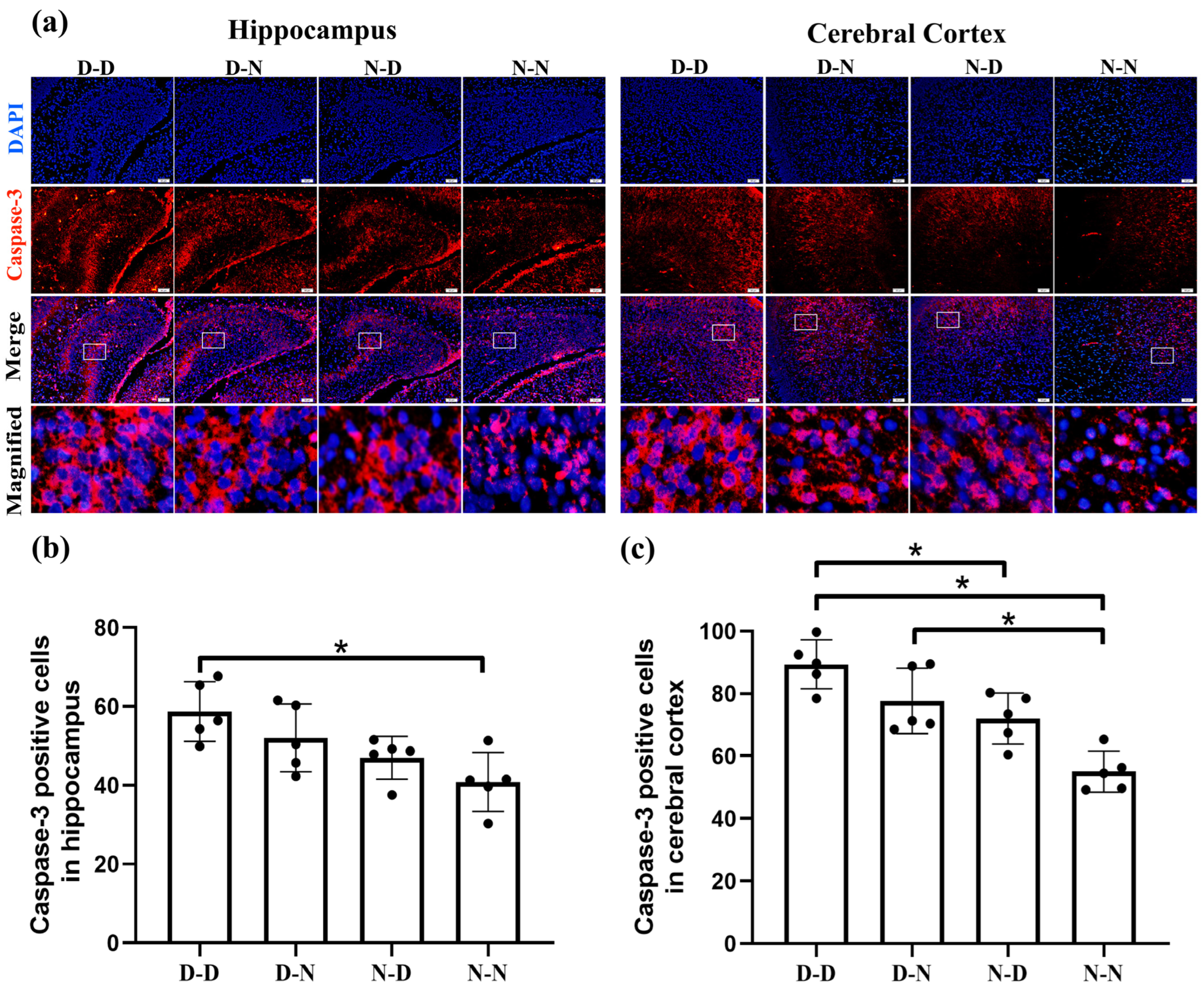
2.2. Parental Folate Deficiency Increased Expression of Caspase-3 of Neural Cells in the Brain of Neonatal Offspring Rats
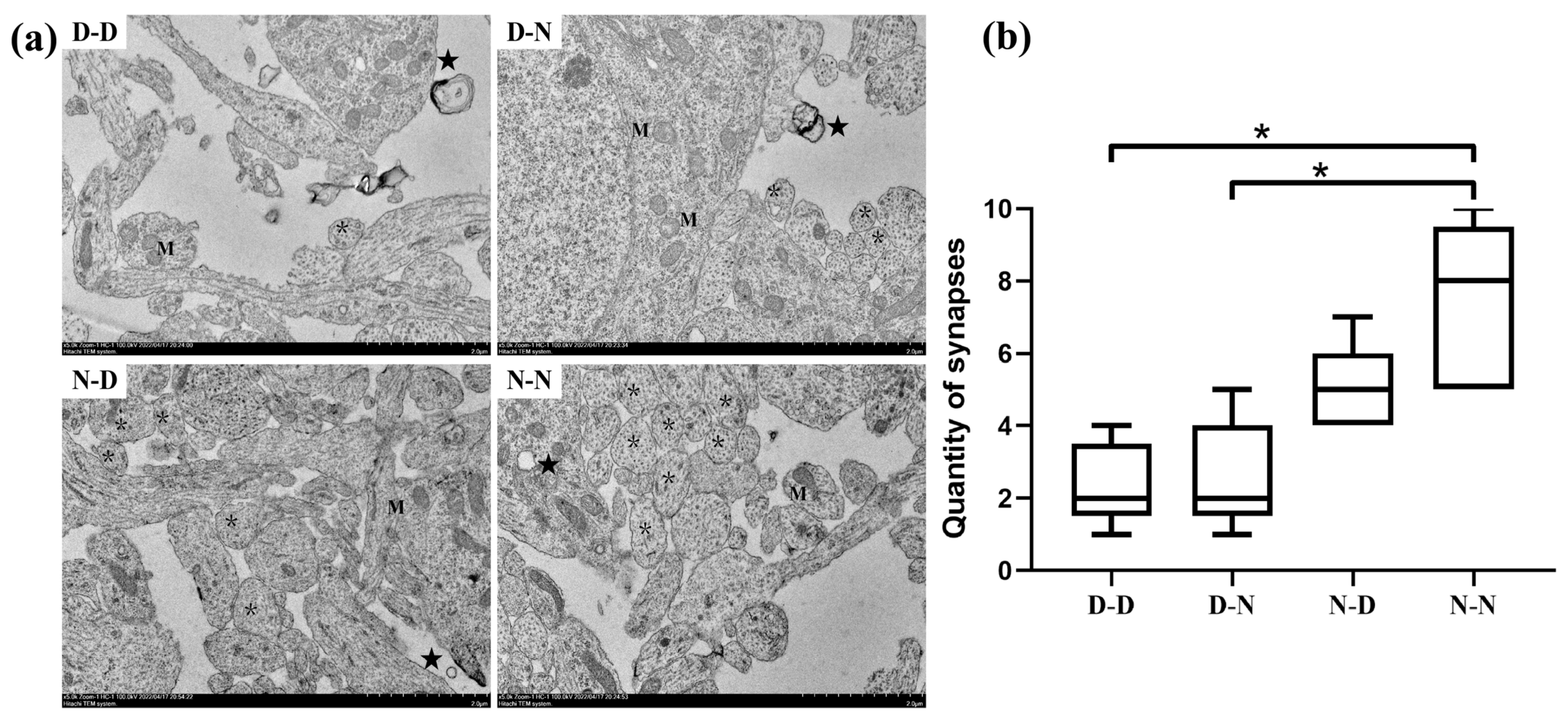
2.3. Parental Folate Deficiency Induced Pathological Changes in the Ultrastructure in Neuronal Cell Bodies of Neonatal Offspring Brain
2.4. SAM and SAH Levels in the Brain Tissue of Offspring Neonatal Rats
3. Discussion
4. Materials and Methods
4.1. Animals and Dietary Treatment
4.2. Immunofluorescence Analysis
4.3. Transmission Electron Microscopy
4.4. SAM and SAH in Brain Tissue Detected by High-Performance Liquid Chromatography (HPLC)
4.5. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Strobbe, S.; Van Der Straeten, D. Folate biofortification in food crops. Curr. Opin. Biotechnol. 2017, 44, 202–211. [Google Scholar] [CrossRef] [PubMed]
- McNulty, H.; Rollins, M.; Cassidy, T.; Caffrey, A.; Marshall, B.; Dornan, J.; McLaughlin, M.; McNulty, B.A.; Ward, M.; Strain, J.J.; et al. Effect of continued folic acid supplementation beyond the first trimester of pregnancy on cognitive performance in the child: A follow-up study from a randomized controlled trial (FASSTT Offspring Trial). BMC Med. 2019, 17, 196. [Google Scholar] [CrossRef] [PubMed]
- McCoy, C.R.; Jackson, N.L.; Brewer, R.L.; Moughnyeh, M.M.; Smith, D.L., Jr.; Clinton, S.M. A paternal methyl donor depleted diet leads to increased anxiety- and depression-like behavior in adult rat offspring. Biosci. Rep. 2018, 38, BSR20180730. [Google Scholar] [CrossRef] [PubMed]
- Hoek, J.; Koster, M.P.H.; Schoenmakers, S.; Willemsen, S.P.; Koning, A.H.J.; Steegers, E.A.P.; Steegers-Theunissen, R.P.M. Does the father matter? The association between the periconceptional paternal folate status and embryonic growth. Fertil. Steril. 2019, 111, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Lennox, A.L.; Mao, H.; Silver, D.L. RNA on the brain: Emerging layers of post-transcriptional regulation in cerebral cortex development. Wiley Interdiscip. Rev. Dev. Biol. 2018, 7, e290. [Google Scholar] [CrossRef]
- Yang, Z.; Geng, Y.; Yao, Z.; Jia, H.; Bai, Y.; Wang, W. Spatiotemporal Expression of Bcl-2/Bax and Neural Cell Apoptosis in the Developing Lumbosacral Spinal Cord of Rat Fetuses with Anorectal Malformations. Neurochem. Res. 2017, 42, 3160–3169. [Google Scholar] [CrossRef] [PubMed]
