1. Introduction
Killer toxins are proteins or glycoproteins able to bind receptors on the surface of the specific target microorganisms [
1]. Their lethal action can be expressed through different and specific modes of action. The killer trait is widespread among the killer toxins’ activity, and it has been reported in over a hundred yeast species, several of them with a potential application in various industrial sectors, such as food, pharmaceutical, agricultural and fermentation, due to their antagonistic activity against pathogens [
2]. In winemaking, killer toxins are interesting biocontrol tools to enhance the wine quality by removing spoilage yeasts with consequent reduction of the use of chemical preservatives [
3,
4].
Several killer toxins secreted by different non-Saccharomyces yeast strains are active against a wide range of spoilage yeasts, including Dekkera/Brettanomyces yeasts, responsible for unpleasant odors produced during wine fermentation, aging and storage, that affect the quality of the final product. Considering the growing interest in reducing sulfur dioxide during winemaking, the killer toxins could represent an interesting alternative.
The potentially effective killer toxins are those that do not negatively interact with Saccharomyces cerevisiae fermenting yeasts, nor with malolactic bacteria correlated to optional malolactic fermentation.
Kluyveromyces wickerhamii,
Pichia anomala,
Pichia membranifaciens (the last two species recently reclassified as
Wickerhamomyces anomalus) and the filamentous fungus
Ustilago maydis, have recently been described as possible alternatives to chemical treatments, to prevent or reduce
Brettanomyces spoilage risk [
5].
The killer activity of these toxins was found to be either fungistatic or fungicidal, depending on their concentration [
6]. Similar results were found by Santos et al. [
7] for the high concentration of the PMKT2 killer toxin, leading to an increasing mortality rate in the three
B. bruxellensis strains tested [
6]. Moreover, lower concentrations of the killer toxin from
U. maydis significantly reduced the amount of 4-ethylphenol produced by
B. bruxellensis, indicating that, in addition to the growth inhibition observed for high concentrations of killer toxins, its small amounts (100 AU) can control flavor defects in wines [
8].
More recently, the killer toxins CpKT1 and CpKT2, isolated from the wine yeast Candida pyralidae, were active and stable under winemaking conditions and appeared to be able to cause cell membrane and cell wall damages in B. bruxellensis.
Villalba and coworkers [
9] recently identified and partially characterized a new killer toxin from
Torulaspora delbrueckii with wide biocontrol spectrum against
B. bruxellensis,
Pichia guilliermondii,
Pichia manshurica and
W. anomalus wine spoilage.
Nowadays, the production of natural antimicrobial compounds, including killer toxins, is a fundamental topic for the modern biotechnology industry [
10]. Although the market for traditional industrial antimicrobials is expanding, the research emphasis on biotech efforts is generating a growing demand for natural alternatives with lower impact on the consumer’s health. The advent of genetic engineering, together with cutting-edge technological processes, is facilitating the large-scale production of such molecules, including proteins with antimicrobial action or industrial enzymes produced naturally only in small quantities [
11]. While this development is particularly evident with regards to the production of therapeutic proteins, in the food sector research it is poorly translated into industrial applications, probably because the level of protein downstream processing depends on its intended application [
12]. The proteins that are potentially interesting in the industry are those produced in bulk and generally requiring little downstream processing and, therefore, relatively crude and inexpensive preparations.
This work is put in such context: a simple technological process of industrial production at the pilot scale, followed by easy downstream steps, to verify the antimicrobial efficacy of killer toxins previously purified and characterized at the laboratory scale, for the control of
B. bruxellensis spoilage yeasts in winemaking [
13,
14,
15,
16].
In particular, three different killer toxins, two excreted by two different
W. anomalus strains and one secreted by
K. wickerhamii yeast, were produced at the pilot scale, semi-purified and lyophilized, following an industrial production step [
13,
14,
15,
16].
Three lyophilized formulations were separately assayed in vitro and in wine to compare their cytocidal activity and their effect on B. bruxellensis. The safety aspects in human intestinal cells were also evaluated.
