

Genome-Wide Identification and Involvement in Response to Biotic and Abiotic Stresses of lncRNAs in Turbot (Scophthalmus maximus)
Abstract
:1. Introduction
2. Results
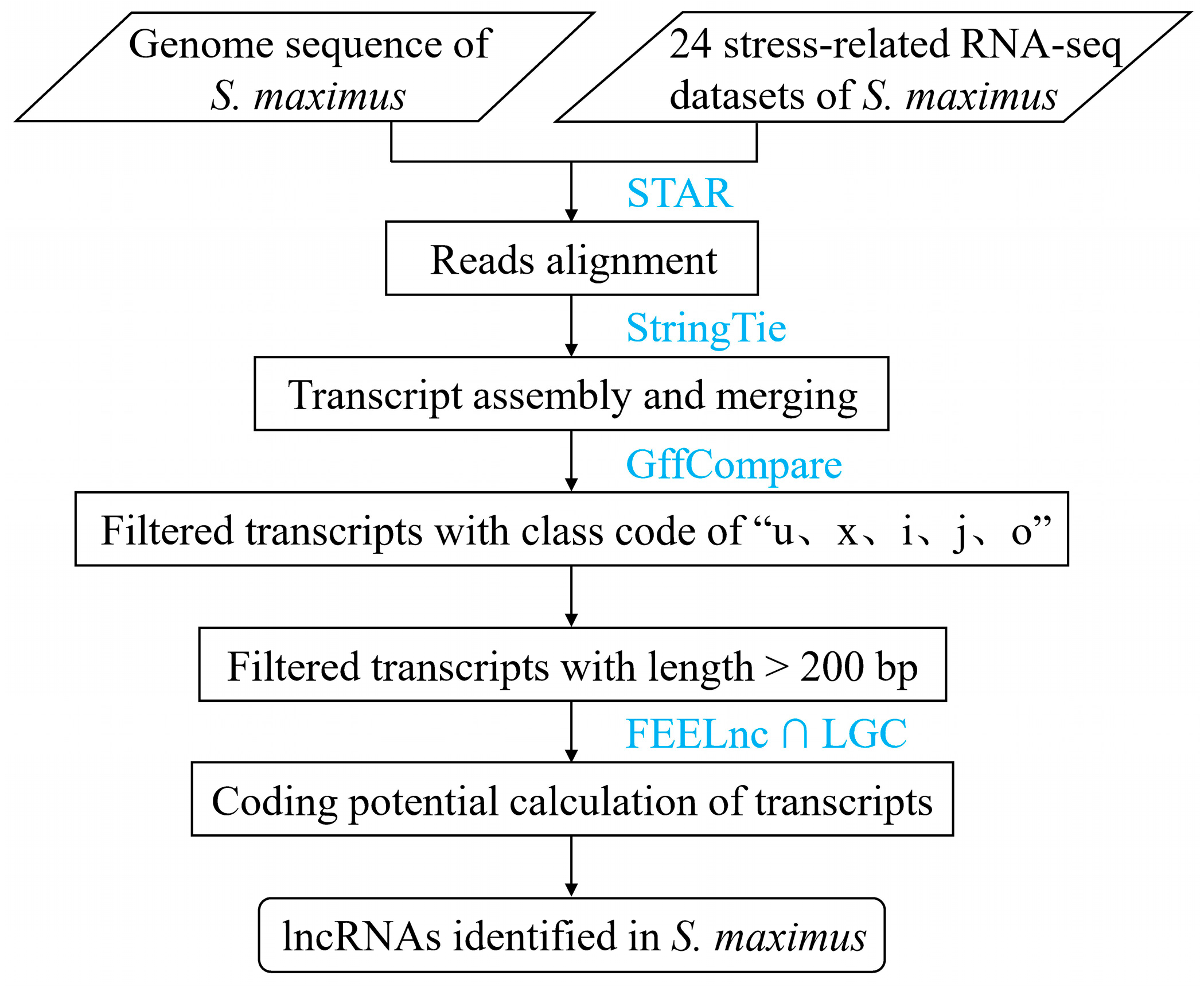
2.1. Genome-Wide Identification of lncRNAs in Turbot

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | SRA Study | Tissue | Number of Samples | Platform (Illumina) | Size (Gb) | Reference | |
|---|---|---|---|---|---|---|---|
| Crowding | - | SRP129900 | kidney, spleen | 12 | HiSeq 4000 | 68.2 | [56] |
| Feeding | myo-inositol | SRP188583 | gill | 15 | HiSeq 4000 | 115.45 | [57] |
| fish meal, soybean meal | SRP074811 | intestine | 2 | NextSeq 500 | 42.56 | [58] | |
| sodium butyrate, soybean meal | SRP275545 | intestine | 6 | HiSeq 2000 | 50.23 | [59] | |
| Heat | 14, 23, 25, 28 °C | SRP152627 | kidney | 10 | HiSeq 4000 | 88.99 | [40] |
| 14, 20, 24, 28 °C | SRP273870 | liver | 12 | HiSeq 2500 | 84.49 | [60] | |
| Oxygen | - | SRP167318 | gill | 9 | HiSeq 2500 | 58.99 | [43] |
| Pathogen | Enteromyxum scophthalmi | SRP308109 | blood | 49 | HiSeq 4000 | 381.62 | [44] |
| SRP255305 | thymus | 10 | HiSeq 4000 | 17.55 | [52] | ||
| SRP065375 | kidney, pyloric caeca, spleen | 12 | HiSeq 2000 | 31.48 | [46] | ||
| SRP050607 | kidney, pyloric caeca, spleen | 12 | HiSeq 2000 | 36.02 | [51] | ||
| Vibrio anguillarum | SRP191266 | intestine | 4 | HiSeq 2500 | 53.34 | [45] | |
| SRP336094 | liver | 2 | NovaSeq 6000 | 12.68 | [47] | ||
| SRP335896 | kidney | 2 | NovaSeq 6000 | 12.47 | [47] | ||
| SRP320422 | gill | 2 | NovaSeq 6000 | 13.58 | [47] | ||
| SRP319434 | spleen | 2 | NovaSeq 6000 | 12.73 | [47] | ||
| Megalocytivirus | SRP347383 | kidney | 18 | HiSeq 2500 | 172.43 | [61] | |
| Salinity | low-salinity | SRP277001 | liver | 6 | HiSeq 4000 | 49.35 | [41] |
| low- and high-salinity | SRP238143 | gill | 9 | HiSeq 2000 | 70.48 | [62] | |
| low- and high-salinity | SRP153594 | kidney | 9 | HiSeq 4000 | 70.86 | [42] | |
| Growth | - | SRP075669 | brain, muscle | 12 | HiSeq 2500 | 36.61 | [63] |
| Sex | - | SRP136753 | testis | 18 | HiSeq X Ten | 120.7 | [64] |
| SRP261889 | testis, ovary | 3 | HiSeq 4000 | 83.12 | - | ||
| SRP287484 | ovary | 12 | HiSeq 2500 | 197.75 | - | ||
| Total | - | 248 | - | 1881.68 | |||
2.2. Sequence Characteristics of lncRNA Transcripts in Turbot
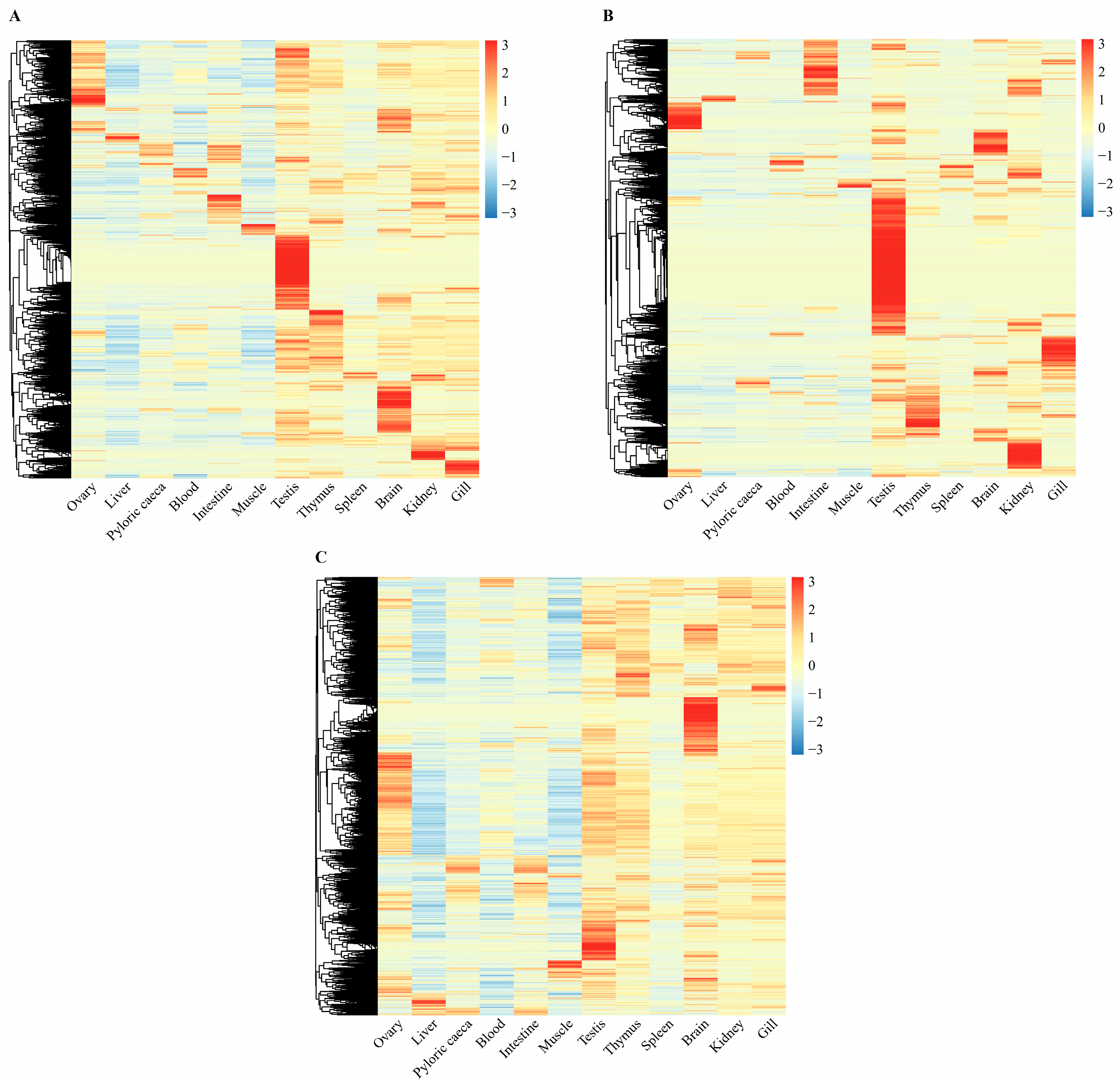
2.3. Tissue-Specific Expression of lncRNAs in Turbot
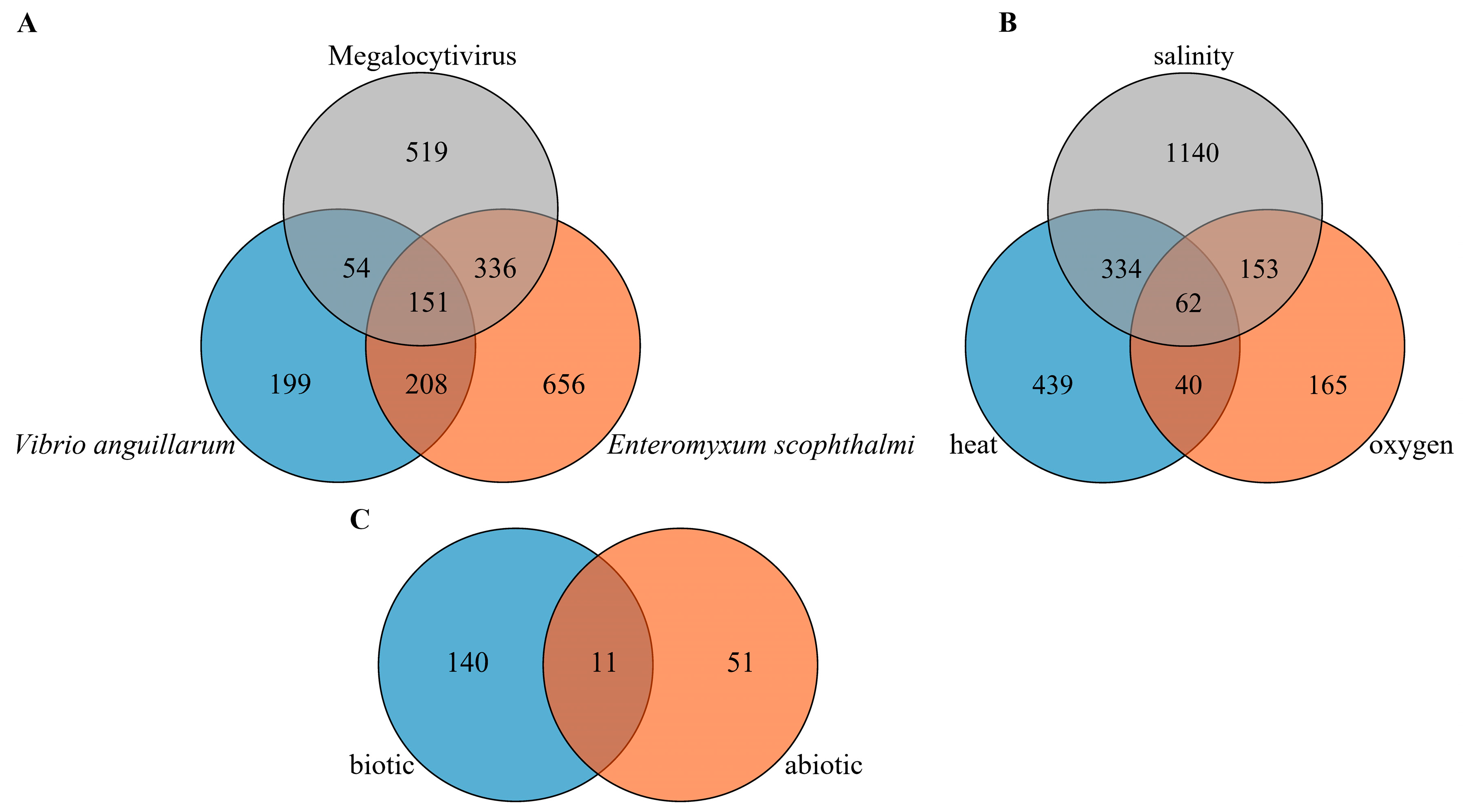
2.4. Differentially Expressed lncRNAs under Biotic Stress Conditions in Turbot
2.5. Differentially Expressed lncRNAs under Abiotic Stress Conditions in Turbot
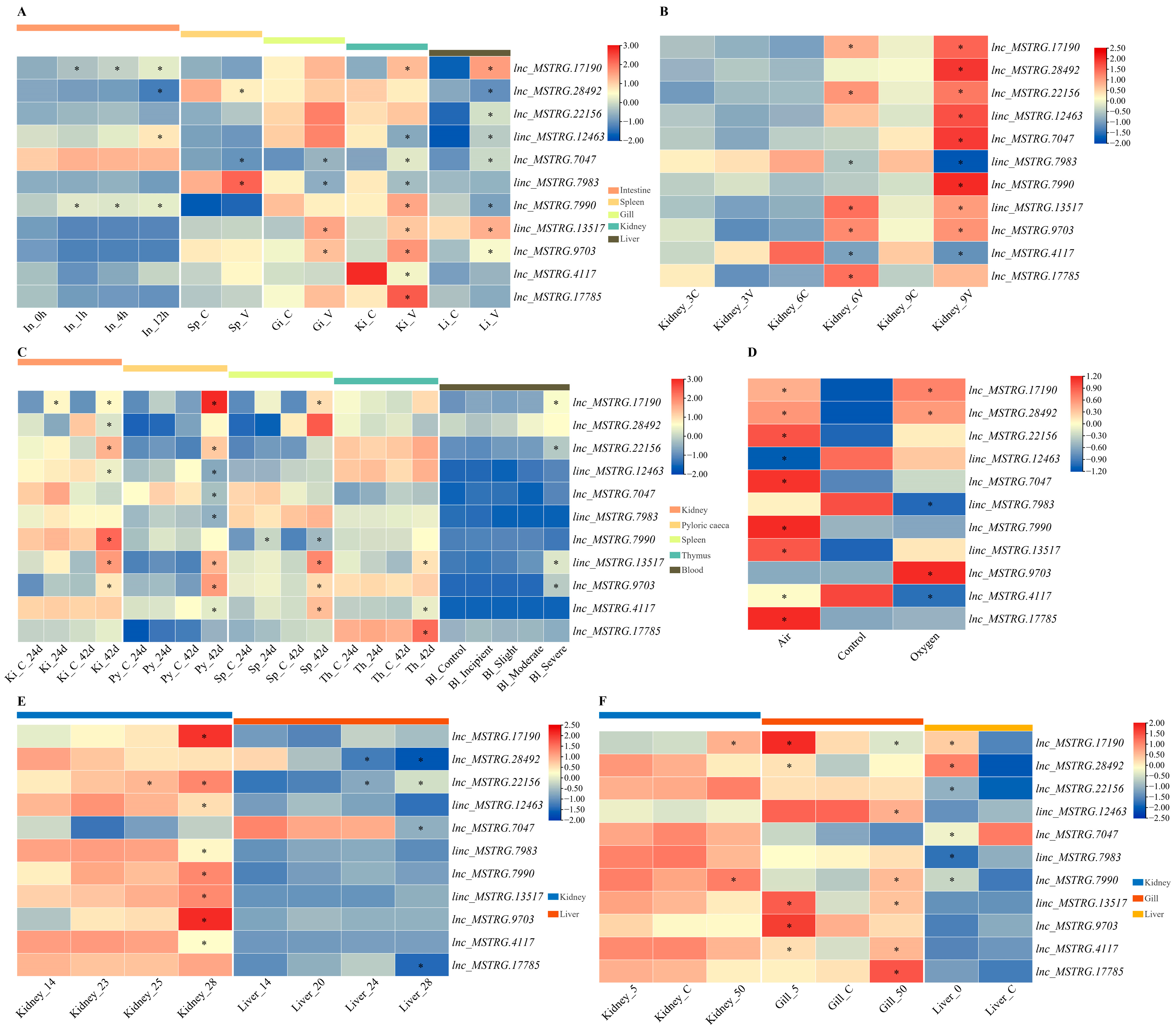
2.6. Expression Patterns of Comprehensive Stress-Responsive lncRNA Candidates under Biotic Stress Conditions
2.7. Expression Patterns of HSP70 Genes under Biotic Stress
