
Hormone Receptor Signaling and Breast Cancer Resistance to Anti-Tumor Immunity
 , ,
, ,
Abstract
:1. Introduction
2. The Estrogen-ER Pathway in BC
2.1. Overview of Estrogen-ER Signalings
2.2. Targeting of the Estrogen-ER Pathway
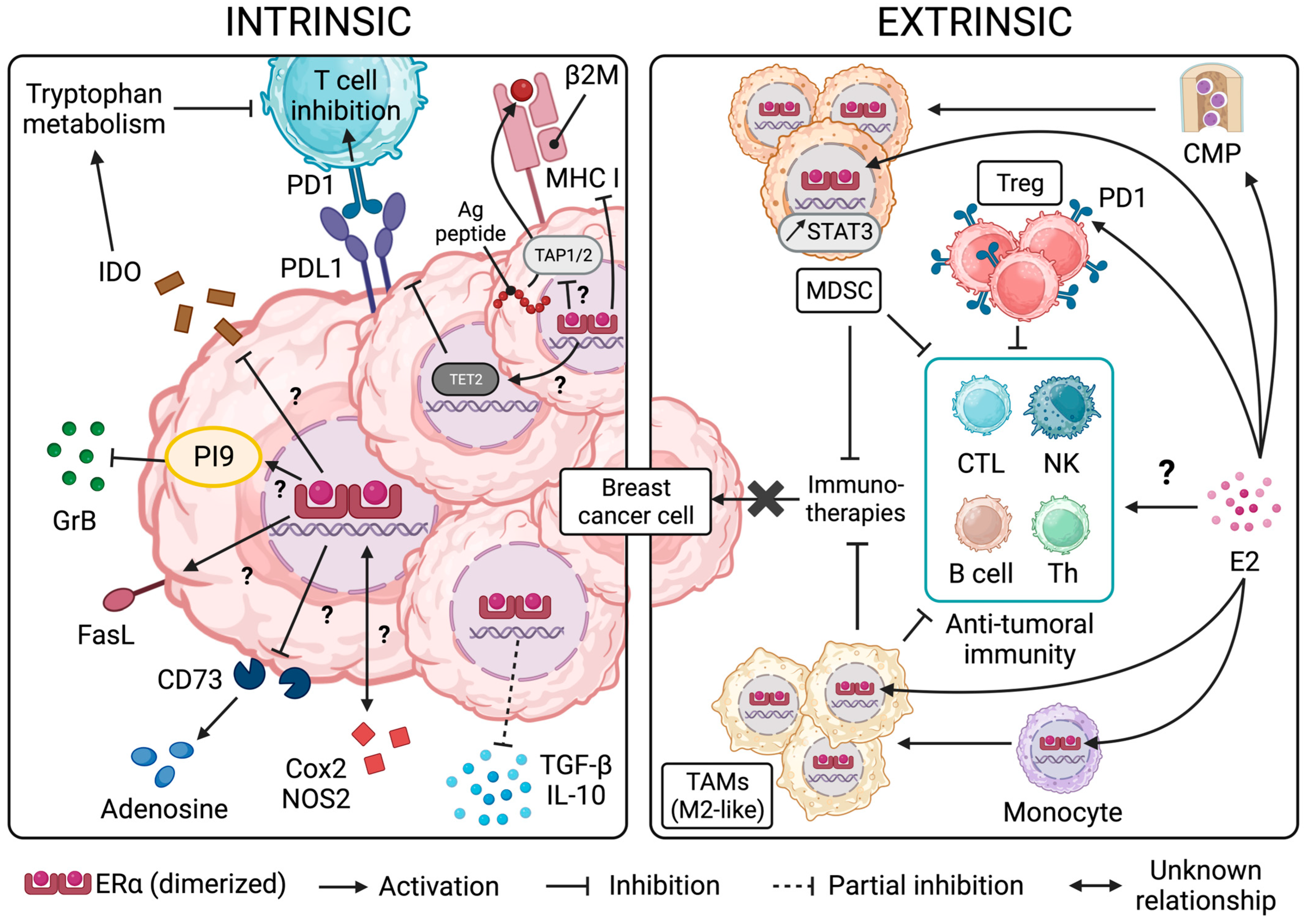
3. ER Signaling and Resistance/Sensitivity of BC Cells to Effector Immune Cells and Immunotherapies
3.1. The Role of ER Signaling in Malignant Cell Resistance to Anti-Tumoral Effectors
3.2. The Link between ER and Immune Inhibitory Receptor Ligand PDL1 in BC
3.3. ER Signaling and the Production of Immunosuppressive Factors by BC Cells
4. The Effects of ER Signaling and the Impact of ER Modulation on Immune Cell Function in the Context of BC
4.1. ER Signaling in the Immune Cells of the Myeloid Lineage
4.2. ER Signaling in Immune Cells of the Lymphoid Lineage
5. Conclusions and Perspectives
Funding
Conflicts of Interest
References
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer–immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Gladney, W.L. Immune Escape Mechanisms as a Guide for Cancer Immunotherapy. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef]
- Clusan, L.; Ferrière, F.; Flouriot, G.; Pakdel, F. A Basic Review on Estrogen Receptor Signaling Pathways in Breast Cancer. Int. J. Mol. Sci. 2023, 24, 6834. [Google Scholar] [CrossRef]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef]
- Wolfson, B.; Padget, M.R.; Schlom, J.; Hodge, J.W. Exploiting off-target effects of estrogen deprivation to sensitize estrogen receptor negative breast cancer to immune killing. J. Immunother. Cancer 2021, 9, e002258. [Google Scholar] [CrossRef]
- Cui, X.; Schiff, R.; Arpino, G.; Osborne, C.K.; Lee, A.V. Biology of Progesterone Receptor Loss in Breast Cancer and Its Implications for Endocrine Therapy. J. Clin. Oncol. 2005, 23, 7721–7735. [Google Scholar] [CrossRef]
- Eroles, P.; Bosch, A.; Alejandro Pérez-Fidalgo, J.; Lluch, A. Molecular biology in breast cancer: Intrinsic subtypes and signaling pathways. Cancer Treat. Rev. 2012, 38, 698–707. [Google Scholar] [CrossRef]
- Goldhirsch, A.; Winer, E.P.; Coates, A.S.; Gelber, R.D.; Piccart-Gebhart, M.; Thürlimann, B.; Senn, H.-J.; Albain, K.S.; André, F.; Bergh, J.; et al. Personalizing the treatment of women with early breast cancer: Highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2013. Ann. Oncol. 2013, 24, 2206–2223. [Google Scholar] [CrossRef] [PubMed]
- Blaye, C.; Boyer, T.; Peyraud, F.; Domblides, C.; Larmonier, N. Beyond Immunosuppression: The Multifaceted Functions of Tumor-Promoting Myeloid Cells in Breast Cancers. Front. Immunol. 2022, 13, 838040. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Cortes, J.; Pusztai, L.; McArthur, H.; Kümmel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; Harbeck, N.; et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2020, 382, 810–821. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.; Cescon, D.W.; Rugo, H.S.; Nowecki, Z.; Im, S.-A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Holgado, E.; et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): A randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet 2020, 396, 1817–1828. [Google Scholar] [CrossRef]
- Goldberg, J.; Pastorello, R.G.; Vallius, T.; Davis, J.; Cui, Y.X.; Agudo, J.; Waks, A.G.; Keenan, T.; McAllister, S.S.; Tolaney, S.M.; et al. The Immunology of Hormone Receptor Positive Breast Cancer. Front. Immunol. 2021, 12, 674192. [Google Scholar] [CrossRef]
- Chung, W.; Eum, H.H.; Lee, H.-O.; Lee, K.-M.; Lee, H.-B.; Kim, K.-T.; Ryu, H.S.; Kim, S.; Lee, J.E.; Park, Y.H.; et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat. Commun. 2017, 8, 15081. [Google Scholar] [CrossRef]
- Azizi, E.; Carr, A.J.; Plitas, G.; Cornish, A.E.; Konopacki, C.; Prabhakaran, S.; Nainys, J.; Wu, K.; Kiseliovas, V.; Setty, M.; et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell 2018, 174, 1293–1308.e36. [Google Scholar] [CrossRef]
- Wagner, J.; Rapsomaniki, M.A.; Chevrier, S.; Anzeneder, T.; Langwieder, C.; Dykgers, A.; Rees, M.; Ramaswamy, A.; Muenst, S.; Soysal, S.D.; et al. A Single-Cell Atlas of the Tumor and Immune Ecosystem of Human Breast Cancer. Cell 2019, 177, 1330–1345.e18. [Google Scholar] [CrossRef]
- Segovia-Mendoza, M.; Morales-Montor, J. Immune Tumor Microenvironment in Breast Cancer and the Participation of Estrogen and Its Receptors in Cancer Physiopathology. Front. Immunol. 2019, 10, 348. [Google Scholar] [CrossRef]