2. Results
2.1. Killer Toxins Industrial Production and Lyophilization
The bioreactor cultivation of
W. anomalus DiSVA2 and DiSVA671, as well as the
K. wickerhamii DiSVA15 strains, confirmed the production of Pikt, WA18 and Kwkt, their respective native killer toxins previously described and characterized (see
Materials and Methods section)
During the pilot scale conditions, the productivity and yields of killer toxins were increased. Indeed, the maximal lab-scale production (in flasks) of each killer toxin was delayed by 6–10 h, if compared to the bioreactor production.
Figure 1 showed the growth of each yeast and the relative production kinetics of each toxin. The maximum concentration of the toxin produced by
W. anomalus DiSVA2 was obtained after 10 hours of incubation, that of
K. wickerhamii DiSVA15 around the 15
th hour and finally
W. anomalus DiSVA671 produced greatest amount of toxin at the 19
th hour.
For each killer toxin, a fermented broth ten-fold concentrated (named broth 10 ×) and partially purified, was obtained by ultrafiltration using tangential flow membranes, as a single downstream step.
The obtained lyophilizates named D2 (from W. anomalus DiSVA2), D15 (from K. wickerhamii DiSVA15) and D18 (from W. anomalus DiSVA671), obtained with yield of 12.3, 22.4 and 55.9 g/L, respectively, were then solubilized in order to be subsequently used After various tests carried out considering the physical solubilization and the economic implications relating to their further use in relation to the production costs, the dose of 100 g/L was chosen as the maximal stock concentration.
The solubilization test of stocks and, therefore, their antimicrobial efficacy was carried out in water, in 0.1 M citrate-phosphate buffer pH 4.4, and wine. The results obtained after well test assays indicated that no differences in the solubility and antimicrobial activity in D2, D15 and D18 in all preparations were observed. Thus, sterile, distilled water was utilized. The D2 lyophilizate showed an increment of 2 mm in the inhibition halo, if compared with the relative ultrafiltered, cell-free supernatant. For D15, the same halos were obtained by testing both the ultrafiltered cell-free supernatant and the lyophilized preparation, while a greater increment of 4 mm was observed for the D18 lyophilized preparation halo (
Table 1).
The evaluation of the long-term stability of all lyophilizates, both in powder and loose form, showed no significant variations of killer activity for up to nine months at 10 °C.
The solubilization of stock lyophilizates at the tested concentrations (100 g/L) showed very low residual sugars, while the protein component increased significantly when compared with the partially concentrated broth, reaching comparable values for all three preparations (about 45 g/L), actually ten-fold higher taking into account the stock dilution factor. The slight increase in the antimicrobial activity of the lyophilizates was consistent with the results of the protein since the halo diameter was in logarithmic function of the concentration of active compounds (
Table 1). An exception was shown by the D2 lyophilized sample, where the diameter of the inhibition halo was already in the limit of sensibility of the well test method.
2.2. Minimum Inhibitory Concentration (MIC) of the Three Killer Toxins
The results of in vitro MIC determinations showed that D15 was the most active against B. bruxellensis with the lowest MIC value of 0.05 mg/mL. D2 also displayed significant in vitro effects but with a double MIC value of 0.1 mg/mL. The highest MIC value obtained against the sensitive strain was exhibited by D18.
The measures of the inhibition halo, corresponding to increasing concentrations of each killer toxin, were reported in
Table 2. Concerning D2 and D15, a progressive decrease in the antimicrobial action was observed (progressive decrease in the inhibition halos). Regarding D18, after the first dilution, showing a small halo, the activity seemed to disappear. This was probably due to the lower initial killer activity that could not be detected by the sensitivity of the well test method.
