
RNA-Based Strategies for Cancer Therapy: In Silico Design and Evaluation of ASOs for Targeted Exon Skipping

 , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. Development of an Integrated Computational Procedure to Support the Design of ES-Based Therapeutic Strategies in Cancer
2.1.1. Classification of a Gene of Interest as an Oncogene or Tumor Suppressor
2.1.2. Identification of Candidate Exons to Be Targeted for Skipping
2.1.3. Analysis of the Mutational Profile
2.1.4. Design and Evaluation of Ad Hoc ASOs to Induce Desired ES Products
2.2. Application of the Bioinformatic Pipeline to Frequently Mutated Cancer Genes
2.2.1. Classifying Role in Cancer for Selected Genes
2.2.2. Identifying Potential Exon Targets for ES in Selected Cancer Genes
2.2.3. Analyzing Exon Mutation Profile of the Selected Cancer Gene Set
2.2.4. Design and Evaluation of the Best Candidate ASO Sequences
2.3. Proof-of-Concept Case Studies
2.3.1. Detecting High-Potential Cancer Genes for Effective Targeted ES Intervention
2.3.2. Harnessing the Potential of Our Pipeline on Well-Studied Cancer Genes: BRAF and TP53
3. Discussion
4. Materials and Methods
- Basic melting temperature (Tm) (°C),
- Salt-adjusted melting temperature (Tm) (°C),
- Nearest-neighbor melting temperature (TmNN) (°C),
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ASO | Antisense oligonucleotide |
| BMD | Becker muscular dystrophy |
| CDS | Coding DNA sequence |
| DMD | Duchenne muscular dystrophy |
| ES | Exon skipping |
| ESE | Exonic splicing enhancer |
| ESS | Exonic splicing silencers |
| FDA | Food and Drug Administration |
| ISE | Intronic splicing enhancer |
| ISS | Intronic splicing silencers |
| NMD | Nonsense-Mediated Decay |
| ONG | Oncogene |
| PTC | Premature termination codon |
| snRNA | Small nuclear RNA |
| TSG | Tumor suppressor gene |
Appendix A

Appendix B

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ASO Drug | Target Exon Ensembl ID | Drug Sequence (Length) Pipeline Matching Hit (Length) |
|---|---|---|
| Eteplirsen | Exon 51 (ENSE00003669071.1) | CTCCAACATCAAGGAAGATGGCATTTCT (28 nt) CTCCAACATCAAGGAAGATGGCATTTCT (25 nt) |
| Golodirsen | Exon 53 (ENSE00001258577.1) | GTTGCCTCCGGTTCTGAAGGTGTTCNNN (25 nt) GTTGCCTCCGGTTCTGAAGGTGTTCNNN (25 nt) |
| Viltolarsen | Exon 53 (ENSE00001258577.1) | CCTCCGGTTCTGAAGGTGTTCNNNTTGT (21 nt) CCTCCGGTTCTGAAGGTGTTCTTGTNNN (25 nt) |
| Casimersen | Exon 45 (ENSE00003988228.1) | GCTGCCCAATGCCATCCTGGAGTTCCTG (22 nt) GCTGCCCAATGCCATCCTGGAGTTCCTG (25 nt) |
Appendix C
| Gene | Exon ID | ASO ID | ASO Sequence |
|---|---|---|---|
| BRAF | ENSE00001907699.1 | H18D (10, ) | GGACAGGAAACGCACCAUAUCCCCC |
| H18D (9, ) | UGGACAGGAAACGCACCAUAUCCCC | ||
| H18D (8, ) | GUGGACAGGAAACGCACCAUAUCCC | ||
| H18D (7, ) | AGUGGACAGGAAACGCACCAUAUCC | ||
| ENSE00003485507.1 | H15D (11, ) | GCCUCAAUUCUUACCAUCCACAAAA | |
| H15D (10, ) | AGCCUCAAUUCUUACCAUCCACAAA | ||
| H15D (9, ) | UAGCCUCAAUUCUUACCAUCCACAA | ||
| H15D (8, ) | AUAGCCUCAAUUCUUACCAUCCACA | ||
| H15D (7, ) | AAUAGCCUCAAUUCUUACCAUCCAC | ||
| ENSE00003521664.1 | H12A (, 9) | CCACAUCACCUAAAAGGCAAUUGUU | |
| H12A (, 10) | GCCACAUCACCUAAAAGGCAAUUGU | ||
| H12A (, 11) | UGCCACAUCACCUAAAAGGCAAUUG | ||