- Patel, H.K.; Bihani, T. Selective estrogen receptor modulators (SERMs) and selective estrogen receptor degraders (SERDs) in cancer treatment. Pharmacol. Ther. 2018, 186, 1–24. [Google Scholar] [CrossRef]
- Kerdivel, G.; Flouriot, G.; Pakdel, F. Modulation of Estrogen Receptor Alpha Activity and Expression During Breast Cancer Progression. In Vitamins & Hormones; Elsevier: Amsterdam, The Netherlands, 2013; pp. 135–160. [Google Scholar]
- Dowsett, M.; Howell, A. Breast cancer: Aromatase inhibitors take on tamoxifen. Nat. Med. 2002, 8, 1341–1344. [Google Scholar] [CrossRef] [PubMed]
- Helguero, L.A.; Faulds, M.H.; Gustafsson, J.A.; Haldosén, L.A. Estrogen receptors alfa (ERa) and beta (ERb) differentially regulate proliferation and apoptosis of the normal murine mammary epithelial cell line HC1. Oncogene 2005, 24, 6605–6616. [Google Scholar] [CrossRef] [PubMed]
- Márquez-Garbán, D.C.; Deng, G.; Comin-Anduix, B.; Garcia, A.J.; Xing, Y.; Chen, H.-W.; Cheung-Lau, G.; Hamilton, N.; Jung, M.E.; Pietras, R.J. Antiestrogens in combination with immune checkpoint inhibitors in breast cancer immunotherapy. J. Steroid Biochem. Mol. Biol. 2019, 193, 105415. [Google Scholar] [CrossRef]
- Hurvitz, S.A.; Pietras, R.J. Rational management of endocrine resistance in breast cancer: A comprehensive review of estrogen receptor biology, treatment options, and future directions. Cancer 2008, 113, 2385–2397. [Google Scholar] [CrossRef]
- Osborne, C.K.; Schiff, R. Mechanisms of Endocrine Resistance in Breast Cancer. Annu. Rev. Med. 2011, 62, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, F.; Penln, F.; Garrido, F.; Ruiz-Cabello, F. Upmodulation by estrogen of HLA class I expression in breast tumor cell lines. Immunogenetics 1994, 39, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Song, I.H.; Park, I.A.; Heo, S.-H.; Kim, Y.-A.; Ahn, J.-H.; Gong, G. Differential expression of major histocompatibility complex class I in subtypes of breast cancer is associated with estrogen receptor and interferon signaling. Oncotarget 2016, 7, 30119–30132. [Google Scholar] [CrossRef]
- Song, I.H.; Kim, Y.-A.; Heo, S.-H.; Bang, W.S.; Park, H.S.; Choi, Y.H.; Lee, H.; Seo, J.-H.; Cho, Y.; Jung, S.W.; et al. The Association of Estrogen Receptor Activity, Interferon Signaling, and MHC Class I Expression in Breast Cancer. Cancer Res. Treat. 2022, 54, 1111–1120. [Google Scholar] [CrossRef]
- Hühn, D.; Martí-Rodrigo, P.; Mouron, S.; Hansel, C.; Tschapalda, K.; Porebski, B.; Häggblad, M.; Lidemalm, L.; Quintela-Fandino, M.; Carreras-Puigvert, J.; et al. Prolonged estrogen deprivation triggers a broad immunosuppressive phenotype in breast cancer cells. Mol. Oncol. 2022, 16, 148–165. [Google Scholar] [CrossRef]
- Li, K.; Du, H.; Lian, X.; Yang, S.; Chai, D.; Wang, C.; Yang, R.; Chen, X. Characterization of β2-microglobulin expression in different types of breast cancer. BMC Cancer 2014, 14, 750. [Google Scholar] [CrossRef]
- Agrawal, S.; Reemtsma, K.; Bagiella, E.; Oluwole, S.F.; Braunstein, N.S. Role of TAP-1 and/or TAP-2 antigen presentation defects in tumorigenicity of mouse melanoma. Cell. Immunol. 2004, 228, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.L.; Gabrilovich, D.; Tampé, R.; Girgis, K.R.; Nadaf, S.; Carbone, D.P. A functionally defective allele of TAP1 results in loss of MHC class I antigen presentation in a human lung cancer. Nat. Genet. 1996, 13, 210–213. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, A.; France, J.; Sy, M.S.; Harding, C.V. Down-regulation of the transporter for antigen presentation, proteasome subunits, and class I major histocompatibility complex in tumor cell lines. Cancer Res. 1998, 58, 3660–3667. [Google Scholar]
- Matsui, M.; Machida, S.; Itani-Yohda, T.; Akatsuka, T. Downregulation of the proteasome subunits, transporter, and antigen presentation in hepatocellular carcinoma, and their restoration by interferon-γ. J. Gastroenterol. Hepatol. 2002, 17, 897–907. [Google Scholar] [CrossRef]
- Yang, T.; McNally, B.A.; Ferrone, S.; Liu, Y.; Zheng, P. A Single-nucleotide Deletion Leads to Rapid Degradation ofTAP-1 mRNA in a Melanoma Cell Line. J. Biol. Chem. 2003, 278, 15291–15296. [Google Scholar] [CrossRef]