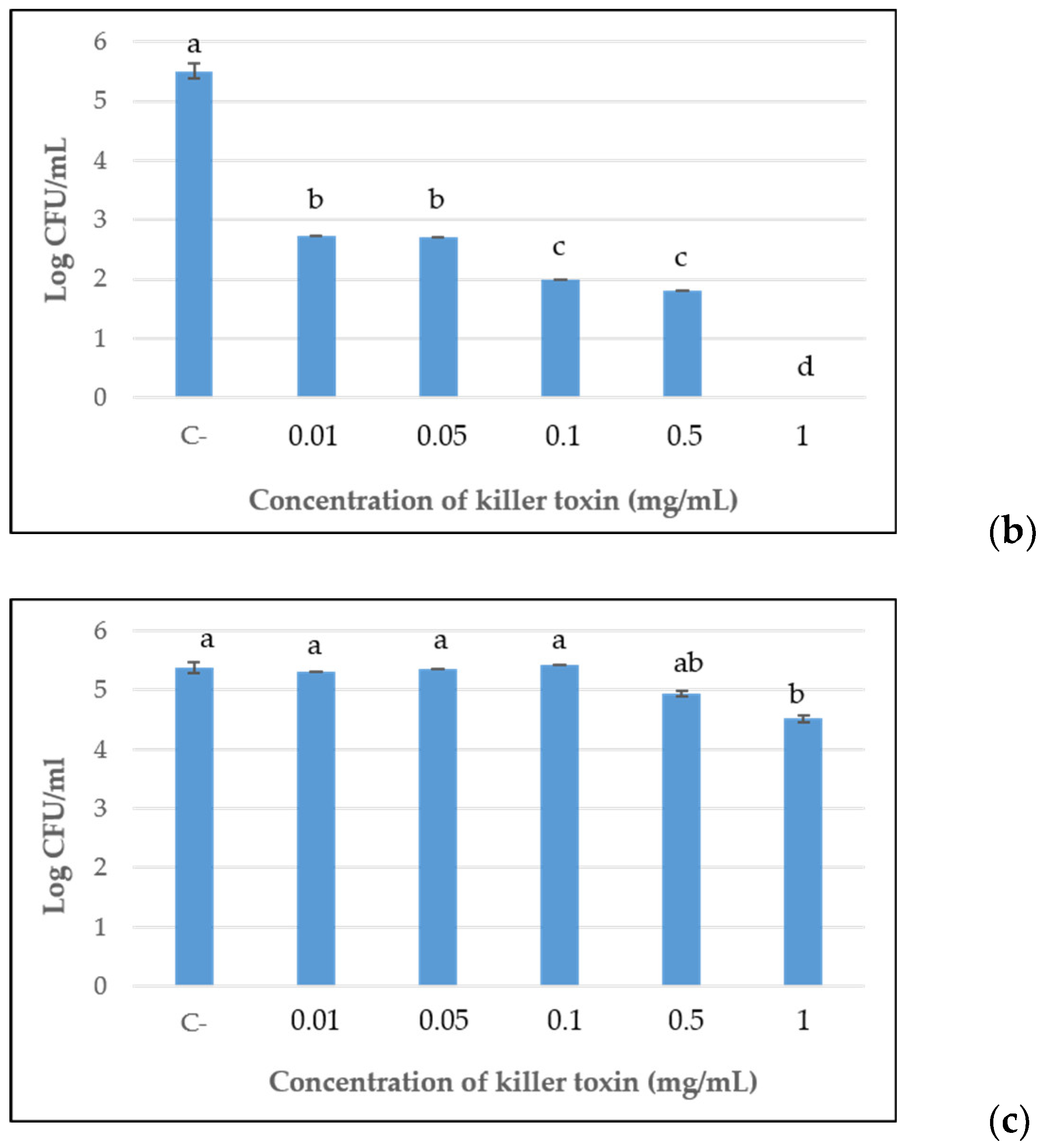
Furthermore, the killer toxins, at each concentration previously assayed for MIC determinations, were tested for their effective lethal activity toward the sensitive strain, after 96 h of incubation, by viable cell counts assay. The results reported in
Figure 2, confirmed the progressive reduction of antimicrobial activity in a dose-dependent manner of the D2 and D15 lyophilized preparations up to 0.1 mg/mL and 0.05 mg/mL, respectively. After that, a constant significant antimicrobial action was shown. For D15, no cultivable cells were detected at the maximum concentration tested (1 mg/mL), while at 0.5 mg/mL and 0.1 mg/mL, the concentrations showed similar results with a reduction of about 3.5 log orders of magnitude, in comparison with the control. A significant containment of the development of
B. bruxellensis was also detected for the doses of 0.05 and 0.01 mg/mL. The same trend was observed for toxin D2, although with less effective containment of yeast growth than D15.
Differently, concerning D18, only the maximal concentration of 1 mg/mL significantly reduced the yeast development compared with the control, by approximately 1 log order of magnitude, while testing 0.5 mg/mL concentration, only a slight reduction was shown.