2.8. Functional Prediction of Comprehensive Stress-Responsive lncRNA Candidates
2.9. qPCR Validation of Comprehensive Stress-Responsive lncRNA Candidates
3. Discussion
4. Materials and Methods
4.1. RNA-seq Datasets Used for the Identification of lncRNAs in Turbot
4.2. Bioinformatics Pipeline for Identifying lncRNAs
4.3. Analysis of Sequence Characteristics of lncRNA Transcripts
4.4. Tissue Expression Analysis of lncRNAs
4.5. Identification of Differentially Expressed lncRNAs under Abiotic and Biotic Stresses
4.6. Expression Patterns of the Comprehensive Stress-Responsive lncRNA Candidates
4.7. Functional Prediction of the Comprehensive Stress-Responsive lncRNA Candidates
4.8. Expression Analyses of HSP70 Genes
4.9. qPCR Validation of the Comprehensive Stress-Responsive lncRNA Candidates
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guttman, M.; Russell, P.; Ingolia, N.T.; Weissman, J.S.; Lander, E.S. Ribosome profiling provides evidence that large noncoding RNAs do not encode proteins. Cell 2013, 154, 240–251. [Google Scholar] [CrossRef]
- Bhartiya, D.; Kapoor, S.; Jalali, S.; Sati, S.; Kaushik, K.; Sachidanandan, C.; Sivasubbu, S.; Scaria, V. Conceptual approaches for lncRNA drug discovery and future strategies. Expert Opin. Drug Discov. 2012, 7, 503–513. [Google Scholar] [CrossRef]
- Liao, Q.; Liu, C.; Yuan, X.; Kang, S.; Miao, R.; Xiao, H.; Zhao, G.; Luo, H.; Bu, D.; Zhao, H.; et al. Large-scale prediction of long non-coding RNA functions in a coding–non-coding gene co-expression network. Nucleic Acids Res. 2011, 39, 3864–3878. [Google Scholar] [CrossRef]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and functions of long noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef]
- Du Toit, A. RNA stability control by Pol II. Nat. Rev. Mol. Cell Biol. 2013, 14, 128–129. [Google Scholar] [CrossRef]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef]
- Wu, J.; Okada, T.; Fukushima, T.; Tsudzuki, T.; Sugiura, M.; Yukawa, Y. A novel hypoxic stress-responsive long non-coding RNA transcribed by RNA polymerase III in Arabidopsis. RNA Biol. 2012, 9, 302–313. [Google Scholar] [CrossRef]
- Rinn, J.L.; Chang, H.Y. Genome Regulation by Long Noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef]
- Mattick, J.S.; Rinn, J.L. Discovery and annotation of long noncoding RNAs. Nat. Struct. Mol. Biol. 2015, 22, 5–7. [Google Scholar] [CrossRef]
- Yadav, V.K.; Sawant, S.V.; Yadav, A.; Jalmi, S.K.; Kerkar, S. Genome-wide analysis of long non-coding RNAs under diel light exhibits role in floral development and the circadian clock in Arabidopsis thaliana. Int. J. Biol. Macromol. 2022, 223, 1693–1704. [Google Scholar] [CrossRef]
- Sun, Z.; Huang, K.; Han, Z.; Wang, P.; Fang, Y. Genome-wide identification of Arabidopsis long noncoding RNAs in response to the blue light. Sci. Rep. 2020, 10, 6229. [Google Scholar] [CrossRef]