| H12A (, 12) | CUGCCACAUCACCUAAAAGGCAAUU | ||
| H12A (, 13) | ACUGCCACAUCACCUAAAAGGCAAU | ||
| H12D (13, ) | ACACAAGCUCACCUGAGUACUCCUA | ||
| H12D (12, ) | CACACAAGCUCACCUGAGUACUCCU | ||
| H12D (11, ) | UCACACAAGCUCACCUGAGUACUCC | ||
| H12D (10, ) | UUCACACAAGCUCACCUGAGUACUC | ||
| H12D (9, ) | AUUCACACAAGCUCACCUGAGUACU | ||
| H12D (8, ) | AAUUCACACAAGCUCACCUGAGUAC | ||
| H12D (7, ) | UAAUUCACACAAGCUCACCUGAGUA | ||
| ENSE00003559218.1 | H11D (13, ) | UUACAUACUUACCAUGCCACUUUCC | |
| H11D (9, ) | CACAUUACAUACUUACCAUGCCACU | ||
| H11D (8, ) | CCACAUUACAUACUUACCAUGCCAC | ||
| H11D (7, ) | ACCACAUUACAUACUUACCAUGCCA | ||
| TP53 | ENSE00003670707.1 | H5A (, 9) | GCAAAACAUCUUGUUGAGGGCAGGG |
| H5A (, 10) | GGCAAAACAUCUUGUUGAGGGCAGG | ||
| H5A (, 11) | UGGCAAAACAUCUUGUUGAGGGCAG | ||
| ENSE00003625790.1 | H3D (13, ) | AGGGCAACUGACCGUGCAAGUCACA | |
| H3D (12, ) | CAGGGCAACUGACCGUGCAAGUCAC | ||
| H3D (11, ) | UCAGGGCAACUGACCGUGCAAGUCA | ||
| H3D (10, ) | CUCAGGGCAACUGACCGUGCAAGUC | ||
| H3D (9, ) | CCUCAGGGCAACUGACCGUGCAAGU |
References
- Chhikara, B.S.; Parang, K. Global Cancer Statistics 2022: The trends projection analysis. Chem. Biol. Lett. 2023, 10, 451. [Google Scholar]
- Tsimberidou, A.M.; Fountzilas, E.; Nikanjam, M.; Kurzrock, R. Review of precision cancer medicine: Evolution of the treatment paradigm. Cancer Treat. Rev. 2020, 86, 102019. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Telser, A. Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002; Volume 18, p. 289. [Google Scholar] [CrossRef]
- Chandrashekar, P.; Ahmadinejad, N.; Wang, J.; Sekulic, A.; Egan, J.B.; Asmann, Y.W.; Kumar, S.; Maley, C.; Liu, L. Somatic selection distinguishes oncogenes and tumor suppressor genes. Bioinformatics 2020, 36, 1712–1717. [Google Scholar] [CrossRef]
- Lyu, J.; Li, J.J.; Su, J.; Peng, F.; Chen, Y.E.; Ge, X.; Li, W. DORGE: Discovery of Oncogenes and tumoR suppressor genes using Genetic and Epigenetic features. Sci. Adv. 2020, 6, eaba6784. [Google Scholar] [CrossRef]
- Pavel, A.B.; Vasile, C.I. Identifying cancer type specific oncogenes and tumor suppressors using limited size data. J. Bioinform. Comput. Biol. 2016, 14, 1650031:1–1650031:16. [Google Scholar] [CrossRef]
- Forbes, S.A.; Beare, D.; Boutselakis, H.; Bamford, S.; Bindal, N.; Tate, J.G.; Cole, C.; Ward, S.; Dawson, E.; Ponting, L.; et al. COSMIC: Somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017, 45, D777–D783. [Google Scholar] [CrossRef]
- Sondka, Z.; Bamford, S.; Cole, C.G.; Ward, S.A.; Dunham, I.; Forbes, S.A. The COSMIC Cancer Gene Census: Describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 2018, 18, 696–705. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.; Sondka, Z.; Beare, D.; Bindal, N.; Boutselakis, H.; Cole, C.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed]
- Kumar-Sinha, C.; Chinnaiyan, A.M. Precision oncology in the age of integrative genomics. Nat. Biotechnol. 2018, 36, 46–60. [Google Scholar] [CrossRef]
- Duffy, M.J.; Crown, J. Drugging “undruggable” genes for cancer treatment: Are we making progress? Int. J. Cancer 2021, 148, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission Possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef]