2.3. Cytofluorimetric Assay
Imaging flow cytometric analyses were performed in order to detect changes induced by the D2, D15 and D18 treatments to sensitive B. bruxellensis cells, previously grown in YM liquid medium and treated for 96 h with each lyophilized killer toxin, separately.
As reported in
Figure 3 and the relative data summarized in
Table 3, in untreated controls, the large majority of the cells (71%) was composed of live cells, while 28% of the cells were clearly propidium iodide (PI) positive (dead cells). The validity of the treatment has been assessed using a positive control, where cells were incubated in ethanol. In these conditions, the percentage of live cells decreased to 0.76%, while PI positive cells accounted for 98.7%. The relative efficacy of the toxins tested is summarized in
Table 3, showing the following degree of cellular toxicity: the highest percentage of PI positive cells was detected in the D2 toxin (73.3%), followed by the D15 toxin (64.1%) and D18 (40.7%). Notably, in sample D15 (
Figure 3b), the percentage of live cells was particularly decreased, and, uniquely in these samples, the partially PI positive cells increased considerably to 30.1% compared to D2 2.7% and D18 0.78%.
The D2 and D15 (
Figure 3a,b) lethal effect was significantly higher when compared to D18 (
Figure 3c) with differences more pronounced among live and dead cell populations. The negative control (
Figure 3d) was also evaluated after 96 h of incubation, similarly to those of lyophilized formulations, thus the resulting dead population represented the fraction of
B. bruxellensis physiologically dead, after 4 days of incubation, probably due to the lack of nutrients.
The percentage of the relative mortality of the cells evaluated with the flow cytometer was compared with the effective number of cells culturable by viable plate counts. Compared with the initial inoculum (Log 4 CFU/mL), the
B. bruxellensis cells after 96 h of incubation reached a value of Log 6.5 CFU/mL. This value was considered equal to 100% of the live cells, as reported in
Table 3, right columns. Similarly, the results of the viable counts of toxin-treated sensitive cells were reported as the relative percentage of the control (100%). The highest concentration tested showed an evident fungicidal activity of all lyophilized formulations towards the sensitive strain.
2.4. Cytotoxicity against Human Intestinal Cells (Caco-2)
The cytotoxic activity of the three killer toxins was tested in vitro against the human intestinal cell line Caco-2. As depicted in
Figure 4, a very low (less than 3%) cytotoxic activity of the killer toxins was observed following 24 h exposure in the experimental conditions.
2.5. D15 Lyophilized Formulate Tested in Wine
Starting from the 100 g/L stock of the most effective lyophilizate D15, different dilutions of 0.5 g/L, 0.2 g/L, 0.1 g/L and 0.05 g/L were tested in a spontaneous contaminated red wine (concentration
Brettanomyces spp., about Log 3 CFU/mL). The efficacy of the D15 formulation was evaluated both by the viable cell counts and through the measurement of the phenols produced after 60 days of D15 addition. The results reported in
Figure 5 clearly showed that all concentrations tested inhibited the growth of
Brettanomyces yeasts, with the exception of the lowest dilution, where a slight
Brettanomyces population survived. Similarly, ethyl phenols significantly decreased if compared with the untreated control (C-).
3. Discussion
The native antimicrobial compounds applied in food and beverages as preservatives combine the protection of the final product with extended shelf life and consumer safety.
The potential roles of many naturally occurring antimicrobial compounds, such as bacteriocins, nisin, plant essential oils, chitosan, have been widely studied and are reported to be effective against spoilage and pathogenic microorganisms. Few of these compounds are commercially available and used in food processing, since their efficacy, consumer acceptance and regulation are not well defined [
17].
In the enological field, although there are numerous genera and species of yeasts able to produce effective killer toxins against the development of
Brettanomyces spp. spoilages, the commercial formulation of such characterized molecules remains a prerogative of the applied studies [
5,
18].
The potential uses proposed for yeast killer toxins have been limited by the recognized skills of these toxins [
19]. The suggested applications are largely limited by the physical and chemical conditions fitting with the stability of killer toxins, mainly low pH and temperature values.