- Einstein, M.H.; Leanza, S.; Chiu, L.G.; Schlecht, N.F.; Goldberg, G.L.; Steinberg, B.M.; Burk, R.D. Genetic Variants in TAP Are Associated with High-Grade Cervical Neoplasia. Clin. Cancer Res. 2009, 15, 1019–1023. [Google Scholar] [CrossRef] [PubMed]
- Hage, F.E.; Durgeau, A.; Mami-Chouaib, F. TAP expression level in tumor cells defines the nature and processing of MHC class I peptides for recognition by tumor-specific cytotoxic T lymphocytes. Ann. N. Y. Acad. Sci. 2013, 1283, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Vitale, M.; Rezzani, R.; Rodella, L.; Zauli, G.; Grigolato, P.; Cadei, M.; Hicklin, D.J.; Ferrone, S. HLA class I antigen and transporter associated with antigen processing (TAP1 and TAP2) down-regulation in high-grade primary breast carcinoma lesions. Cancer Res. 1998, 58, 737–742. [Google Scholar] [PubMed]
- Kaklamanis, L.; Leek, R.; Koukourakis, M.; Gatter, K.C.; Harris, A.L. Loss of transporter in antigen processing 1 transport protein and major histocompatibility complex class I molecules in metastatic versus primary breast cancer. Cancer Res. 1995, 55, 5191–5194. [Google Scholar]
- Liu, Y.; Komohara, Y.; Domenick, N.; Ohno, M.; Ikeura, M.; Hamilton, R.L.; Horbinski, C.; Wang, X.; Ferrone, S.; Okada, H. Expression of antigen processing and presenting molecules in brain metastasis of breast cancer. Cancer Immunol. Immunother. 2012, 61, 789–801. [Google Scholar] [CrossRef]
- Henle, A.M.; Nassar, A.; Puglisi-Knutson, D.; Youssef, B.; Knutson, K.L. Downregulation of TAP1 and TAP2 in early stage breast cancer. PLoS ONE 2017, 12, e0187323. [Google Scholar] [CrossRef] [PubMed]
- Medema, J.P.; De Jong, J.; Peltenburg, L.T.C.; Verdegaal, E.M.E.; Gorter, A.; Bres, S.A.; Franken, K.L.M.C.; Hahne, M.; Albar, J.P.; Melief, C.J.M.; et al. Blockade of the granzyme B/perforin pathway through overexpression of the serine protease inhibitor PI-9/SPI-6 constitutes a mechanism for immune escape by tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 11515–11520. [Google Scholar] [CrossRef] [PubMed]
- Bots, M.; VAN Bostelen, L.; Rademaker, M.T.; Offringa, R.; Medema, J.P. Serpins prevent granzyme-induced death in a species-specific manner. Immunol. Cell Biol. 2006, 84, 79–86. [Google Scholar] [CrossRef]
- Kanamori, H.; Krieg, S.; Mao, C.; Di Pippo, V.A.; Wang, S.; Zajchowski, D.A.; Shapiro, D.J. Proteinase Inhibitor 9, an Inhibitor of Granzyme B-Mediated Apoptosis, Is a Primary Estrogen-Inducible Gene in Human Liver Cells. J. Biol. Chem. 2000, 275, 5867–5873. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Ellison, S.J.; Alarid, E.T.; Shapiro, D.J. Interplay between the levels of estrogen and estrogen receptor controls the level of the granzyme inhibitor, proteinase inhibitor 9 and susceptibility to immune surveillance by natural killer cells. Oncogene 2007, 26, 4106–4114. [Google Scholar] [CrossRef] [PubMed]
- Lauricella, M.; Carlisi, D.; Giuliano, M.; Calvaruso, G.; Cernigliaro, C.; Vento, R.; D’Anneo, A. The analysis of estrogen receptor-α positive breast cancer stem-like cells unveils a high expression of the serpin proteinase inhibitor PI-9: Possible regulatory mechanisms. Int. J. Oncol. 2016, 49, 352–360. [Google Scholar] [CrossRef]
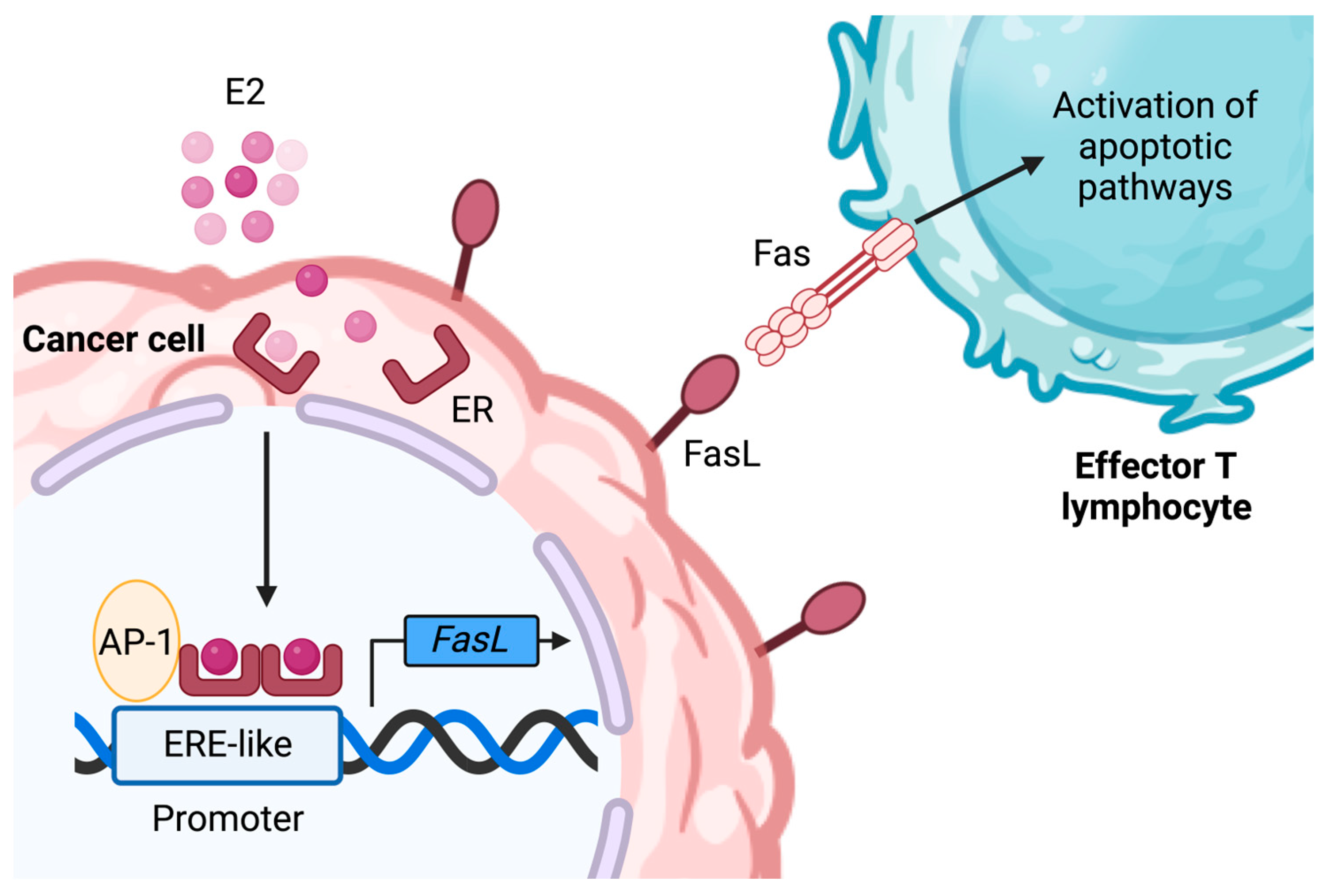
- Gutierrez, L.S.; Eliza, M.; Niven-Fairchild, T.; Naftolin, F.; Mor, G. The Fas/Fas-ligand system: A mechanism for immune evasion in human breast carcinomas. Breast Cancer Res. Treat. 1999, 54, 245–253. [Google Scholar] [CrossRef]
- Mor, G.; Kohen, F.; Garcia-Velasco, J.; Nilsen, J.; Brown, W.; Song, J.; Naftolin, F. Regulation of Fas ligand expression in breast cancer cells by estrogen: Functional differences between estradiol and tamoxifen. J. Steroid Biochem. Mol. Biol. 2000, 73, 185–194. [Google Scholar] [CrossRef]
- Song, R.X.-D.; Mor, G.; Naftolin, F.; McPherson, R.A.; Song, J.; Zhang, Z.; Yue, W.; Wang, J.; Santen, R.J. Effect of Long-Term Estrogen Deprivation on Apoptotic Responses of Breast Cancer Cells to 17β-Estradiol. JNCI J. Natl. Cancer Inst. 2001, 93, 1714–1723. [Google Scholar] [CrossRef]
- METABRIC Group; Curtis, C.; Shah, S.P.; Chin, S.-F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar]
- Sabatier, R.; Finetti, P.; Mamessier, E.; Adelaide, J.; Chaffanet, M.; Ali, H.R.; Viens, P.; Caldas, C.; Birnbaum, D.; Bertucci, F. Prognostic and predictive value of PDL1 expression in breast cancer. Oncotarget 2015, 6, 5449–5464. [Google Scholar] [CrossRef] [PubMed]