- Priestley, P.; Baber, J.; Lolkema, M.P.; Steeghs, N.; de Bruijn, E.; Shale, C.; Duyvesteyn, K.; Haidari, S.; van Hoeck, A.; Onstenk, W.; et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 2019, 575, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Leroy, B.; Anderson, M.; Soussi, T. TP53 Mutations in Human Cancer: Database Reassessment and Prospects for the Next Decade. Hum. Mutat. 2014, 35, 672–688. [Google Scholar] [CrossRef] [PubMed]
- Coleman, N.; Rodon, J. Taking Aim at the Undruggable. Am. Soc. Clin. Oncol. 2021, 41, e145–e152. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, M.; Ashizawa, A.T. The Challenges and Strategies of Antisense Oligonucleotide Drug Delivery. Biomedicines 2021, 9, 433. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef] [PubMed]
- Setten, R.L.; Rossi, J.J.; Han, S.p. The current state and future directions of RNAi-based therapeutics. Nat. Rev. Drug Discov. 2019, 18, 421–446. [Google Scholar] [CrossRef]
- Xiong, H.; Veedu, R.N.; Diermeier, S.D. Recent Advances in Oligonucleotide Therapeutics in Oncology. Int. J. Mol. Sci. 2021, 22, 3295. [Google Scholar] [CrossRef]
- Havens, M.A.; Hastings, M.L. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016, 44, 6549–6563. [Google Scholar] [CrossRef]
- Will, C.L.; Luhrmann, R. Spliceosome Structure and Function. Cold Spring Harb. Perspect. Biol. 2011, 3, a003707. [Google Scholar] [CrossRef]
- Scotti, M.M.; Swanson, M.S. RNA mis-splicing in disease. Nat. Rev. Genet 2016, 17, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; van Ommen, G.J.B. Antisense-mediated exon skipping: A versatile tool with therapeutic and research applications. RNA 2007, 13, 1609–1624. [Google Scholar] [CrossRef] [PubMed]
- Leoni, G.; Le Pera, L.; Ferrè, F.; Raimondo, D.; Tramontano, A. Coding potential of the products of alternative splicing in human. Genome Biol. 2011, 12, R9. [Google Scholar] [CrossRef] [PubMed]
- Shimo, T.; Maruyama, R.; Yokota, T. Designing Effective Antisense Oligonucleotides for Exon Skipping; Springer: Berlin/Heidelberg, Germany, 2018. [Google Scholar] [CrossRef]
- Kim, Y. Drug Discovery Perspectives of Antisense Oligonucleotides. Biomol. Ther. 2023, 31, 241–252. [Google Scholar] [CrossRef]
- Lim, K.R.Q.; Maruyama, R.; Yokota, T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des. Dev. Ther. 2017, 11, 533–545. [Google Scholar]
- Roshmi, R.R.; Yokota, T. Viltolarsen for the treatment of Duchenne muscular dystrophy. Drugs Today 2019, 55, 627. [Google Scholar] [CrossRef]
- Anwar, S.; Yokota, T. Golodirsen for Duchenne muscular dystrophy. Drugs Today 2020, 56, 491. [Google Scholar] [CrossRef]
- Shirley, M. Casimersen: First Approval. Drugs 2021, 81, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.C.; Sheehan, D.W.; Prochoroff, A.; Birnkrant, D.J. Muscular Dystrophies. Clin. Chest Med. 2018, 39, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Bladen, C.L.; Salgado, D.; Monges, S.; Foncuberta, M.E.; Kekou, K.; Kosma, K.; Dawkins, H.; Lamont, L.; Roy, A.J.; Chamova, T.; et al. The TREAT-NMD DMD Global Database: Analysis of More than 7000 Duchenne Muscular Dystrophy Mutations. Hum. Mutat. 