- Gopisetty, G.; Ramachandran, K.; Singal, R. DNA methylation and apoptosis. Mol. Immunol. 2006, 43, 1729–1740. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Fairbrother, W.J.; Leverson, J.D.; Souers, A.J. From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat. Rev. Drug Discov. 2017, 16, 273–284. [Google Scholar] [CrossRef]
- Kim, J.S.; Kim, J.M.; Jung, H.C.; Song, I.S. Caspase-3 activity and expression of Bcl-2 family in human neutrophils by Helicobacter pylori water-soluble proteins. Helicobacter 2001, 6, 207–215. [Google Scholar] [CrossRef]
- Cheng, E.H.; Kirsch, D.G.; Clem, R.J.; Ravi, R.; Kastan, M.B.; Bedi, A.; Ueno, K.; Hardwick, J.M. Conversion of Bcl-2 to a Bax-like death effector by caspases. Science 1997, 278, 1966–1968. [Google Scholar] [CrossRef]
- Shao, Y.; Tan, B.; Shi, J.; Zhou, Q. Methotrexate induces astrocyte apoptosis by disrupting folate metabolism in the mouse juvenile central nervous system. Toxicol. Lett. 2019, 301, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Jadavji, N.M.; Deng, L.; Malysheva, O.; Caudill, M.A.; Rozen, R. MTHFR deficiency or reduced intake of folate or choline in pregnant mice results in impaired short-term memory and increased apoptosis in the hippocampus of wild-type offspring. Neuroscience 2015, 300, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Blaise, S.A.; Nédélec, E.; Schroeder, H.; Alberto, J.M.; Bossenmeyer-Pourié, C.; Guéant, J.L.; Daval, J.L. Gestational vitamin B deficiency leads to homocysteine-associated brain apoptosis and alters neurobehavioral development in rats. Am. J. Pathol. 2007, 170, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Bravo, A.C.; Aguilera, M.N.L.; Marziali, N.R.; Moritz, L.; Wingert, V.; Klotz, K.; Schumann, A.; Grünert, S.C.; Spiekerkoetter, U.; Berger, U.; et al. Analysis of S-Adenosylmethionine and S-Adenosylhomocysteine: Method Optimisation and Profiling in Healthy Adults upon Short-Term Dietary Intervention. Metabolites 2022, 12, 373. [Google Scholar] [CrossRef] [PubMed]
- Halsted, C.H.; Villanueva, J.A.; Devlin, A.M.; Niemelä, O.; Parkkila, S.; Garrow, T.A.; Wallock, L.M.; Shigenaga, M.K.; Melnyk, S.; James, S.J. Folate deficiency disturbs hepatic methionine metabolism and promotes liver injury in the ethanol-fed micropig. Proc. Natl. Acad. Sci. USA 2002, 99, 10072–10077. [Google Scholar] [CrossRef]
- Menezo, Y.; Elder, K.; Clement, A.; Clement, P. Folic Acid, Folinic Acid, 5 Methyl TetraHydroFolate Supplementation for Mutations That Affect Epigenesis through the Folate and One-Carbon Cycles. Biomolecules 2022, 12, 197. [Google Scholar] [CrossRef]