Although the use of killer yeasts is effective at limiting the risk of the onset of
Brettanomyces spp., their antimicrobial activity could represent a preventive action during alcoholic fermentation, but it is not suitable to solve in highly compromised situations or to be used in the advanced stages of production when the yeast inoculum is not easy to be performed. By contrast, the formulation of purified products to be added at concentrations able to guarantee greater efficacy, associated with a higher similarity with conventional chemical analogues, is limited due to the high production costs and low stability of the active compounds [
6,
20,
21].
Recently, novel agents based on natural antimicrobials (chitosan, essential oil compounds) or metal nanoparticles (silver nanoparticles) have been suggested to replace or reduce the use of SO
2 in winemaking. Although these strategies improve the antimicrobial characteristics of essential oils, the high production costs, related to the complex down-stream steps, limits the commercialization of these natural antimicrobials [
22].
In the present work, the formulated ready-to-use killer toxins, denominated D2, D15 and D18, were characterized and compared for their zymocidial effect against Brettanomyces yeasts, a most relevant spoilage agent in winemaking.
The determination of viability using image-based flow cytometry, coupled with PI staining, allowed for the precise determination of live (-PI) and dead (+PI) cells. Compared with other flow cytometric determinations, the use of these instruments decreases potential artifacts associated with the erroneous classification of debris, with the images of each event acquired and used for the separation of the populations [
23,
24,
25]. In this work, the data highlighted a remarkable increase in the percentage of dead cells in the D2-treated samples, while a lower effect was observed in the D18 samples. The samples treated with D15 showed a particular distribution: while the percentage of dead cells did not increase remarkably compared with other samples, the live (-PI) population was basically lowered to a residual 2.06% population, highlighting the particular toxicity of this compound. This behavior, in the experimental time frame, led a very evident shift of the distribution toward higher levels of PI fluorescence, stressing the particular cytotoxicity of the killer toxin D15.
The viable counts, carried out after 96 h of inoculation of the lyophilizates, allowed us to validate the actual death of the sensitive cells observed using imaging flow cytometric determination, in order to exclude a possible state of VBNC (viable but not culturable), typical of the
Brettanomyces/
Dekkera spp. strains [
26]. Indeed, in the VBNC state, as a result of some treatments with low SO
2 doses or chitosan, the production of 4-ethylphenol stopped and the possible wine conditions changed (e.g., concentration of molecular SO
2 decreases), so the spoilage yeasts could theoretically regrow and re-infect the wine [
27,
28]. Here, the results of D15 in the real condition in wine confirmed the long-term control of
Brettanomyces yeasts and, at the same time, the control on the ethyl phenols production.
Another fundamental characterization of antimicrobial compounds for a possible application concerns the evaluation of their cytotoxicity. Here, the exclusion of a cytotoxic effect by the killer toxins tested, assessed on human intestinal epithelial cells [
29], suggests their possible safe use in food production after the necessary validation of the formulations. Therefore, in agreement with the safety of the yeast killer toxin on animal systems [
30], our results are compatible with the hypothesis that, in the tested concentrations, the three killer toxins should not represent a potential health risk for human consumption.
In conclusion, similarly to the antiseptic action performed by sulfur dioxide, the use these three killer toxins pilot-scale produced and lyophilized could represent an effective natural strategy for controlling the onset or the proliferation of Brettanomyces/Dekkera spoilage yeasts, as the possible substitution of chemical additives. Depending on the concentration, all preparations greatly reduced the B. bruxellensis populations; in particular, D15 was also efficient during the industrial winemaking step, revealing a healing power even when the Brettanomyces spp. infection was already established. Therefore, D15 represents a strong candidate as a natural antifungal agent for the wine industry.
4. Materials and Methods
4.1. Killer Toxins Production and Characterization
Three killer toxins, named Pikt, Kwkt and WA18, were previously described and characterized, and the main features are shown in
Table 4. They were produced by three different yeast strains coming from the Yeast Collection of the Department of Life and Environmental Sciences (DiSVA) of the Polytechnic University of Marche (Italy), such as
Wickerhamomyces anomalus DiSVA2,
Kluyveromyces wickerhamii DiSVA15 and
Wickerhamomyces anomalus DiSVA671, for Pikt, Kwkt and WA18, respectively. On the basis of the results previously obtained at laboratory scale, here the killer toxins were produced at the pilot scale using bioreactors, partially purified and then lyophilized.