2015, 36, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Koenig, M.; Hoffman, E.P.; Bertelson, C.J.; Monaco, A.P.; Feener, C.; Kunkel, L.M. Complete cloning of the duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 1987, 50, 509–517. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Deutekom, J.C.T.V.; Fokkema, I.F.; Ommen, G.J.B.V.; Dunnen, J.T.D. Entries in the Leiden Duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 2006, 34, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Shen, L.; Zhang, Z.; Xie, X. Therapeutic Strategies for Duchenne Muscular Dystrophy: An Update. Genes 2020, 11, 837. [Google Scholar] [CrossRef] [PubMed]
- Monaco, A.P.; Bertelson, C.J.; Liechti-Gallati, S.; Moser, H.; Kunkel, L.M. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 1988, 2, 90–95. [Google Scholar] [CrossRef]
- M, M. Antisense Oligonucleotide-Mediated Exon-skipping Therapies: Precision Medicine Spreading from Duchenne Muscular Dystrophy. JMA J. 2021, 4, 232–240. [Google Scholar] [CrossRef]
- Echevarría, L.; Aupy, P.; Goyenvalle, A. Exon-skipping advances for Duchenne muscular dystrophy. Hum. Mol. Genet 2018, 27, R163–R172. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Hobson, L.; Perry, L.; Clark, B.; Heavey, S.; Haider, A.; Sridhar, A.; Shaw, G.; Kelly, J.; Freeman, A.; et al. Targeting the ERG oncogene with splice-switching oligonucleotides as a novel therapeutic strategy in prostate cancer. Br. J. Cancer 2020, 123, 1024–1032. [Google Scholar] [CrossRef]
- Ma, W.K.; Voss, D.M.; Scharner, J.; Costa, A.S.H.; Lin, K.T.; Jeon, H.Y.; Wilkinson, J.E.; Jackson, M.; Rigo, F.; Bennett, C.F.; et al. ASO-Based PKM Splice-Switching Therapy Inhibits Hepatocellular Carcinoma Growth. Cancer Res. 2022, 82, 900–915. [Google Scholar] [CrossRef]
- Scharnhorst, V.; van der Eb, A.J.; Jochemsen, A.G. WT1 proteins: Functions in growth and differentiation. Gene 2001, 273, 141–161. [Google Scholar] [CrossRef] [PubMed]
- Renshaw, J.; Orr, R.M.; Walton, M.I.; te Poele, R.; Williams, R.D.; Wancewicz, E.V.; Monia, B.P.; Workman, P.; Pritchard-Jones, K. Disruption of WT1 gene expression and exon 5 splicing following cytotoxic drug treatment: Antisense down-regulation of exon 5 alters target gene expression and inhibits cell survival. Mol. Cancer Ther. 2004, 3, 1467–1484. [Google Scholar] [CrossRef]
- Kibbe, W.A. OligoCalc: An online oligonucleotide properties calculator. Nucleic Acids Res. 2007, 35, 43–46. [Google Scholar] [CrossRef]
- Lorenz, R.; Bernhart, S.H.; zu Siederdissen, C.H.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Reuter, J.S.; Mathews, D.H. RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinform. 2010, 11, 129. [Google Scholar] [CrossRef] [PubMed]
- Piva, F.; Giulietti, M.; Burini, A.B.; Principato, G. SpliceAid 2: A database of human splicing factors expression data and RNA target motifs. Hum. Mutat. 2012, 33, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Sciabola, S.; Xi, H.; Cruz, D.; Cao, Q.; Lawrence, C.; Zhang, T.; Rotstein, S.; Hughes, J.D.; Caffrey, D.R.; Stanton, R.V. PFRED: A computational platform for siRNA and antisense oligonucleotides design. PLoS ONE 2021, 16, e0238753. [Google Scholar] [CrossRef]