- Mittendorf, E.A.; Philips, A.V.; Meric-Bernstam, F.; Qiao, N.; Wu, Y.; Harrington, S.; Su, X.; Wang, Y.; Gonzalez-Angulo, A.M.; Akcakanat, A.; et al. PD-L1 Expression in Triple-Negative Breast Cancer. Cancer Immunol. Res. 2014, 2, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Bertucci, F.; Finetti, P.; Birnbaum, D.; Mamessier, E. The PD1/PDL1 axis, a promising therapeutic target in aggressive breast cancers. OncoImmunology 2016, 5, e1085148. [Google Scholar] [CrossRef] [PubMed]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef] [PubMed]
- Reiss, K.A.; Forde, P.M.; Brahmer, J.R. Harnessing the power of the immune system via blockade of PD-1 and PD-L1: A promising new anticancer strategy. Immunotherapy 2014, 6, 459–475. [Google Scholar] [CrossRef]
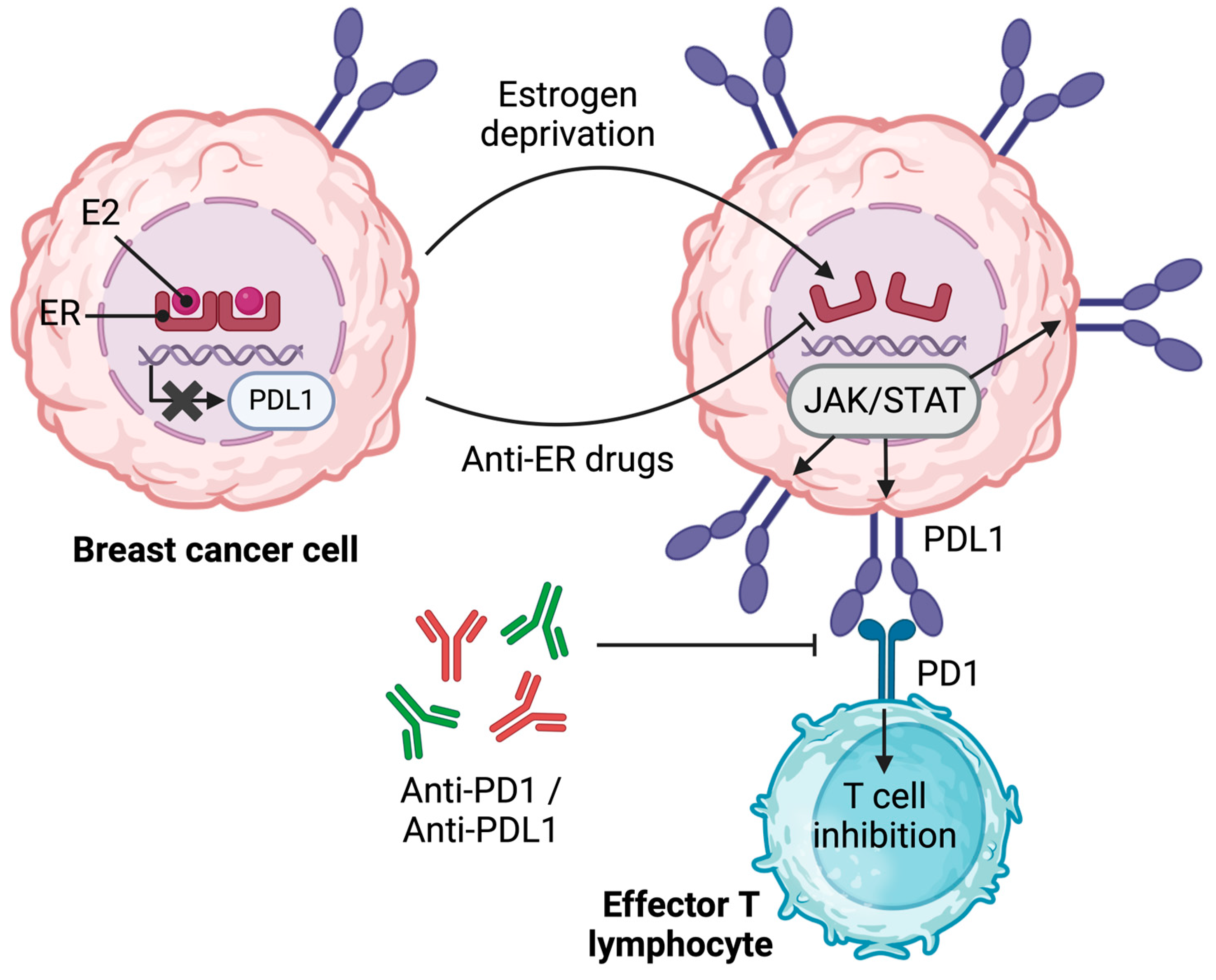
- Liu, L.; Shen, Y.; Zhu, X.; Lv, R.; Li, S.; Zhang, Z.; Shi, Y.G.; Tan, L. ERα is a negative regulator of PD-L1 gene transcription in breast cancer. Biochem. Biophys. Res. Commun. 2018, 505, 157–161. [Google Scholar] [CrossRef]
- Yang, L.; Huang, F.; Mei, J.; Wang, X.; Zhang, Q.; Wang, H.; Xi, M.; You, Z. Posttranscriptional Control of PD-L1 Expression by 17β-Estradiol via PI3K/Akt Signaling Pathway in ERα-Positive Cancer Cell Lines. Int. J. Gynecol. Cancer 2017, 27, 196–205. [Google Scholar] [CrossRef]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef]
- Pereira, B.; Chin, S.-F.; Rueda, O.M.; Vollan, H.-K.M.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.-J.; et al. The somatic mutation profiles of 2,433 breast cancers refine their genomic and transcriptomic landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef]
- Rueda, O.M.; Sammut, S.-J.; Seoane, J.A.; Chin, S.-F.; Caswell-Jin, J.L.; Callari, M.; Batra, R.; Pereira, B.; Bruna, A.; Ali, H.R.; et al. Dynamics of breast-cancer relapse reveal late-recurring ER-positive genomic subgroups. Nature 2019, 567, 399–404. [Google Scholar] [CrossRef]
- Kumar, S.; Sharawat, S.K. Epigenetic regulators of programmed death-ligand 1 expression in human cancers. Transl. Res. 2018, 202, 129–145. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Xiong, L.; Lyu, R.; Shen, Y.; Liu, L.; Li, S.; Argueta, C.; Tan, L. Regulation of TET2 gene expression and 5mC oxidation in breast cancer cells by estrogen signaling. Biochem. Biophys. Res. Commun. 2022, 589, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Liu, L.; Wang, M.; Xu, B.; Lyu, R.; Shi, Y.G.; Tan, L. TET2 Inhibits PD-L1 Gene Expression in Breast Cancer Cells through Histone Deacetylation. Cancers 2021, 13, 2207. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Chen, Y.; Bian, C.; Fujiki, R.; Yu, X. TET2 promotes histone O-GlcNAcylation during gene transcription. Nature 2013, 493, 561–564. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhao, K.; Shen, Q.; Han, Y.; Gu, Y.; Li, X.; Zhao, D.; Liu, Y.; Wang, C.; Zhang, X.; et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 2015, 525, 389–393. [Google Scholar] [CrossRef]
- Tanaka, S.; Ise, W.; Inoue, T.; Ito, A.; Ono, C.; Shima, Y.; Sakakibara, S.; Nakayama, M.; Fujii, K.; Miura, I.; et al. Tet2 and Tet3 in B cells are required to repress CD86 and prevent autoimmunity. Nat. Immunol. 2020, 21, 950–961. [Google Scholar] [CrossRef]
- Prendergast, G.C. Immune escape as a fundamental trait of cancer: Focus on IDO. Oncogene 2008, 27, 3889–3900. [Google Scholar] [CrossRef]
- Soliman, H.; Rawal, B.; Fulp, J.; Lee, J.-H.; Lopez, A.; Bui, M.M.; Khalil, F.; Antonia, S.; Yfantis, H.G.; Lee, D.H.; et al. Analysis of indoleamine 2–3 dioxygenase (IDO1) expression in breast cancer tissue by immunohistochemistry. Cancer Immunol. Immunother. 2013, 62, 829–837. [Google Scholar] [CrossRef]
- Jacquemier, J.; Bertucci, F.; Finetti, P.; Esterni, B.; Charafe-Jauffret, E.; Thibult, M.-L.; Houvenaeghel, G.; Van Den Eynde, B.; Birnbaum, D.; Olive, D.; et al. High expression of indoleamine 2,3-dioxygenase in the tumour is associated with medullary features and favourable outcome in basal-like breast carcinoma. Int. J. Cancer 2012, 130, 96–104. [Google Scholar] [CrossRef]
- Dewi, D.L.; Mohapatra, S.R.; Blanco Cabañes, S.; Adam, I.; Somarribas Patterson, L.F.; Berdel, B.; Kahloon, M.; Thürmann, L.; Loth, S.; Heilmann, K.; et al. Suppression of indoleamine-2,3-dioxygenase 1 expression by promoter hypermethylation in ER-positive breast cancer. OncoImmunology 2017, 6, e1274477. [Google Scholar] [CrossRef]