- Chen, B.; Zhang, Y.; Zhang, X.; Jia, S.; Chen, S.; Kang, L. Genome-wide identification and developmental expression profiling of long noncoding RNAs during Drosophila metamorphosis. Sci. Rep. 2016, 6, 23330. [Google Scholar] [CrossRef]
- Gopalakrishnan, K.; Kumarasamy, S.; Mell, B.; Joe, B. Genome-Wide Identification of Long Noncoding RNAs in Rat Models of Cardiovascular and Renal Disease. Hypertension 2015, 65, 200–210. [Google Scholar] [CrossRef]
- Jia, H.; Osak, M.; Bogu, G.K.; Stanton, L.W.; Johnson, R.; Lipovich, L. Genome-wide computational identification and manual annotation of human long noncoding RNA genes. RNA 2010, 16, 1478–1487. [Google Scholar] [CrossRef]
- Quan, J.; Kang, Y.; Luo, Z.; Zhao, G.; Ma, F.; Li, L.; Liu, Z. Identification and characterization of long noncoding RNAs provide insight into the regulation of gene expression in response to heat stress in rainbow trout (Oncorhynchus mykiss). Comp. Biochem. Physiol. Part D Genom. Proteom. 2020, 36, 100707. [Google Scholar] [CrossRef]
- Al-Tobasei, R.; Paneru, B.; Salem, M. Genome-Wide Discovery of Long Non-Coding RNAs in Rainbow Trout. PLoS ONE 2016, 11, e0148940. [Google Scholar] [CrossRef]
- Dettleff, P.; Hormazabal, E.; Aedo, J.; Fuentes, M.; Meneses, C.; Molina, A.; Valdes, J.A. Identification and Evaluation of Long Noncoding RNAs in Response to Handling Stress in Red Cusk-Eel (Genypterus chilensis) via RNA-seq. Mar. Biotechnol. 2020, 22, 94–108. [Google Scholar] [CrossRef]
- Tarifeño-Saldivia, E.; Valenzuela-Miranda, D.; Gallardo-Escárate, C. In the shadow: The emerging role of long non-coding RNAs in the immune response of Atlantic salmon. Dev. Comp. Immunol. 2017, 73, 193–205. [Google Scholar] [CrossRef]
- Li, B.J.; Jiang, D.L.; Meng, Z.N.; Zhang, Y.; Zhu, Z.X.; Lin, H.R.; Xia, J.H. Genome-wide identification and differentially expression analysis of lncRNAs in tilapia. BMC Genom. 2018, 19, 729. [Google Scholar] [CrossRef]
- Wang, X.; Lin, J.; Li, F.; Zhang, C.; Li, J.; Wang, C.; Dahlgren, R.A.; Zhang, H.; Wang, H. Screening and functional identification of lncRNAs under β-diketone antibiotic exposure to zebrafish (Danio rerio) using high-throughput sequencing. Aquat. Toxicol. 2017, 182, 214–225. [Google Scholar] [CrossRef]
- Jiang, L.; Liu, W.; Zhu, A.; Zhang, J.; Zhou, J.; Wu, C. Transcriptome analysis demonstrate widespread differential expression of long noncoding RNAs involve in Larimichthys crocea immune response. Fish Shellfish Immunol. 2016, 51, 1–8. [Google Scholar] [CrossRef]
- Leiva, F.; Rojas-Herrera, M.; Reyes, D.; Bravo, S.; Garcia, K.K.; Moya, J.; Vidal, R. Identification and characterization of miRNAs and lncRNAs of coho salmon (Oncorhynchus kisutch) in normal immune organs. Genomics 2020, 112, 45–54. [Google Scholar] [CrossRef]