- Chiba, S.; Lim, K.; Sheri, N.; Anwar, S.; Erkut, E.; Shah, M.; Aslesh, T.; Woo, S.; Sheikh, O.; Maruyama, R.; et al. eSkip-Finder: A machine learning-based web application and database to identify the optimal sequences of antisense oligonucleotides for exon skipping. Nucleic Acids Res. 2021, 49, W193–W198. [Google Scholar] [CrossRef] [PubMed]
- Morcos, P.A. Achieving targeted and quantifiable alteration of mRNA splicing with Morpholino oligos. Biochem. Biophys. Res. Commun. 2007, 358, 521–527. [Google Scholar] [CrossRef]
- Moulton, J.D.; Yan, Y. Using Morpholinos to Control Gene Expression. Curr. Protoc. Mol. Biol. 2008, 83, 26.8.1–26.8.29. [Google Scholar] [CrossRef]
- Frankish, A.; Diekhans, M.; Jungreis, I.; Lagarde, J.; Loveland, J.E.; Mudge, J.M.; Sisu, C.; Wright, J.C.; Armstrong, J.; Barnes, I.; et al. GENCODE 2021. Nucleic Acids Res. 2021, 49, D916–D923. [Google Scholar] [CrossRef]
- Hug, N.; Longman, D.; Cáceres, J.F. Mechanism and regulation of the nonsense-mediated decay pathway. Nucleic Acids Res. 2016, 44, 1483–1495. [Google Scholar] [CrossRef] [PubMed]
- Le Pera, L.; Marcatili, P.; Tramontano, A. PICMI: Mapping point mutations on genomes. Bioinformatics 2010, 26, 2904–2905. [Google Scholar] [CrossRef] [PubMed]
- Futreal, P.A.; Coin, L.; Marshall, M.; Down, T.; Hubbard, T.; Wooster, R.; Rahman, N.; Stratton, M.R. A census of human cancer genes. Nat. Rev. Cancer 2004, 4, 177–183. [Google Scholar] [CrossRef]
- Tokheim, C.J.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; Karchin, R. Evaluating the evaluation of cancer driver genes. Proc. Natl. Acad. Sci. USA 2016, 113, 14330–14335. [Google Scholar] [CrossRef]
- Mann, C.J.; Honeyman, K.; McClorey, G.; Fletcher, S.; Wilton, S.D. Improved antisense oligonucleotide induced exon skipping in themdx mouse model of muscular dystrophy. J. Gene Med. 2002, 4, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Randic, T.; Kozar, I.; Margue, C.; Utikal, J.; Kreis, S. NRAS mutant melanoma: Towards better therapies. Cancer Treat. Rev. 2021, 99, 102238. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Chen, K.; Wang, C.; Chen, Z.; Meng, Z.; Wang, P. Targeting NRAS-Mutant Cancers with the Selective STK19 Kinase Inhibitor Chelidonine. Clin. Cancer Res. 2020, 26, 3408–3419. [Google Scholar] [CrossRef] [PubMed]
- Gossage, L.; Eisen, T.; Maher, E.R. VHL, the story of a tumour suppressor gene. Nat. Rev. Cancer 2015, 15, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Takamori, H.; Yamasaki, T.; Kitadai, R.; Minamishima, Y.A.; Nakamura, E. Development of drugs targeting hypoxia-inducible factor against tumor cells with VHL mutation: Story of 127 years. Cancer Sci. 2023, 114, 1208–1217. [Google Scholar] [CrossRef] [PubMed]
- Zaman, A.; Wu, W.; Bivona, T.G. Targeting Oncogenic BRAF: Past, Present, and Future. Cancers 2019, 11, 1197. [Google Scholar] [CrossRef] [PubMed]
- Hassin, O.; Oren, M. Drugging p53 in cancer: One protein, many targets. Nat. Rev. Drug Discov. 2023, 22, 127–144. [Google Scholar] [CrossRef]
- Kahraman, A.; Karakulak, T.; Szklarczyk, D.; von Mering, C. Pathogenic impact of transcript isoform switching in 1209 cancer samples covering 27 cancer types using an isoform-specific interaction network. Sci. Rep. 2020, 10, 14453. [Google Scholar] [CrossRef] [PubMed]