- Tuo, L.J.; Song, X.Y.; Zhu, Y.Y.; He, H.N.; Song, Y.P.; Chen, D.Z.; Zheng, X.M.; Zhang, H.; Xu, D.X. Gestational folic acid supplement prevents vitamin D deficiency-induced depression-like behavior by reversing cortical DNA hypomethylation in adult offspring. J. Steroid Biochem. Mol. Biol. 2023, 231, 106313. [Google Scholar] [CrossRef]
- Yang, X.; Fan, G.; Wang, Z.; Li, S.; Qin, H.; Wang, Y.; Ma, X.; Ji, W.; Wang, Y. Study on the relationship between genetic polymorphism of reductive folic acid carrier and the risk of neural tube defects. Child’s Nerv. Syst. 2023, 39, 1711–1718. [Google Scholar] [CrossRef]
- Bai, J.; Gao, Y.; Chen, L.; Yin, Q.; Lou, F.; Wang, Z.; Xu, Z.; Zhou, H.; Li, Q.; Cai, W.; et al. Identification of a natural inhibitor of methionine adenosyltransferase 2A regulating one-carbon metabolism in keratinocytes. EBioMedicine 2019, 39, 575–590. [Google Scholar] [CrossRef]
- de Queiroz, K.B.; Cavalcante-Silva, V.; Lopes, F.L.; Rocha, G.A.; D’Almeida, V.; Coimbra, R.S. Vitamin B(12) is neuroprotective in experimental pneumococcal meningitis through modulation of hippocampal DNA methylation. J. Neuroinflamm. 2020, 17, 96. [Google Scholar] [CrossRef]
- Islam, A.; Shaukat, Z.; Newman, D.L.; Hussain, R.; Ricos, M.G.; Dibbens, L.; Gregory, S.L. Chromosomal Instability Causes Sensitivity to Polyamines and One-Carbon Metabolism. Metabolites 2023, 13, 642. [Google Scholar] [CrossRef] [PubMed]
- Bekdash, R.A. Methyl Donors, Epigenetic Alterations, and Brain Health: Understanding the Connection. Int. J. Mol. Sci. 2023, 24, 2346. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.; Shea, T.B. Dietary and genetically-induced oxidative stress alter tau phosphorylation: Influence of folate and apolipoprotein E deficiency. J. Alzheimer’s Dis. 2006, 9, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Ren, Q.; Zhang, G.; Dong, C.; Li, Z.; Zhou, D.; Huang, L.; Li, W.; Huang, G.; Yan, J. Parental Folate Deficiency Inhibits Proliferation and Increases Apoptosis of Neural Stem Cells in Rat Offspring: Aggravating Telomere Attrition as a Potential Mechanism. Nutrients 2023, 15, 2843. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Suo, L.; Liu, Y.; Li, H.; Xue, W. Protective effects of ginsenoside Rg1 against oxygen-glucose-deprivation-induced apoptosis in neural stem cells. J. Neurol. Sci. 2017, 373, 107–112. [Google Scholar] [CrossRef]
- Virdi, S.; Jadavji, N.M. The Impact of Maternal Folates on Brain Development and Function after Birth. Metabolites 2022, 12, 876. [Google Scholar] [CrossRef]