- Liu, B.; Qu, L.; Yan, S. Cyclooxygenase-2 promotes tumor growth and suppresses tumor immunity. Cancer Cell Int. 2015, 15, 106. [Google Scholar] [CrossRef] [PubMed]
- Maturu, P.; Jones, D.; Ruteshouser, E.C.; Hu, Q.; Reynolds, J.M.; Hicks, J.; Putluri, N.; Ekmekcioglu, S.; Grimm, E.A.; Dong, C.; et al. Role of Cyclooxygenase-2 Pathway in Creating an Immunosuppressive Microenvironment and in Initiation and Progression of Wilms’ Tumor. Neoplasia 2017, 19, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Parrett, M.; Harris, R.; Joarder, F.; Ross, M.; Clausen, K.; Robertson, F. Cyclooxygenase-2 gene expression in human breast cancer. Int. J. Oncol. 1997, 10, 503–507. [Google Scholar] [CrossRef]
- Hwang, D.; Byrne, J.; Scollard, D.; Levine, E. Expression of Cyclooxygenase-1 and Cyclooxygenase-2 in Human Breast Cancer. JNCI J. Natl. Cancer Inst. 1998, 90, 455–460. [Google Scholar] [CrossRef]
- Brueggemeier, R.W.; Quinn, A.L.; Parrett, M.L.; Joarder, F.S.; Harris, R.E.; Robertson, F.M. Correlation of aromatase and cyclooxygenase gene expression in human breast cancer specimens. Cancer Lett. 1999, 140, 27–35. [Google Scholar] [CrossRef]
- Soslow, R.A.; Dannenberg, A.J.; Rush, D.; Woerner, B.M.; Khan, K.N.; Masferrer, J.; Koki, A.T. COX-2 is expressed in human pulmonary, colonic, and mammary tumors. Cancer 2000, 89, 2637–2645. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.A.; Bae, J.W.; Woo, S.U.; Kim, H.; Kim, C.H. Correlation between COX-2 Expression and Hormone Receptors in Invasive Ductal Breast Cancer. J. Korean Surg. Soc. 2010, 78, 140. [Google Scholar] [CrossRef]
- Kang, H.J.; Gong, G.; Jang, S.J.; Jung, P.J.; Park, C.K. Expression of Cyclooxygenase-2 in Human Breast Carcinoma: Relevance to Tumor Angiogenesis and Expression of Estrogen Receptor. Cancer Res. Treat. 2001, 33, 286–295. [Google Scholar] [CrossRef]
- Solanki, R.; Agrawal, N.; Ansari, M.; Jain, S.; Jindal, A. COX-2 Expression in Breast Carcinoma with Correlation to Clinicopathological Parameters. Asian Pac. J. Cancer Prev. 2018, 19, 1971. [Google Scholar] [CrossRef]
- Rozenowicz, R.D.L.; Santos, R.E.D.; Silva, M.A.L.G.; Rodrigues, F.F.O.; Oliveira, A.L.D.; Ulson, L.B.; Oliveira, V.M.; Aoki, T. Cox-2 e sua associação com fatores prognósticos e resposta à quimioterapia primária em pacientes com câncer de mama. Rev. Col. Bras. Cir. 2010, 37, 323–327. [Google Scholar] [CrossRef]
- Ristimaki, A.; Sivula, A.; Lundin, J.; Lundin, M.; Salminen, T.; Haglund, C.; Joensuu, H.; Isola, J. Prognostic Significance of Elevated Cyclooxygenase-2 Expression in Breast Cancer. Cancer Res. 2002, 62, 632–635. [Google Scholar] [PubMed]
- Jana, D.; Sarkar, D.K.; Ganguly, S.; Saha, S.; Sa, G.; Manna, A.K.; Banerjee, A.; Mandal, S. Role of Cyclooxygenase 2 (COX-2) in Prognosis of Breast Cancer. Indian. J. Surg. Oncol. 2014, 5, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Basudhar, D.; Glynn, S.A.; Greer, M.; Somasundaram, V.; No, J.H.; Scheiblin, D.A.; Garrido, P.; Heinz, W.F.; Ryan, A.E.; Weiss, J.M.; et al. Coexpression of NOS2 and COX2 accelerates tumor growth and reduces survival in estrogen receptor-negative breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, 13030–13035. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, D.; Young, A.; Teng, M.W.L.; Smyth, M.J. Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer 2017, 17, 709–724. [Google Scholar] [CrossRef]
- Spychala, J.; Lazarowski, E.; Ostapkowicz, A.; Ayscue, L.H.; Jin, A.; Mitchell, B.S. Role of Estrogen Receptor in the Regulation of Ecto-5′-Nucleotidase and Adenosine in Breast Cancer. Clin. Cancer Res. 2004, 10, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Spychala, J.; Zimmermann, A.G.; Mitchell, B.S. Tissue-specific Regulation of the Ecto-5′-nucleotidase Promoter. J. Biol. Chem. 1999, 274, 22705–22712. [Google Scholar] [CrossRef]
- Chavey, C.; Bibeau, F.; Gourgou-Bourgade, S.; Burlinchon, S.; Boissière, F.; Laune, D.; Roques, S.; Lazennec, G. Oestrogen receptor negative breast cancers exhibit high cytokine content. Breast Cancer Res. 2007, 9, R15. [Google Scholar] [CrossRef]
- Chu, P.-Y.; Wang, S.-M.; Chen, P.-M.; Tang, F.-Y.; Chiang, E.-P.I. Expression of MTDH and IL-10 Is an Independent Predictor of Worse Prognosis in ER-Negative or PR-Negative Breast Cancer Patients. J. Clin. Med. 2020, 9, 3153. [Google Scholar] [CrossRef]