- Zhuhong, H.; Zhenyu, B.; Xiangyuan, C.; Tingzhen, X.; Libin, S. Genome-wide isoform-level analysis reveals tumor-specific isoforms for lung adenocarcinoma diagnosis and prognosis. Cancer Genet 2019, 230, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Barrett, C.L.; DeBoever, C.; Jepsen, K.; Saenz, C.C.; Carson, D.A.; Frazer, K.A. Systematic transcriptome analysis reveals tumor-specific isoforms for ovarian cancer diagnosis and therapy. Proc. Natl. Acad. Sci. USA 2015, 112, E3050–E3057. [Google Scholar] [CrossRef]
- Sterne-Weiler, T.; Howard, J.; Mort, M.; Cooper, D.N.; Sanford, J.R. Loss of exon identity is a common mechanism of human inherited disease. Genome Res. 2011, 21, 1563–1571. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Consuegra, I.; Rubio, J.C.; Nogales-Gadea, G.; Bautista, J.; Jimenez, S.; Cabello, A.; Lucia, A.; Andreu, A.L.; Arenas, J.; Martin, M.A. Novel mutations in patients with McArdle disease by analysis of skeletal muscle mRNA. J. Med. Genet 2009, 46, 198–202. [Google Scholar] [CrossRef]
- Shiraishi, Y.; Okada, A.; Chiba, K.; Kawachi, A.; Omori, I.; Mateos, R.N.; Iida, N.; Yamauchi, H.; Kosaki, K.; Yoshimi, A. Systematic identification of intron retention associated variants from massive publicly available transcriptome sequencing data. Nat. Commun. 2022, 13, 5357. [Google Scholar] [CrossRef] [PubMed]
- Dawes, R.; Bournazos, A.M.; Bryen, S.J.; Bommireddipalli, S.; Marchant, R.G.; Joshi, H.; Cooper, S.T. SpliceVault predicts the precise nature of variant-associated mis-splicing. Nat. Genet. 2023, 55, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Desmet, F.O.; Hamroun, D.; Lalande, M.; Collod-Béroud, G.; Claustres, M.; Béroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef]
- Nelson, K.K.; Green, M.R. Mechanism for cryptic splice site activation during pre-mRNA splicing. Proc. Natl. Acad. Sci. USA 1990, 87, 6253–6257. [Google Scholar] [CrossRef]
- Alimonti, A.; Carracedo, A.; Clohessy, J.G.; Trotman, L.C.; Nardella, C.; Egia, A.; Salmena, L.; Sampieri, K.; Haveman, W.J.; Brogi, E.; et al. Subtle variations in Pten dose determine cancer susceptibility. Nat. Genet 2010, 42, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.C.; Wood, M.J.A.; Davies, K.E. Therapeutic approaches for Duchenne muscular dystrophy. Nat. Rev. Drug Discov. 2023. [Google Scholar] [CrossRef] [PubMed]
- Dowdy, S.F. Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol. 2017, 35, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Prakash, T.P.; Mullick, A.E.; Lee, R.G.; Yu, J.; Yeh, S.T.; Low, A.; Chappell, A.E.; Østergaard, M.E.; Murray, S.; Gaus, H.J.; et al. Fatty acid conjugation enhances potency of antisense oligonucleotides in muscle. Nucleic Acids Res. 2019, 47, 6029–6044. [Google Scholar] [CrossRef]
- Shadid, M.; Badawi, M.; Abulrob, A. Antisense oligonucleotides: Absorption, distribution, metabolism, and excretion. Expert Opin. Drug Metab. Toxicol. 2021, 17, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- Lindeboom, R.G.H.; Vermeulen, M.; Lehner, B.; Supek, F. The impact of nonsense-mediated mRNA decay on genetic disease, gene editing and cancer immunotherapy. Nat. Genet. 2019, 51, 1645–1651. [Google Scholar] [CrossRef]