- Valenzuela-Muñoz, V.; Valenzuela-Miranda, D.; Gallardo-Escárate, C. Comparative analysis of long non-coding RNAs in Atlantic and Coho salmon reveals divergent transcriptome responses associated with immunity and tissue repair during sea lice infestation. Dev. Comp. Immunol. 2018, 87, 36–50. [Google Scholar] [CrossRef]
- Basu, S.; Hadzhiev, Y.; Petrosino, G.; Nepal, C.; Gehrig, J.; Armant, O.; Ferg, M.; Strahle, U.; Sanges, R.; Müller, F. The Tetraodon nigroviridis reference transcriptome: Developmental transition, length retention and microsynteny of long non-coding RNAs in a compact vertebrate genome. Sci. Rep. 2016, 6, 33210. [Google Scholar] [CrossRef]
- Hu, F.; Xu, K.; Zhou, Y.; Wu, C.; Wang, S.; Xiao, J.; Wen, M.; Zhao, R.; Luo, K.; Tao, M.; et al. Different expression patterns of sperm motility-related genes in testis of diploid and tetraploid cyprinid fish. Biol. Reprod. 2017, 96, 907–920. [Google Scholar] [CrossRef]
- Mallory, A.C.; Shkumatava, A. LncRNAs in vertebrates: Advances and challenges. Biochimie 2015, 117, 3–14. [Google Scholar] [CrossRef]
- Long, Y.; Wang, X.; Youmans, D.T.; Cech, T.R. How do lncRNAs regulate transcription? Sci. Adv. 2017, 3, eaao2110. [Google Scholar] [CrossRef]
- Yoon, J.-H.; Abdelmohsen, K.; Gorospe, M. Posttranscriptional Gene Regulation by Long Noncoding RNA. J. Mol. Biol. 2013, 425, 3723–3730. [Google Scholar] [CrossRef]
- Mercer, T.R.; Mattick, J.S. Structure and function of long noncoding RNAs in epigenetic regulation. Nat. Struct. Mol. Biol. 2013, 20, 300–307. [Google Scholar] [CrossRef]
- Li, J.; Tian, H.; Yang, J.; Gong, Z. Long Noncoding RNAs Regulate Cell Growth, Proliferation, and Apoptosis. DNA Cell Biol. 2016, 35, 459–470. [Google Scholar] [CrossRef]
- Kim, C.; Kang, D.; Lee, E.K.; Lee, J.-S. Long Noncoding RNAs and RNA-Binding Proteins in Oxidative Stress, Cellular Senescence, and Age-Related Diseases. Oxidative Med. Cell. Longev. 2017, 2017, 2062384. [Google Scholar] [CrossRef]
- Montes, M.; Lund, A.H. Emerging roles of lncRNAs in senescence. FEBS J. 2016, 283, 2414–2426. [Google Scholar] [CrossRef]
- Walther, K.; Schulte, L.N. The role of lncRNAs in innate immunity and inflammation. RNA Biol. 2021, 18, 587–603. [Google Scholar] [CrossRef]
- Ali, A.; Al-Tobasei, R.; Kenney, B.; Leeds, T.D.; Salem, M. Integrated analysis of lncRNA and mRNA expression in rainbow trout families showing variation in muscle growth and fillet quality traits. Sci. Rep. 2018, 8, 12111. [Google Scholar] [CrossRef]
- Valadkhan, S.; Valencia-Hipólito, A. lncRNAs in Stress Response. In Current Topics in Microbiology and Immunology; Morris, K.V., Ed.; Springer International Publishing: Cham, Germany, 2016; Volume 394, pp. 203–236. [Google Scholar]