- Ewan, K.B.R.; Oketch-Rabah, H.A.; Ravani, S.A.; Shyamala, G.; Moses, H.L.; Barcellos-Hoff, M.H. Proliferation of Estrogen Receptor-α-Positive Mammary Epithelial Cells Is Restrained by Transforming Growth Factor-β1 in Adult Mice. Am. J. Pathol. 2005, 167, 409–417. [Google Scholar] [CrossRef]
- Knabbe, C.; Lippman, M.E.; Wakefield, L.M.; Flanders, K.C.; Kasid, A.; Derynck, R.; Dickson, R.B. Evidence that transforming growth factor-β is a hormonally regulated negative growth factor in human breast cancer cells. Cell 1987, 48, 417–428. [Google Scholar] [CrossRef]
- Knabbe, C.; Kopp, A.; Hilgers, W.; Lang, D.; Müller, V.; Zugmaier, G.; Jonat, W. Regulation and Role of TGF? Production in Breast Cancer. Ann. N. Y. Acad. Sci. 1996, 784, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Joffroy, C.M.; Buck, M.B.; Stope, M.B.; Popp, S.L.; Pfizenmaier, K.; Knabbe, C. Antiestrogens Induce Transforming Growth Factor β–Mediated Immunosuppression in Breast Cancer. Cancer Res. 2010, 70, 1314–1322. [Google Scholar] [CrossRef] [PubMed]
- Banka, C.L.; Lund, C.V.; Nguyen, M.T.N.; Pakchoian, A.J.; Mueller, B.M.; Eliceiri, B.P. Estrogen induces lung metastasis through a host compartment-specific response. Cancer Res. 2006, 66, 3667–3672. [Google Scholar] [CrossRef] [PubMed]
- Curran, E.M.; Judy, B.M.; Duru, N.A.; Wang, H.-Q.; Vergara, L.A.; Lubahn, D.B.; Estes, D.M. Estrogenic regulation of host immunity against an estrogen receptor-negative human breast cancer. Clin. Cancer Res. 2006, 12, 5641–5647. [Google Scholar] [CrossRef]
- Gupta, P.B.; Proia, D.; Cingoz, O.; Weremowicz, J.; Naber, S.P.; Weinberg, R.A.; Kuperwasser, C. Systemic stromal effects of estrogen promote the growth of estrogen receptor-negative cancers. Cancer Res. 2007, 67, 2062–2071. [Google Scholar] [CrossRef]
- Iyer, V.; Klebba, I.; McCready, J.; Arendt, L.M.; Betancur-Boissel, M.; Wu, M.-F.; Zhang, X.; Lewis, M.T.; Kuperwasser, C. Estrogen promotes ER-negative tumor growth and angiogenesis through mobilization of bone marrow-derived monocytes. Cancer Res. 2012, 72, 2705–2713. [Google Scholar] [CrossRef]
- Yang, X.; Belosay, A.; Du, M.; Fan, T.M.; Turner, R.T.; Iwaniec, U.T.; Helferich, W.G. Estradiol increases ER-negative breast cancer metastasis in an experimental model. Clin. Exp. Metastasis 2013, 30, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, L.; Chang, W.; Fang, B.; Qin, J.; Qu, X.; Cheng, F. Estrogen-induced SDF-1α production promotes the progression of ER-negative breast cancer via the accumulation of MDSCs in the tumor microenvironment. Sci. Rep. 2016, 6, 39541. [Google Scholar] [CrossRef]
- Svoronos, N.; Perales-Puchalt, A.; Allegrezza, M.J.; Rutkowski, M.R.; Payne, K.K.; Tesone, A.J.; Nguyen, J.M.; Curiel, T.J.; Cadungog, M.G.; Singhal, S.; et al. Tumor Cell-Independent Estrogen Signaling Drives Disease Progression through Mobilization of Myeloid-Derived Suppressor Cells. Cancer Discov. 2017, 7, 72–85. [Google Scholar] [CrossRef]
- Tower, H.; Dall, G.; Davey, A.; Stewart, M.; Lanteri, P.; Ruppert, M.; Lambouras, M.; Nasir, I.; Yeow, S.; Darcy, P.K.; et al. Estrogen-induced immune changes within the normal mammary gland. Sci. Rep. 2022, 12, 18986. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Condamine, T.; Ramachandran, I.; Youn, J.-I.; Gabrilovich, D.I. Regulation of Tumor Metastasis by Myeloid-Derived Suppressor Cells. Annu. Rev. Med. 2015, 66, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Ouzounova, M.; Lee, E.; Piranlioglu, R.; El Andaloussi, A.; Kolhe, R.; Demirci, M.F.; Marasco, D.; Asm, I.; Chadli, A.; Hassan, K.A.; et al. Monocytic and granulocytic myeloid derived suppressor cells differentially regulate spatiotemporal tumour plasticity during metastatic cascade. Nat. Commun. 2017, 8, 14979. [Google Scholar] [CrossRef]
- Nalbandian, G.; Paharkova-Vatchkova, V.; Mao, A.; Nale, S.; Kovats, S. The Selective Estrogen Receptor Modulators, Tamoxifen and Raloxifene, Impair Dendritic Cell Differentiation and Activation. J. Immunol. 2005, 175, 2666–2675. [Google Scholar] [CrossRef] [PubMed]