4.2. Set Up of Pilot Scale Killer Toxins Production
On the basis of the preliminary results, here the pilot scale production (300 L) of the three killer toxins was set up. The medium used has the following composition: yeast extract 10 g/L; malt extract 5 g/L and casein peptone 5 g/L, buffered to pH 4.4 with 0.1 M citric acid/dibasic sodium phosphate. The glucose concentrations were 30 g/L for Pikt and 40 g/L for both Kwkt and WA18, to avoid the presence of residual sugars at the end of the process.
Fresh cultures of the yeasts K. wickerhamii DiSVA15, W. anomalus DiSVA2 and W. anomalus DiSVA671 were used to inoculate the medium (equal to 5% of the final volume of the growth process) for the production of each killer toxin. The growth conditions for the process were: pH 4.4, temperature 25 °C, pO2 20% and stirring 250 rpm. The process was followed for 16 h, 10 h and 9 h for Kwkt, Pikt and WA18, respectively. At the end of the process, the supernatant of each culture was microfiltered (Ø 0.2 µm cut-off) and the acellular broth was subjected to ultrafiltration (BioFlex 50, Schleicher & Schuell, Whatman GmbH, GE Healthcare, Dassel, Germany) using a membrane with 10 KDa cut-off, with the purpose to concentrate the broth 10-fold, then consequently concentrate the protein killer. Afterwards, each concentrated broth underwent a dialysis process (4-fold) using the same ultrafiltration system and 0.1 M citric acid/dibasic sodium phosphate buffer, obtaining a broth 10 × partially purified for each toxin. The purpose was to remove the residual sugars and unnecessary proteins/metabolites to obtain a greater purification of the concentrated killer protein.
The cell-free supernatants at the end of each growth process and the broths 10 × partially purified were tested for the presence of killer activity against a common wine spoilage yeast,
Brettanomyces bruxellensis DiSVA 46 (Yeast Collection of the Department of Life and Environmental Sciences of the Polytechnic University of Marche, Italy), through the well test screening method, as described by Comitini et al. [
13]. Briefly, an aliquot of the sensitive
B. bruxellensis DiSVA46 pure culture was suspended in sterile water to obtain a concentration of about 10
6 cells/mL. In total, 1 mL of this suspension was put inside the 90 mm Petri dish with 20 mL of buffered Malt Agar (2.7% malt extract, 1.8% agar, buffered to pH 4.4 with 0.1 M citric acid/dibasic sodium phosphate) still liquid, and well-distributed. Prior to the agar solidification, sterile steel wells, with 6 mm of diameter, were placed on the agar. Left to solidify the medium, the wells were removed with sterile forceps with the aim to create a space then filled with 70 µL of each killer toxin suspension.
The plates were incubated at 22 °C for about five days and the killer activity of the lyophilized toxins against the sensitive
B. bruxellensis strain was evaluated by measuring the diameter of the halo around the wells, corresponding to the zone of growth inhibition of the sensitive yeast [
32].
Moreover, the glucose content and total protein content of all cell-free supernatants and broths 10 × partially purified were determined. The glucose concentration was determined using a specific enzymatic kit (Megazyme International, Ireland, Wicklow, Ireland), while the total protein content was expressed as the gallic acid equivalents, following the method described by Singleton et al. [
33].
4.3. Lyophilization Step
The broths 10 × partially purified, concerning each Pikt, Kwkt and WA18 killer toxins, were subjected to the lyophilization process, obtaining three preparations named D2, D15 and D18, respectively. The lyophilization was carried out frozen for each broth 10 × partially purified at −20 °C, then transferred to the lyophilizer for 48 h, temperature −53.2 °C, pressure 1 mbar.
The lyophilized preparations have been assayed for their killer activity against a common spoilage yeast of wine,
B. bruxellensis DiSVA46, following the well test screening method, as described in
Section 4.2. The lyophilizates D2, D15 and D18 were subjected to a solubilization test and, therefore, their antimicrobial efficacy was evaluated. In total, 100 g of each lyophilizate was rehydrated in 1 L of H
2O, 0.1 M citrate-phosphate buffer (pH 4.4) or wine and sterilized by 0.45 µm pore-size filters, in order to establish the possible differences in solubilization and killer activity, depending on the solvent used.