- Wang, J.; Koganti, P.P.; Yao, J.; Wei, S.; Cleveland, B. Comprehensive analysis of lncRNAs and mRNAs in skeletal muscle of rainbow trout (Oncorhynchus mykiss) exposed to estradiol. Sci. Rep. 2017, 7, 11780. [Google Scholar] [CrossRef]
- Liu, X.; Li, W.; Jiang, L.; Lü, Z.; Liu, M.; Gong, L.; Liu, B.; Liu, L.; Yin, X. Immunity-associated long non-coding RNA and expression in response to bacterial infection in large yellow croaker (Larimichthys crocea). Fish Shellfish Immunol. 2019, 94, 634–642. [Google Scholar] [CrossRef]
- Boltaña, S.; Valenzuela-Miranda, D.; Aguilar, A.; Mackenzie, S.; Gallardo-Escárate, C. Long noncoding RNAs (lncRNAs) dynamics evidence immunomodulation during ISAV-Infected Atlantic salmon (Salmo salar). Sci. Rep. 2016, 6, 22698. [Google Scholar] [CrossRef]
- Lei, J.L.; Liu, X.F.; Guan, C.T. Turbot culture in China for two decades: Achievements and prospect. Prog. Fish. Sci. 2012, 33, 123–130. [Google Scholar] [CrossRef]
- Huang, Z.; Ma, A.; Yang, S.; Liu, X.; Zhao, T.; Zhang, J.; Wang, X.A.; Sun, Z.; Liu, Z.; Xu, R. Transcriptome analysis and weighted gene co-expression network reveals potential genes responses to heat stress in turbot Scophthalmus maximus. Comp. Biochem. Physiol. D Genom. Proteom. 2020, 33, 100632. [Google Scholar] [CrossRef]
- Liu, Z.; Ma, A.; Yuan, C.; Zhao, T.; Chang, H.; Zhang, J. Transcriptome analysis of liver lipid metabolism disorders of the turbot Scophthalmus maximus in response to low salinity stress. Aquaculture 2021, 534, 736273. [Google Scholar] [CrossRef]
- Cui, W.; Ma, A.; Huang, Z.; Wang, X.A.; Sun, Z.; Liu, Z.; Zhang, W.; Yang, J.; Zhang, J.; Qu, J. Transcriptomic analysis reveals putative osmoregulation mechanisms in the kidney of euryhaline turbot Scophthalmus maximus responded to hypo-saline seawater. J. Oceanol. Limnol. 2019, 38, 467–479. [Google Scholar] [CrossRef]
- Nie, X.; Zhang, C.; Jiang, C.; Li, S.; Hong, W.; Chen, S.; Zhang, Y. Characterizing transcriptome changes in gill tissue of turbot (Scophthalmus maximus) for waterless preservation. Aquaculture 2020, 518, 734830. [Google Scholar] [CrossRef]
- Ronza, P.; Alvarez-Dios, J.A.; Robledo, D.; Losada, A.P.; Romero, R.; Bermudez, R.; Pardo, B.G.; Martinez, P.; Quiroga, M.I. Blood transcriptomics of turbot Scophthalmus maximus: A tool for health monitoring and disease studies. Animal 2021, 11, 1296. [Google Scholar] [CrossRef]
- Gao, C.; Cai, X.; Cao, M.; Fu, Q.; Yang, N.; Liu, X.; Wang, B.; Li, C. Comparative analysis of the miRNA-mRNA regulation networks in turbot (Scophthalmus maximus L.) following Vibrio anguillarum infection. Dev. Comp. Immunol. 2021, 124, 104164. [Google Scholar] [CrossRef]
- Ronza, P.; Robledo, D.; Bermudez, R.; Losada, A.P.; Pardo, B.G.; Sitja-Bobadilla, A.; Quiroga, M.I.; Martinez, P. RNA-seq analysis of early enteromyxosis in turbot (Scophthalmus maximus): New insights into parasite invasion and immune evasion strategies. Int. J. Parasitol. 2016, 46, 507–517. [Google Scholar] [CrossRef]