- Pelekanou, V.; Kampa, M.; Kiagiadaki, F.; Deli, A.; Theodoropoulos, P.; Agrogiannis, G.; Patsouris, E.; Tsapis, A.; Castanas, E.; Notas, G. Estrogen anti-inflammatory activity on human monocytes is mediated through cross-talk between estrogen receptor ERα36 and GPR30/GPER1. J. Leukoc. Biol. 2016, 99, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Gide, T.N.; Quek, C.; Menzies, A.M.; Tasker, A.T.; Shang, P.; Holst, J.; Madore, J.; Lim, S.Y.; Velickovic, R.; Wongchenko, M.; et al. Distinct Immune Cell Populations Define Response to Anti-PD-1 Monotherapy and Anti-PD-1/Anti-CTLA-4 Combined Therapy. Cancer Cell 2019, 35, 238–255.e6. [Google Scholar] [CrossRef]
- Chakraborty, B.; Byemerwa, J.; Shepherd, J.; Haines, C.N.; Baldi, R.; Gong, W.; Liu, W.; Mukherjee, D.; Artham, S.; Lim, F.; et al. Inhibition of estrogen signaling in myeloid cells increases tumor immunity in melanoma. J. Clin. Investig. 2021, 131, e151347. [Google Scholar] [CrossRef]
- Straub, R.H. The Complex Role of Estrogens in Inflammation. Endocr. Rev. 2007, 28, 521–574. [Google Scholar] [CrossRef]
- Kovats, S. Estrogen receptors regulate innate immune cells and signaling pathways. Cell. Immunol. 2015, 294, 63–69. [Google Scholar] [CrossRef]
- Gilmore, W.; Weiner, L.P.; Correale, J. Effect of estradiol on cytokine secretion by proteolipid protein-specific T cell clones isolated from multiple sclerosis patients and normal control subjects. J. Immunol. 1997, 158, 446–451. [Google Scholar] [CrossRef]
- Correale, J.; Arias, M.; Gilmore, W. Steroid hormone regulation of cytokine secretion by proteolipid protein-specific CD4+ T cell clones isolated from multiple sclerosis patients and normal control subjects. J. Immunol. 1998, 161, 3365–3374. [Google Scholar] [CrossRef] [PubMed]
- Bao, M.; Yang, Y.; Jun, H.-S.; Yoon, J.-W. Molecular mechanisms for gender differences in susceptibility to T cell-mediated autoimmune diabetes in nonobese diabetic mice. J. Immunol. 2002, 168, 5369–5375. [Google Scholar] [CrossRef] [PubMed]
- Maret, A.; Coudert, J.D.; Garidou, L.; Foucras, G.; Gourdy, P.; Krust, A.; Dupont, S.; Chambon, P.; Druet, P.; Bayard, F.; et al. Estradiol enhances primary antigen-specific CD4 T cell responses and Th1 development in vivo. Essential role of estrogen receptor alpha expression in hematopoietic cells. Eur. J. Immunol. 2003, 33, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Purohit, A.; Newman, S.P.; Reed, M.J. The role of cytokines in regulating estrogen synthesis: Implications for the etiology of breast cancer. Breast Cancer Res. 2002, 4, 65. [Google Scholar] [CrossRef]
- Polanczyk, M.J.; Hopke, C.; Huan, J.; Vandenbark, A.A.; Offner, H. Enhanced FoxP3 expression and Treg cell function in pregnant and estrogen-treated mice. J. Neuroimmunol. 2005, 170, 85–92. [Google Scholar] [CrossRef]
- Polanczyk, M.J.; Carson, B.D.; Subramanian, S.; Afentoulis, M.; Vandenbark, A.A.; Ziegler, S.F.; Offner, H. Cutting Edge: Estrogen Drives Expansion of the CD4+ CD25+ Regulatory T Cell Compartment. J. Immunol. 2004, 173, 2227–2230. [Google Scholar] [CrossRef]
- Polanczyk, M.J.; Hopke, C.; Vandenbark, A.A.; Offner, H. Treg suppressive activity involves estrogen-dependent expression of programmed death-1 (PD-1). Int. Immunol. 2007, 19, 337–343. [Google Scholar] [CrossRef]
- Hao, S.; Zhao, J.; Zhou, J.; Zhao, S.; Hu, Y.; Hou, Y. Modulation of 17β-estradiol on the number and cytotoxicity of NK cells in vivo related to MCM and activating receptors. Int. Immunopharmacol. 2007, 7, 1765–1775. [Google Scholar] [CrossRef]
- Gabrilovac, J.; Zadjelović, J.; Osmak, M.; Suchanek, E.; Zupanović, Z.; Boranić, M. NK cell activity and estrogen hormone levels during normal human pregnancy. Gynecol. Obs. Investig. 1988, 25, 165–172. [Google Scholar] [CrossRef]
- Ahmed, S.A.; Aufdemorte, T.B.; Chen, J.-R.; Montoya, A.I.; Olive, D.; Talal, N. Estrogen induces the development of autoantibodies and promotes salivary gland lymphoid infiltrates in normal mice. J. Autoimmun. 1989, 2, 543–552. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Hanker, A.B.; Sudhan, D.R.; Arteaga, C.L. Overcoming Endocrine Resistance in Breast Cancer. Cancer Cell 2020, 37, 496–513. [Google Scholar] [CrossRef] [PubMed]
- Rugo, H.S.; Delord, J.-P.; Im, S.-A.; Ott, P.A.; Piha-Paul, S.A.; Bedard, P.L.; Sachdev, J.; Tourneau, C.L.; Van Brummelen, E.M.J.; Varga, A.; et al. Safety and Antitumor Activity of Pembrolizumab in Patients with Estrogen Receptor–Positive/Human Epidermal Growth Factor Receptor 2–Negative Advanced Breast Cancer. Clin. Cancer Res. 2018, 24, 2804–2811. [Google Scholar] [CrossRef] [PubMed]