The well test plates were incubated at 22 °C for about five days and the killer activity of the lyophilized toxins against the sensitive
B. bruxellensis strain was evaluated by measuring the diameter of the halo around the wells, corresponding to the zone of growth inhibition of the sensitive yeast [
32]. The glucose and protein content of each lyophilized product (100 g/L in water) were determined as described above.
4.4. Determination of Minimum Inhibitory Concentration (MIC) of the Three Killer Toxins
The determination of the MIC regarding the three killer toxins against sensitive
B. bruxellensis was carried out following the procedure reported by the Clinical and Laboratory Standards Institute guidelines [
34], with some adaptations. Briefly, a 96-well plate was filled with Yeast Mold broth (yeast extract 3 g/L, malt extract 3 g/L, peptone 5 g/L, glucose 10 g/L) buffered to pH 4.4 with 0.1 M citric acid/dibasic sodium phosphate and inoculated with 1 × 10
4 cell/mL of
B. bruxellensis DiSVA46 fresh culture. Then, five different concentrations of each killer toxin were tested, ranging from 1 mg/mL to 0.01 mg/mL. Each test was performed in duplicate and the medium inoculated with
B. bruxellensis DiSVA46, and without the toxins was used as a control. The 96-well plate was incubated at 22 °C for 96 h. Then, viable cell counts were carried out to determine the presumed reduction of yeast viability due to the killer toxin activity. Hence, 100 µL of suspension from each microplate well was spread on Yeast Mold Agar plates and compared with the control.
4.5. Citotoxicity of Killer Toxins for B. bruxellensis-Imaging Cytofluorimetric Assay
The three lyophilized killer toxins (D2, D15, D18) were tested through cytofluorimetric assay, with the aim to compare their cytotoxicity against B. bruxellensis DiSVA46. Here, the sensitive yeast was cultured in the Yeast Mold broth buffered to pH 4.4 with 0.1 M citric acid/dibasic sodium phosphate for 48 h. The fresh culture was centrifuged and washed twice with 0.9% NaCl and used to obtain flasks containing a cell suspension of about 1 × 104 cell/mL in 0.9% NaCl and inoculated with 1 mg/mL of each toxin. The flasks were incubated at 22 °C for 96 h prior to imaging cytofluorimetric assay. A negative control was conducted without the killer toxins (live cells), while 80% ethanol was added to ensure the obtaining of dead cells (positive control).
The cytotoxicity of Brettanomyces was evaluated using Imaging Flow Cytometer (IFC) FlowSight (Luminex, Austin, TX, USA) with propidium iodide (PI) probe.
The (IFC) assay was performed with 300 000 yeast cells obtained by centrifugation at 600 g × 6 min of 10 mL of Brettanomyces at 30 000 cells per mL. After that, the yeast pellet was resuspended in 100 µL of PBS and cell suspension was stained with 10 µL of PI (1 mg/mL), before acquisition was performed by FlowSight.
The samples were excited with a 488 nm laser with the following laser powers: 488 nm 5.00 mW; 785 nm 3.75 mW. The imaging flow cytometry analysis was performed on 8000 focused and singled cells gates (R2) set up in aspect ratio and area dot plot in the Channel 1 (Ch1) mask. Within the R2 gate, the emission of PI was reported as the Fluorescence intensity of Channel 4 (Ch4).
After the acquisition phase, before analyzing the live and dead cells, the raw data files (rif) were processed by the IDEAS software (Luminex, USA) to discriminate the Brettanomyces population from cellular debris.
Arbitrary gates were defined in the control untreated cells (
Figure 2d) in order to identify propidium negative cells (live) and propidium positive cells (dead). The histogram of distribution also shows an intermediate population with partial permeability to PI that accounts for the 100 − (% live + % dead). This partial positivity to PI is suggestive of potential apoptotic processes.
Moreover, to confirm the cytofluorimetric results, it was also carried out the microbiological assay by viable plate counting.