- Song, Y.; Dong, X.; Hu, G. Transcriptome analysis of turbot (Scophthalmus maximus) head kidney and liver reveals immune mechanism in response to Vibrio anguillarum infection. J. Fish Dis. 2022, 45, 1045–1057. [Google Scholar] [CrossRef]
- Zheng, W.; Xu, X.-w.; E, Z.; Liu, Y.; Chen, S. Genome-wide identification of the MAPK gene family in turbot and its involvement in abiotic and biotic stress responses. Front. Mar. Sci. 2022, 9, 1005401. [Google Scholar] [CrossRef]
- Zheng, W.; Xu, X.; Chen, Y.; Wang, J.; Zhang, T.; E, Z.; Chen, S.; Liu, Y. Genome-Wide Identification, Molecular Characterization, and Involvement in Response to Abiotic and Biotic Stresses of the HSP70 Gene Family in Turbot (Scophthalmus maximus). Int. J. Mol. Sci. 2023, 24, 6025. [Google Scholar] [CrossRef]
- Xu, X.-w.; Zheng, W.; Meng, Z.; Xu, W.; Liu, Y.; Chen, S. Identification of stress-related genes by co-expression network analysis based on the improved turbot genome. Sci. Data 2022, 9, 374. [Google Scholar] [CrossRef]
- Robledo, D.; Ronza, P.; Harrison, P.W.; Losada, A.P.; Bermudez, R.; Pardo, B.G.; Redondo, M.J.; Sitja-Bobadilla, A.; Quiroga, M.I.; Martinez, P. RNA-seq analysis reveals significant transcriptome changes in turbot (Scophthalmus maximus) suffering severe enteromyxosis. BMC Genom. 2014, 15, 1149. [Google Scholar] [CrossRef]
- Ronza, P.; Robledo, D.; Losada, A.P.; Bermudez, R.; Pardo, B.G.; Martinez, P.; Quiroga, M.I. The teleost thymus in health and disease: New insights from transcriptomic and histopathological analyses of turbot, Scophthalmus maximus. Biology 2020, 9, 221. [Google Scholar] [CrossRef]
- Pertea, G.; Pertea, M. GFF Utilities: GffRead and GffCompare [version 2; peer review: 3 approved]. F1000Research 2020, 9, 304. [Google Scholar] [CrossRef]
- Wang, G.; Yin, H.; Li, B.; Yu, C.; Wang, F.; Xu, X.; Cao, J.; Bao, Y.; Wang, L.; Abbasi, A.A.; et al. Characterization and identification of long non-coding RNAs based on feature relationship. Bioinformatics 2019, 35, 2949–2956. [Google Scholar] [CrossRef]
- Wucher, V.; Legeai, F.; Hédan, B.; Rizk, G.; Lagoutte, L.; Leeb, T.; Jagannathan, V.; Cadieu, E.; David, A.; Lohi, H.; et al. FEELnc: A tool for long non-coding RNA annotation and its application to the dog transcriptome. Nucleic Acids Res. 2017, 45, e57. [Google Scholar] [CrossRef]
- Huo, H.; Gao, X.; Fei, F.; Qin, F.; Huang, B.; Liu, B. Transcriptomic profiling of the immune response to crowding stress in juvenile turbot (Scophthalmus maximus). J. Ocean Univ. China 2020, 19, 911–922. [Google Scholar] [CrossRef]
- Cui, W.; Ma, A.; Huang, Z.; Liu, Z.; Yang, K.; Zhang, W. myo-inositol facilitates salinity tolerance by modulating multiple physiological functions in the turbot Scophthalmus maximus. Aquaculture 2020, 527, 735451. [Google Scholar] [CrossRef]