- Craciunescu, C.N.; Brown, E.C.; Mar, M.H.; Albright, C.D.; Nadeau, M.R.; Zeisel, S.H. Folic acid deficiency during late gestation decreases progenitor cell proliferation and increases apoptosis in fetal mouse brain. J. Nutr. 2004, 134, 162–166. [Google Scholar] [CrossRef]
- Xiao, S.; Hansen, D.K.; Horsley, E.T.; Tang, Y.S.; Khan, R.A.; Stabler, S.P.; Jayaram, H.N.; Antony, A.C. Maternal folate deficiency results in selective upregulation of folate receptors and heterogeneous nuclear ribonucleoprotein-E1 associated with multiple subtle aberrations in fetal tissues. Birth Defects Research. Part A Clin. Mol. Teratol. 2005, 73, 6–28. [Google Scholar] [CrossRef]
- Li, W.; Li, Z.; Zhou, D.; Zhang, X.; Yan, J.; Huang, G. Maternal folic acid deficiency stimulates neural cell apoptosis via miR-34a associated with Bcl-2 in the rat foetal brain. Int. J. Dev. Neurosci. 2019, 72, 6–12. [Google Scholar] [CrossRef]
- Harlan De Crescenzo, A.; Panoutsopoulos, A.A.; Tat, L.; Schaaf, Z.; Racherla, S.; Henderson, L.; Leung, K.Y.; Greene, N.D.E.; Green, R.; Zarbalis, K.S. Deficient or Excess Folic Acid Supply During Pregnancy Alter Cortical Neurodevelopment in Mouse Offspring. Cereb. Cortex 2021, 31, 635–649. [Google Scholar] [CrossRef]
- Wang, X.; Li, W.; Li, S.; Yan, J.; Wilson, J.X.; Huang, G. Maternal Folic Acid Supplementation During Pregnancy Improves Neurobehavioral Development in Rat Offspring. Mol. Neurobiol. 2018, 55, 2676–2684. [Google Scholar] [CrossRef] [PubMed]
- Glier, M.B.; Green, T.J.; Devlin, A.M. Methyl nutrients, DNA methylation, and cardiovascular disease. Mol. Nutr. Food Res. 2014, 58, 172–182. [Google Scholar] [CrossRef]
- Irwin, R.E.; Pentieva, K.; Cassidy, T.; Lees-Murdock, D.J.; McLaughlin, M.; Prasad, G.; McNulty, H.; Walsh, C.P. The interplay between DNA methylation, folate and neurocognitive development. Epigenomics 2016, 8, 863–879. [Google Scholar] [CrossRef] [PubMed]
- McGarel, C.; Pentieva, K.; Strain, J.J.; McNulty, H. Emerging roles for folate and related B-vitamins in brain health across the lifecycle. Proc. Nutr. Soc. 2015, 74, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Hong, K.; Lee, J.H.; Lee, S.; Chang, N. Effect of folate deficiency on placental DNA methylation in hyperhomocysteinemic rats. J. Nutr. Biochem. 2009, 20, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Guerra-Shinohara, E.M.; Morita, O.E.; Peres, S.; Pagliusi, R.A.; Sampaio Neto, L.F.; D’Almeida, V.; Irazusta, S.P.; Allen, R.H.; Stabler, S.P. Low ratio of S-adenosylmethionine to S-adenosylhomocysteine is associated with vitamin deficiency in Brazilian pregnant women and newborns. Am. J. Clin. Nutr. 2004, 80, 1312–1321. [Google Scholar] [CrossRef]