4.6. Cytotoxicity Assay against Human Intestinal Cell Line Caco-2
The cytotoxic potential of the three lyophilized killer toxins (D2, D15, D18) was tested against the human intestinal cell line Caco-2 derived from human colorectal adenocarcinoma. To this aim, the cells were seeded in 96 well plates at a density of 2 × 104 cells/well in high glucose DMEM added with 2 mM L-glutamine, 1% non-essential amino acids and 10% FBS. The cells were incubated for 24 h at 37 °C in a humidified atmosphere containing 5.5% CO2 to reach approximately 90 to 100% confluence. Following a wash to remove non-adherent cells, adherent cells were added with various concentrations of the three killer toxins (D15: 0.05, 0.15, 0.5 mg/mL; D2 and D18: 0.15, 0.45, 1.5 mg/mL), with the complete medium only (negative control), or with 0.01% Triton X-100 in PBS (positive control for cytotoxicity) in the humidified atmosphere containing 5.5% CO2 and incubated for 24 h at 37 °C. After incubation, the adherent cells (previously washed twice with warm PBS) were detached with Trypsin/EDTA treatment. The pellets were washed with PBS (8 min 400× g) and were resuspended in the PBS. Following 5 min staining with 1 µg/mL PI at room temperature, 50,000 events were acquired ungated by using a flow cytometer (BD Accuri C6, BD biosciences, Franklin Lakes, NJ, USA). The percent of PI positive, dead Caco-2 cells weas calculated by computer-assisted analyses (BD Accuri C6 software, BD Biosciences). Four independent experiments with duplicates were carried out.
4.7. Application of D15 in Wine
Wine naturally contaminated with Brettanomyces yeasts was treated with different concentrations of D15. The 4-ethylphenol and 4-ethylguaiacol were evaluated using a HSPME (headspace-solid-phase microextraction) technique and analyzed by GC. The fiber used was: divinyl-benzene/carboxen/polydimethylsiloxane (DVB/CAR/PDMS), 50 to 30 μm, Stable Flex/SS, 1 cm (Supelco, Bellefonte, PA, USA). The sample was prepared as follows: 5 mL of wine was placed into a 10-mL vial with 2.5 g of NaCl and a magnetic stirrer. The samples, after equilibration for 10 min at 25 °C, the DVB/CAR/PDMS fiber was inserted through the vial septum and exposed for 40 min at 55 °C. The 3-octanol was used as an internal standard (1.6 mg/L). A GC-2014 gas chromatograph (Shimadzu, Kyoto, Japan) equipped with a flame ionization detector and a glass Supelcowax-10 column (60 m × 0.32 mm × 0.25 mm) from Supelco was used. GC conditions: carrier gas (flow rate of 3.74 mL/min); split/splitless modality: 60 s splitless; injection and detector temperature, 230 °C. The column program was: 50 °C for 1 min, and increased 2 °C/min to 200 °C and maintained at 200 °C for 20 min. The compounds were identified and quantified by comparison with internal calibration curves of known compounds. After 60 days of treatment of the wine with the D15 formulation, the viable plate counts were carried out on the Yeast Mold Agar.
4.8. Statistical Analyses
Experimental data were reported as the mean values ± standard deviations or error bars. Regarding viable cell counts, data were subjected to analysis of variance (ANOVA). The significant differences were determined using Duncan tests with associated p-values < 0.05.

 ,
,  ), (
), ( ), (
), ( ) indicate values of OD600 nm for W. anomalus DiSVA2, K. wickerhamii DiSVA15 and W. anomalus DiSVA671. Similarly, lines (
) indicate values of OD600 nm for W. anomalus DiSVA2, K. wickerhamii DiSVA15 and W. anomalus DiSVA671. Similarly, lines ( ), (
), ( ), (
), ( ) indicate the values of diameter. Microbial growth (bars) was reported as mean values ± standard deviations.
) indicate the values of diameter. Microbial growth (bars) was reported as mean values ± standard deviations.





 ), (
), ( ) and (
) and ( ) represent 4-ethyl guaiacol and 4-ethyl phenol concentrations and Log CFU/mL, respectively. C- represent the negative control, where no killer toxin was added. Phenols determinations are reported as mean value ± standard deviations (error bars).
) represent 4-ethyl guaiacol and 4-ethyl phenol concentrations and Log CFU/mL, respectively. C- represent the negative control, where no killer toxin was added. Phenols determinations are reported as mean value ± standard deviations (error bars).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}