- Rigiracciolo, D.C.; Santolla, M.F.; Lappano, R.; Vivacqua, A.; Cirillo, F.; Galli, G.R.; Talia, M.; Muglia, L.; Pellegrino, M.; Nohata, N.; et al. Focal adhesion kinase (FAK) activation by estrogens involves GPER in triple-negative breast cancer cells. J. Exp. Clin. Cancer Res. 2019, 38, 58. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Ma, D.; Chen, S.; Tang, R.; Yang, J.; Meng, C.; Feng, Y.; Liu, L.; Wang, J.; Luo, H.; et al. High GPER expression in triple-negative breast cancer is linked to pro-metastatic pathways and predicts poor patient outcomes. NPJ Breast Cancer 2022, 8, 100. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecular Actor Investigated | Pre-Clinical Observations | Clinical Observations | References | |
|---|---|---|---|---|
| In Vitro | In Vivo | |||
| MHC I (HLA-I) presentation | HLA-I expression is enhanced by E2 in MCF-7 (ER+) but not in MDA-MB-231/MDA-MB-435s (ER−) Inverse correlation between ER status and HLA-A,B,C gene expression in BC cell lines ER antagonizing increases HLA-A,B,C proteins in ER+ MCF-7 and T47D BC cell lines | No studies performed to date | HLA-A,B,C genes negatively correlate with ER in treatment-naïve patients with primary BC HLA-A,B,C increased following treatment with estrogen modulators (tamoxifen, goserelin) | [23,24,25] |
| β-2- microglobulin | No studies performed to date | Reduced protein expression in ER+ patients; no difference of expression at the mRNA level | [27] | |
| TAP1/2 | No studies performed to date | No clear association with ER status | [36,37,38] | |
| PI-9 | E2 and tamoxifen are inducers of the protease, while raloxifene and fulvestrant block its production in MCF-7 cells MCF-7 CSC expresses a higher level of PI-9 | No studies performed to date | No studies performed to date | [42,43] |
| FasL | E2 increases FasL mRNA in MCF-7 and T47D cells; tamoxifen decreases it | No studies performed to date | No studies performed to date | [45,46] |
| Immune checkpoint molecules | PDL1 protein is downregulated in ER+ BC cells lines (MCF-7, T47D, CAMA-1, ZR-75-1, BT-474) but not ER− ones (MDA-MB-231, HCC1937, BT-549) | Tamoxifen treatment of tumor-bearing MMTV-PyMT upregulates tumor PDL1 expression | PDL1 mRNA inversely correlates with ERα (TCGA) Patients treated with anti-hormonal therapy have an increased mRNA expression of PDL1, PDL1, LGALS9, CD86, and CD48 | [26,47,53,54,56,57] |
| IDO | Prominent methylation of the IDO1 promoter in ER+ (MCF-7, ZR-75-1, BT-474) compared to ER− (MDA-MB-231) BC cells | No studies performed to date | One study shows increased IDO protein in ER+ BC [65] IDO mRNA is found in higher quantities in ER− BC specimens; there is a negative correlation between ESR1 and IDO1 | [65,66,67] |
| Cox2 NOS2 | No studies performed to date | No studies performed to date | Contradictory observations have been reported | [74,75,76,77,78,79] |
| CD73 (eN) | Low to undetectable levels in ER+ BC cells (MCF-7, ZR-75-1), which is reversed by tamoxifen or fulvestrant treatment High activity of the enzyme in ER− BC cells (MDA-MD-231, BT-549) | No studies performed to date | No studies performed to date | [82,83] |
| Immunosuppressive cytokines | No studies performed to date | No studies performed to date | Contradictory observations have been reported | [84,85] |
| TGF-β secretion by MCF-7 is enhanced following tamoxifen and fulvestrant treatment | No studies performed to date | No studies performed to date | [88,89] | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moisand, A.; Madéry, M.; Boyer, T.; Domblides, C.; Blaye, C.; Larmonier, N. Hormone Receptor Signaling and Breast Cancer Resistance to Anti-Tumor Immunity. Int. J. Mol. Sci. 2023, 24, 15048. https://doi.org/10.3390/ijms242015048
Moisand A, Madéry M, Boyer T, Domblides C, Blaye C, Larmonier N. Hormone Receptor Signaling and Breast Cancer Resistance to Anti-Tumor Immunity. International Journal of Molecular Sciences. 2023; 24(20):15048. https://doi.org/10.3390/ijms242015048
Chicago/Turabian StyleMoisand, Alexandra, Mathilde Madéry, Thomas Boyer, Charlotte Domblides, Céline Blaye, and Nicolas Larmonier. 2023. "Hormone Receptor Signaling and Breast Cancer Resistance to Anti-Tumor Immunity" International Journal of Molecular Sciences 24, no. 20: 15048. https://doi.org/10.3390/ijms242015048