- Coppen, A.; Bolander-Gouaille, C. Treatment of depression: Time to consider folic acid and vitamin B12. J. Psychopharmacol. 2005, 19, 59–65. [Google Scholar] [CrossRef]
- Barve, A.; Vega, A.; Shah, P.P.; Ghare, S.; Casson, L.; Wunderlich, M.; Siskind, L.J.; Beverly, L.J. Perturbation of Methionine/S-adenosylmethionine Metabolism as a Novel Vulnerability in MLL Rearranged Leukemia. Cells 2019, 8, 1322. [Google Scholar] [CrossRef]
- Yang, A.; Sun, Y.; Mao, C.; Yang, S.; Huang, M.; Deng, M.; Ding, N.; Yang, X.; Zhang, M.; Jin, S.; et al. Folate Protects Hepatocytes of Hyperhomocysteinemia Mice From Apoptosis via Cystic Fibrosis Transmembrane Conductance Regulator (CFTR)-Activated Endoplasmic Reticulum Stress. J. Cell. Biochem. 2017, 118, 2921–2932. [Google Scholar] [CrossRef]
- Sertorio, M.N.; César, H.; de Souza, E.A.; Mennitti, L.V.; Santamarina, A.B.; De Souza Mesquita, L.M.; Jucá, A.; Casagrande, B.P.; Estadella, D.; Aguiar, O., Jr.; et al. Parental High-Fat High-Sugar Diet Intake Programming Inflammatory and Oxidative Parameters of Reproductive Health in Male Offspring. Front. Cell Dev. Biol. 2022, 10, 867127. [Google Scholar] [CrossRef]
- De-Regil, L.M.; Fernández-Gaxiola, A.C.; Dowswell, T.; Peña-Rosas, J.P. Effects and safety of periconceptional folate supplementation for preventing birth defects. Cochrane Database Syst. Rev. 2010, 12, Cd007950. [Google Scholar] [CrossRef]
- Naninck, E.F.G.; Stijger, P.C.; Brouwer-Brolsma, E.M. The Importance of Maternal Folate Status for Brain Development and Function of Offspring. Adv. Nutr. 2019, 10, 502–519. [Google Scholar] [CrossRef] [PubMed]
- Caffrey, A.; McNulty, H.; Rollins, M.; Prasad, G.; Gaur, P.; Talcott, J.B.; Witton, C.; Cassidy, T.; Marshall, B.; Dornan, J.; et al. Effects of maternal folic acid supplementation during the second and third trimesters of pregnancy on neurocognitive development in the child: An 11-year follow-up from a randomised controlled trial. BMC Med. 2021, 19, 73. [Google Scholar] [CrossRef] [PubMed]