- Gu, M.; Bai, N.; Zhang, Y.; Krogdahl, Å. Soybean meal induces enteritis in turbot Scophthalmus maximus at high supplementation levels. Aquaculture 2016, 464, 286–295. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, Z.; Dai, J.; Yang, P.; Xu, W.; Ai, Q.; Zhang, W.; Zhang, Y.; Zhang, Y.; Mai, K. Sodium butyrate supplementation in high-soybean meal diets for turbot (Scophthalmus maximus L.): Effects on inflammatory status, mucosal barriers and microbiota in the intestine. Fish Shellfish Immunol. 2019, 88, 65–75. [Google Scholar] [CrossRef]
- Zhao, T.; Ma, A.; Huang, Z.; Liu, Z.; Sun, Z.; Zhu, C.; Yang, J.; Li, Y.; Wang, Q.; Qiao, X.; et al. Transcriptome analysis reveals that high temperatures alter modes of lipid metabolism in juvenile turbot (Scophthalmus maximus) liver. Comp. Biochem. Physiol. D Genom. Proteom. 2021, 40, 100887. [Google Scholar] [CrossRef]
- Xu, X.; Liu, L.; Feng, J.; Li, X.; Zhang, J. Comparative transcriptome analysis reveals potential anti-viral immune pathways of turbot (Scophthalmus maximus) subverted by megalocytivirus RBIV-C1 for immune evasion. Fish Shellfish Immunol. 2022, 122, 153–161. [Google Scholar] [CrossRef]
- Cui, W.; Ma, A.; Wang, X. Response of the PI3K-AKT signalling pathway to low salinity and the effect of its inhibition mediated by wortmannin on ion channels in turbot Scophthalmus maximus. Aquac. Res. 2020, 51, 2676–2686. [Google Scholar] [CrossRef]
- Robledo, D.; Rubiolo, J.A.; Cabaleiro, S.; Martínez, P.; Bouza, C. Differential gene expression and SNP association between fast- and slow-growing turbot (Scophthalmus maximus). Sci. Rep. 2017, 7, 12105. [Google Scholar] [CrossRef]
- Wang, X.; Liu, Q.; Xu, S.; Xiao, Y.; Wang, Y.; Feng, C.; Xue, R.; Zhao, H.; Song, Z.; Li, J. Transcriptome Dynamics during Turbot Spermatogenesis Predicting the Potential Key Genes Regulating Male Germ Cell Proliferation and Maturation. Sci. Rep. 2018, 8, 15825. [Google Scholar] [CrossRef]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef]
- Niazi, F.; Valadkhan, S. Computational analysis of functional long noncoding RNAs reveals lack of peptide-coding capacity and parallels with 3′ UTRs. RNA 2012, 18, 825–843. [Google Scholar] [CrossRef]
- Kumar, H.; Srikanth, K.; Park, W.; Lee, S.-H.; Choi, B.-H.; Kim, H.; Kim, Y.-M.; Cho, E.-S.; Kim, J.H.; Lee, J.H.; et al. Transcriptome analysis to identify long non coding RNA (lncRNA) and characterize their functional role in back fat tissue of pig. Gene 2019, 703, 71–82. [Google Scholar] [CrossRef]
- Pauli, A.; Valen, E.; Lin, M.F.; Garber, M.; Vastenhouw, N.L.; Levin, J.Z.; Fan, L.; Sandelin, A.; Rinn, J.L.; Regev, A.; et al. Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome Res. 2012, 22, 577–591. [Google Scholar] [CrossRef]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011, 25, 1915–1927. [Google Scholar] [CrossRef]