- Nagy, E.; Maquat, L.E. A rule for termination-codon position within intron-containing genes: When nonsense affects RNA abundance. Trends Biochem. Sci. 1998, 23, 198–199. [Google Scholar] [CrossRef]
- Carrier, L.; Schlossarek, S.; Willis, M.S.; Eschenhagen, T. The ubiquitin-proteasome system and nonsense-mediated mRNA decay in hypertrophic cardiomyopathy. Cardiovasc. Res. 2010, 85, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Sun, X.; Qian, Y.; LaDuca, J.P.; Maquat, L.E. At Least One Intron Is Required for the Nonsense-Mediated Decay of Triosephosphate Isomerase mRNA: A Possible Link between Nuclear Splicing and Cytoplasmic Translation. Mol. Cell. Biol. 1998, 18, 5272–5283. [Google Scholar] [CrossRef] [PubMed]
- Nickless, A.; Bailis, J.M.; You, Z. Control of gene expression through the nonsense-mediated RNA decay pathway. Cell Biosci. 2017, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Coban-Akdemir, Z.; White, J.J.; Song, X.; Jhangiani, S.N.; Fatih, J.M.; Gambin, T.; Bayram, Y.; Chinn, I.K.; Karaca, E.; Punetha, J.; et al. Identifying Genes Whose Mutant Transcripts Cause Dominant Disease Traits by Potential Gain-of-Function Alleles. Am. J. Hum. Genet. 2018, 103, 171–187. [Google Scholar] [CrossRef]
- Hsu, M.K.; Lin, H.Y.; Chen, F.C. NMD Classifier: A reliable and systematic classification tool for nonsense-mediated decay events. PLoS ONE 2017, 12, e0174798. [Google Scholar] [CrossRef]
- Popp, M.W.; Maquat, L.E. Leveraging Rules of Nonsense-Mediated mRNA Decay for Genome Engineering and Personalized Medicine. Cell 2016, 165, 1319–1322. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Lehner, B.; Lindeboom, R.G.H. To NMD or Not To NMD: Nonsense-Mediated mRNA Decay in Cancer and Other Genetic Diseases. Trends Genet 2021, 37, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.; Stupack, D.G.; Wilkinson, M.F. Nonsense-mediated RNA decay: An emerging modulator of malignancy. Nat. Rev. Cancer 2022, 22, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Piva, F.; Giulietti, M.; Nocchi, L.; Principato, G. SpliceAid: A database of experimental RNA target motifs bound by splicing proteins in humans. Bioinformatics 2009, 25, 1211–1213. [Google Scholar] [CrossRef]
- Breslauer, K.J.; Frank, R.; Blöcker, H.; Marky, L.A. Predicting DNA duplex stability from the base sequence. Proc. Natl. Acad. Sci. USA 1986, 83, 3746–3750. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, N.; Nakano, S.i.; Yoneyama, M.; Honda, K.i. Improved Thermodynamic Parameters and Helix Initiation Factor to Predict Stability of DNA Duplexes. Nucleic Acids Res. 1996, 24, 4501–4505. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; SantaLucia, J.; Burkard, M.E.; Kierzek, R.; Schroeder, S.J.; Jiao, X.; Cox, C.; Turner, D.H. Thermodynamic Parameters for an Expanded Nearest-Neighbor Model for Formation of RNA Duplexes with Watson−Crick Base Pairs. Biochemistry 1998, 37, 14719–14735. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]



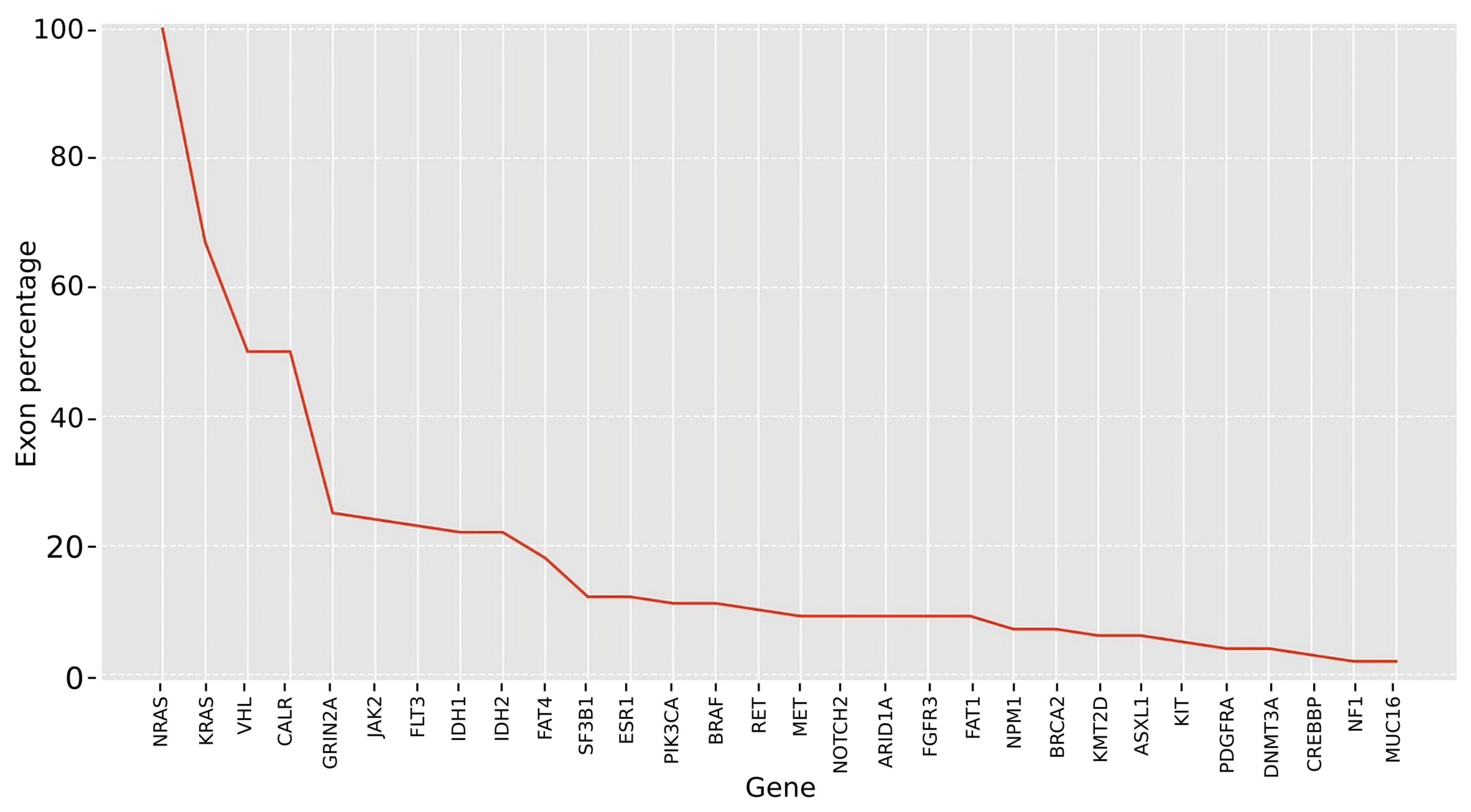