- Bekdash, R.A. Early Life Nutrition and Mental Health: The Role of DNA Methylation. Nutrients 2021, 13, 3111. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, X.; Wang, Y.; Zhao, X.; Zhang, L.; Li, J.; Zhang, Y.; Wang, P.; Liang, H. Dietary Folic Acid Supplementation Attenuates Maternal High-Fat Diet-Induced Fetal Intrauterine Growth Retarded via Ameliorating Placental Inflammation and Oxidative Stress in Rats. Nutrients 2023, 15, 3263. [Google Scholar] [CrossRef] [PubMed]
- Priliani, L.; Oktavianthi, S.; Prado, E.L.; Malik, S.G.; Shankar, A.H. Maternal biomarker patterns for metabolism and inflammation in pregnancy are influenced by multiple micronutrient supplementation and associated with child biomarker patterns and nutritional status at 9–12 years of age. PLoS ONE 2020, 15, e0216848. [Google Scholar] [CrossRef]
- Lambrot, R.; Xu, C.; Saint-Phar, S.; Chountalos, G.; Cohen, T.; Paquet, M.; Suderman, M.; Hallett, M.; Kimmins, S. Low paternal dietary folate alters the mouse sperm epigenome and is associated with negative pregnancy outcomes. Nat. Commun. 2013, 4, 2889. [Google Scholar] [CrossRef]
- Mahajan, A.; Sapehia, D.; Thakur, S.; Mohanraj, P.S.; Bagga, R.; Kaur, J. Effect of imbalance in folate and vitamin B12 in maternal/parental diet on global methylation and regulatory miRNAs. Sci. Rep. 2019, 9, 17602. [Google Scholar] [CrossRef]
- Hong, Y.; Gu, L.; Huang, Y.; Jin, F.; Chen, Q.; Tao, H.; Zhang, F.; Zhu, Y.; Xu, J.; Li, G. Reproductive performance in SD rats and offspring growth and development. Occupation 2014, 30, 3215–3217. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, Q.; Zhang, G.; Yan, R.; Zhou, D.; Huang, L.; Zhang, Q.; Li, W.; Huang, G.; Li, Z.; Yan, J. SAM/SAH Mediates Parental Folate Deficiency-Induced Neural Cell Apoptosis in Neonatal Rat Offspring: The Expression of Bcl-2, Bax, and Caspase-3. Int. J. Mol. Sci. 2023, 24, 14508. https://doi.org/10.3390/ijms241914508
Ren Q, Zhang G, Yan R, Zhou D, Huang L, Zhang Q, Li W, Huang G, Li Z, Yan J. SAM/SAH Mediates Parental Folate Deficiency-Induced Neural Cell Apoptosis in Neonatal Rat Offspring: The Expression of Bcl-2, Bax, and Caspase-3. International Journal of Molecular Sciences. 2023; 24(19):14508. https://doi.org/10.3390/ijms241914508
Chicago/Turabian StyleRen, Qinghan, Guoquan Zhang, Ruiting Yan, Dezheng Zhou, Li Huang, Qianwen Zhang, Wen Li, Guowei Huang, Zhenshu Li, and Jing Yan. 2023. "SAM/SAH Mediates Parental Folate Deficiency-Induced Neural Cell Apoptosis in Neonatal Rat Offspring: The Expression of Bcl-2, Bax, and Caspase-3" International Journal of Molecular Sciences 24, no. 19: 14508. https://doi.org/10.3390/ijms241914508