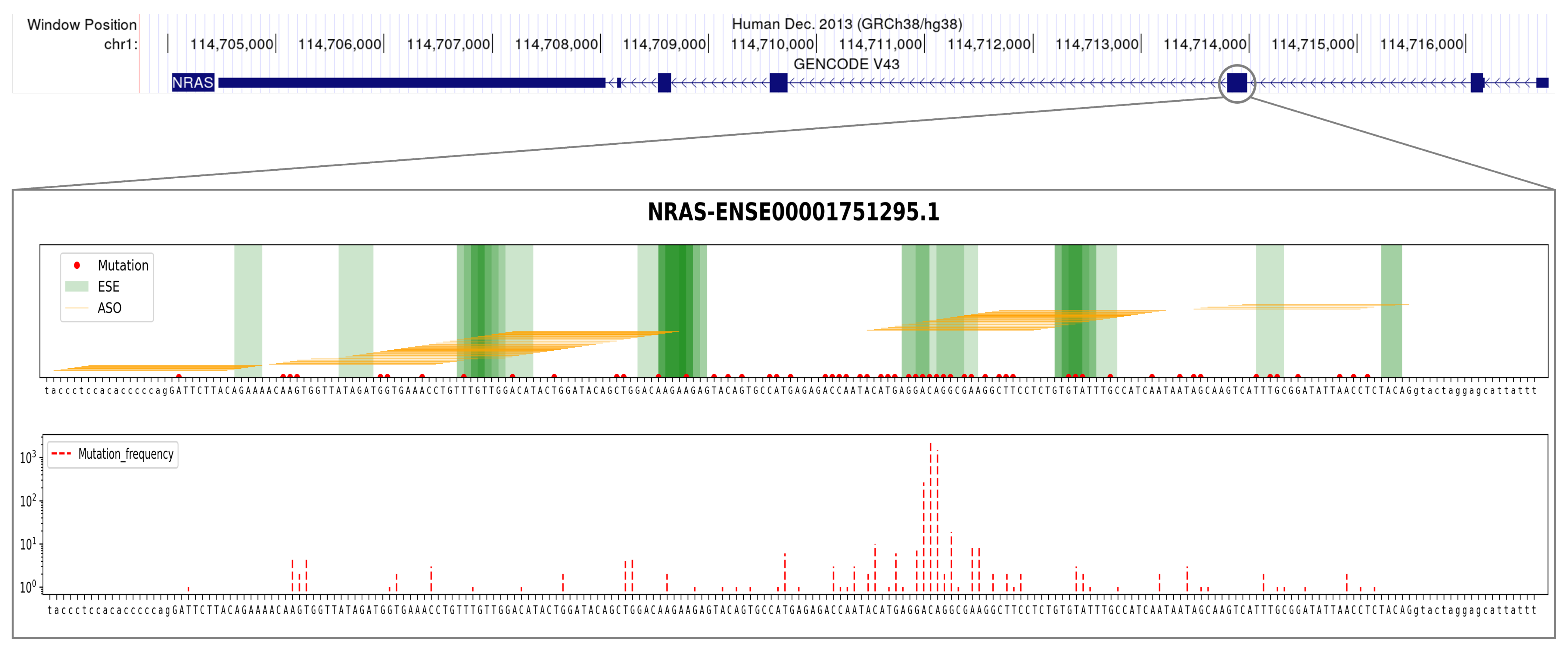

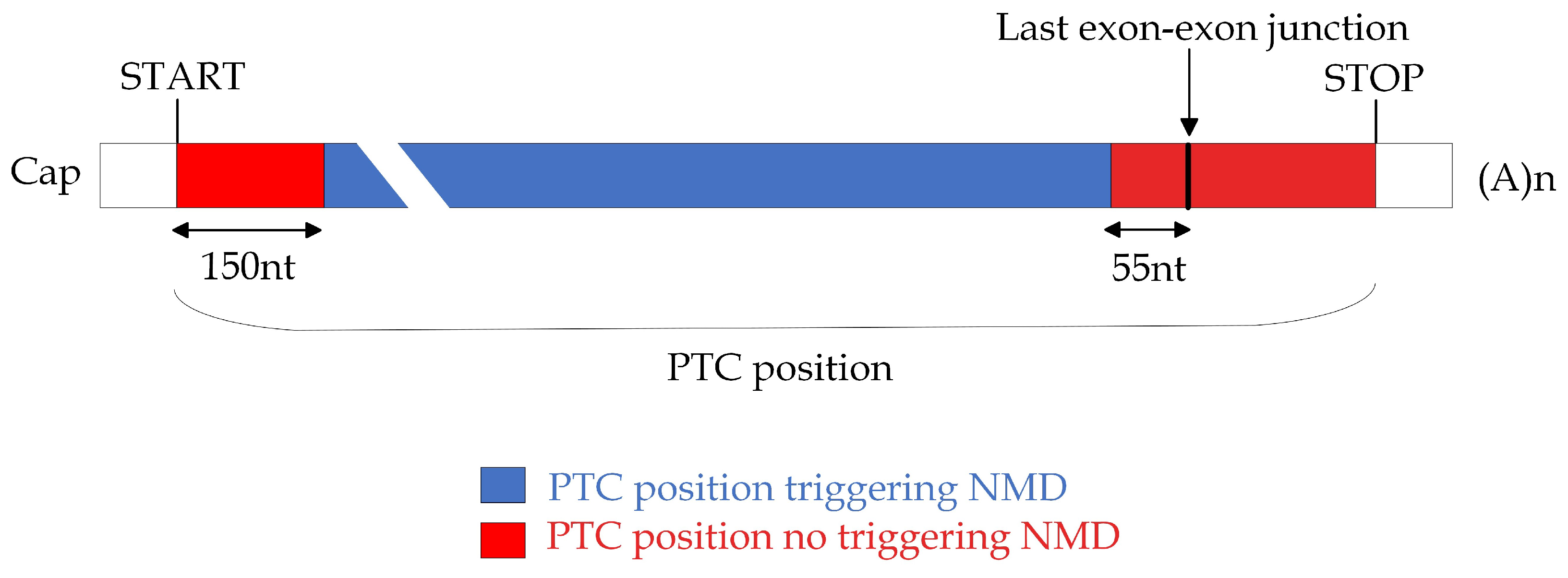
| Gene | Exon ID | ASO ID | ASO Sequence |
|---|---|---|---|
| NRAS | ENSE00001751295.1 | H3D (13, ) | UGCUCCUAGUACCUGUAGAGGUUAA |
| H3D (12, ) | AUGCUCCUAGUACCUGUAGAGGUUA | ||
| H3D (11, ) | AAUGCUCCUAGUACCUGUAGAGGUU | ||
| H3D (10, ) | UAAUGCUCCUAGUACCUGUAGAGGU | ||
| H3D (9, ) | AUAAUGCUCCUAGUACCUGUAGAGG | ||
| H3D (8, ) | AAUAAUGCUCCUAGUACCUGUAGAG |
| Gene | Exon ID | ASO ID | ASO Sequence |
|---|---|---|---|
| VHL | ENSE00003504189.1 | H2A (, 7) | AGGUGACCUAUCGGGACAAGCAAAG |
| H2A (, 8) | AAGGUGACCUAUCGGGACAAGCAAA | ||
| H2A (, 9) | AAAGGUGACCUAUCGGGACAAGCAA | ||
| H2A (, 10) | CAAAGGUGACCUAUCGGGACAAGCA | ||
| H2A (, 11) | CCAAAGGUGACCUAUCGGGACAAGC | ||
| H2A (, 12) | GCCAAAGGUGACCUAUCGGGACAAG | ||
| H2A (, 13) | AGCCAAAGGUGACCUAUCGGGACAA | ||
| H2D (13, ) | AAACGUCAGUACCUGGCAGUGUGAU | ||
| H2D (12, | AAAACGUCAGUACCUGGCAGUGUGA | ||
| H2D (11, ) | UAAAACGUCAGUACCUGGCAGUGUG | ||
| H2D (10, ) | GUAAAACGUCAGUACCUGGCAGUGU | ||
| H2D (9, ) | AGUAAAACGUCAGUACCUGGCAGUG | ||
| H2D (8, ) | AAGUAAAACGUCAGUACCUGGCAGU | ||
| H2D (7, ) | AAAGUAAAACGUCAGUACCUGGCAG |
| Gene | Classification | Top 10 Mutated Exons | IN/OUT Frame | ASO-E | ASO-J |
|---|---|---|---|---|---|
| BRAF | Oncogene | ENSE00003485507.1 | OUT | 4 | 5 |
| ENSE00003559218.1 | OUT | 13 | 4 | ||
| ENSE00003569635.1 | OUT | 16 | 0 | ||
| ENSE00003587655.1 | IN | 18 | 14 | ||
| ENSE00001035295.1 | IN | 34 | 4 | ||
| ENSE00001907699.1 | OUT | 3 | 4 | ||
| ENSE00003527888.1 | IN | 12 | 7 | ||
| ENSE00003521664.1 | OUT | 15 | 12 | ||
| ENSE00003487759.1 | IN | 13 | 13 | ||
| ENSE00003687908.1 | OUT | 21 | 0 | ||
| TP53 | Tumor Suppressor | ENSE00003518480.1 | OUT | 59 | 4 |
| ENSE00003725258.1 | OUT | 57 | 13 | ||
| ENSE00003712342.1 | OUT | 30 | 6 | ||
| ENSE00002048269.1 | OUT | 25 | 5 | ||
| ENSE00003723991.1 | OUT | 56 | 10 | ||
| ENSE00002073243.1 | OUT | 25 | 7 | ||
| ENSE00003625790.1 | IN | 97 | 5 | ||
| ENSE00003670707.1 | IN | 10 | 3 | ||
| ENSE00003545950.1 | OUT | 46 | 5 | ||
| ENSE00003786593.1 | OUT | 18 | 7 |
| ONG Score ( × 100) | TSG Score ( × 100) | Classification |
|---|---|---|
| >20% | <=5% | Oncogene |
| >20% | >5% | Tumor suppressor |
| <20% | >20% | Tumor suppressor |
| <20% | <20% | Unclassified |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pacelli, C.; Rossi, A.; Milella, M.; Colombo, T.; Le Pera, L. RNA-Based Strategies for Cancer Therapy: In Silico Design and Evaluation of ASOs for Targeted Exon Skipping. Int. J. Mol. Sci. 2023, 24, 14862. https://doi.org/10.3390/ijms241914862
Pacelli C, Rossi A, Milella M, Colombo T, Le Pera L. RNA-Based Strategies for Cancer Therapy: In Silico Design and Evaluation of ASOs for Targeted Exon Skipping. International Journal of Molecular Sciences. 2023; 24(19):14862. https://doi.org/10.3390/ijms241914862
Chicago/Turabian StylePacelli, Chiara, Alice Rossi, Michele Milella, Teresa Colombo, and Loredana Le Pera. 2023. "RNA-Based Strategies for Cancer Therapy: In Silico Design and Evaluation of ASOs for Targeted Exon Skipping" International Journal of Molecular Sciences 24, no. 19: 14862. https://doi.org/10.3390/ijms241914862