- Zhang, K.; Huang, K.; Luo, Y.; Li, S. Identification and functional analysis of long non-coding RNAs in mouse cleavage stage embryonic development based on single cell transcriptome data. BMC Genom. 2014, 15, 845. [Google Scholar] [CrossRef]
- Valenzuela-Muñoz, V.; Pereiro, P.; Álvarez-Rodríguez, M.; Gallardo-Escárate, C.; Figueras, A.; Novoa, B. Comparative modulation of lncRNAs in wild-type and rag1-heterozygous mutant zebrafish exposed to immune challenge with spring viraemia of carp virus (SVCV). Sci. Rep. 2019, 9, 14174. [Google Scholar] [CrossRef]
- Liu, S.; Pan, C.; Tang, Y.; Chen, F.; Yang, M.; Wang, K.-J. Identification of novel long non-coding RNAs involved in bisphenol A induced immunotoxicity in fish primary macrophages. Fish Shellfish Immunol. 2020, 100, 152–160. [Google Scholar] [CrossRef]
- Shen, Y.; Liang, W.; Lin, Y.; Yang, H.; Chen, X.; Feng, P.; Zhang, B.; Zhu, J.; Zhang, Y.; Luo, H. Single molecule real-time sequencing and RNA-seq unravel the role of long non-coding and circular RNA in the regulatory network during Nile tilapia (Oreochromis niloticus) infection with Streptococcus agalactiae. Fish Shellfish Immunol. 2020, 104, 640–653. [Google Scholar] [CrossRef]
- Banks, C.A.; Boanca, G.; Lee, Z.T.; Eubanks, C.G.; Hattem, G.L.; Peak, A.; Weems, L.E.; Conkright, J.J.; Florens, L.; Washburn, M.P. TNIP2 is a Hub Protein in the NF-κB Network with Both Protein and RNA Mediated Interactions. Mol. Cell. Proteom. 2016, 15, 3435–3449. [Google Scholar] [CrossRef]
- Camps, M.; Nichols, A.; Arkinstall, S. Dual specificity phosphatases: A gene family for control of MAP kinase function. FASEB J. 2000, 14, 6–16. [Google Scholar] [CrossRef]
- Lanaspa, M.A.; Andres-Hernando, A.; Rivard, C.J.; Dai, Y.; Berl, T. Hypertonic stress increases claudin-4 expression and tight junction integrity in association with MUPP1 in IMCD3 cells. Proc. Natl. Acad. Sci. USA 2008, 105, 15797–15802. [Google Scholar] [CrossRef]
- Team, SRA Toolkit Development. SRAtoolkit Version 2.11.0; National Center for Biotechnology Information: Bethesda, MD, USA, 2021. [Google Scholar]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Shumate, A.; Wong, B.; Pertea, G.; Pertea, M. Improved transcriptome assembly using a hybrid of long and short reads with StringTie. PLoS Comput. Biol. 2022, 18, e1009730. [Google Scholar] [CrossRef]
- Vera Alvarez, R.; Pongor, L.S.; Mariño-Ramírez, L.; Landsman, D. TPMCalculator: One-step software to quantify mRNA abundance of genomic features. Bioinformatics 2019, 35, 1960–1962. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. The Subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013, 41, e108. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]


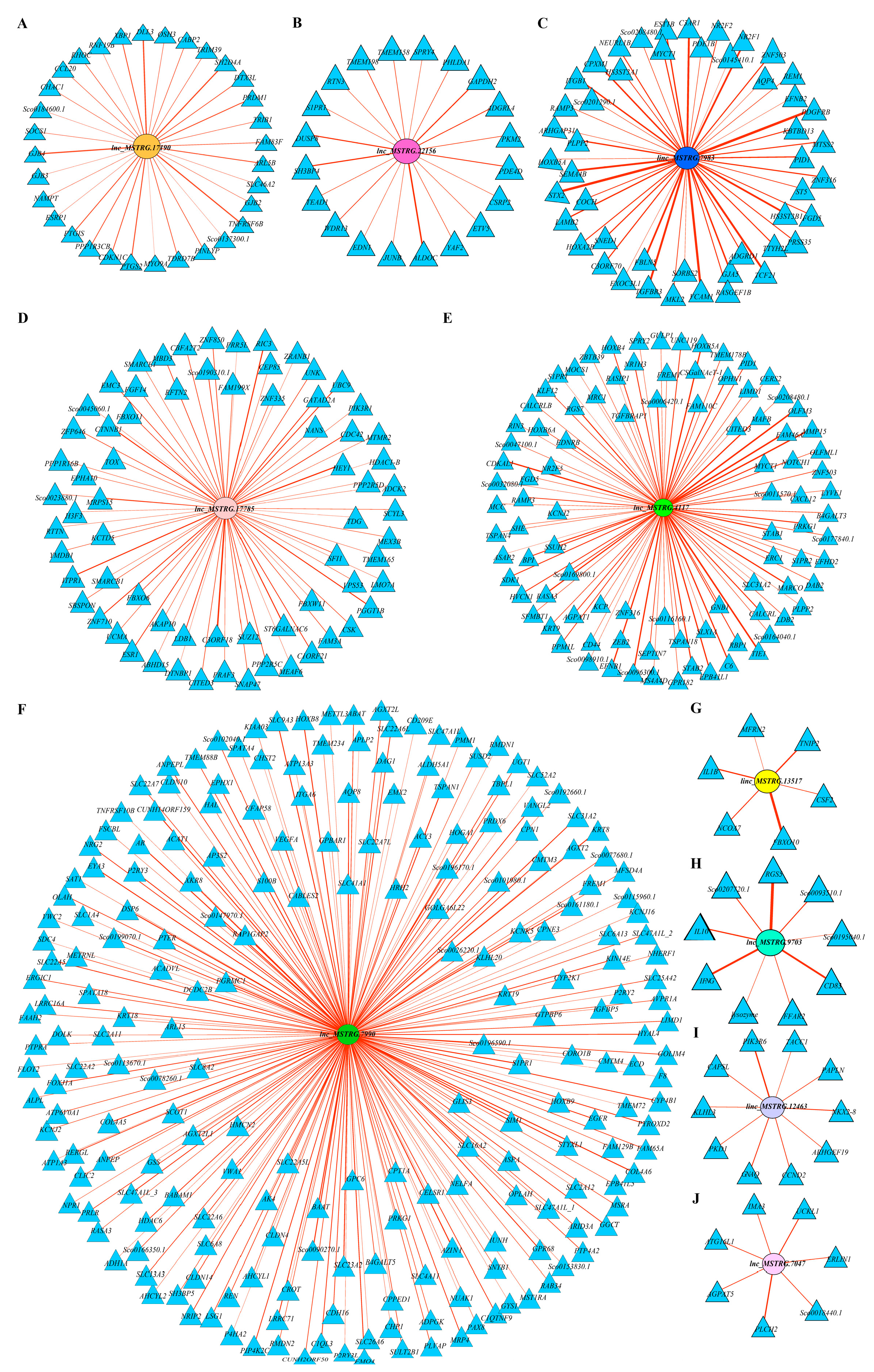

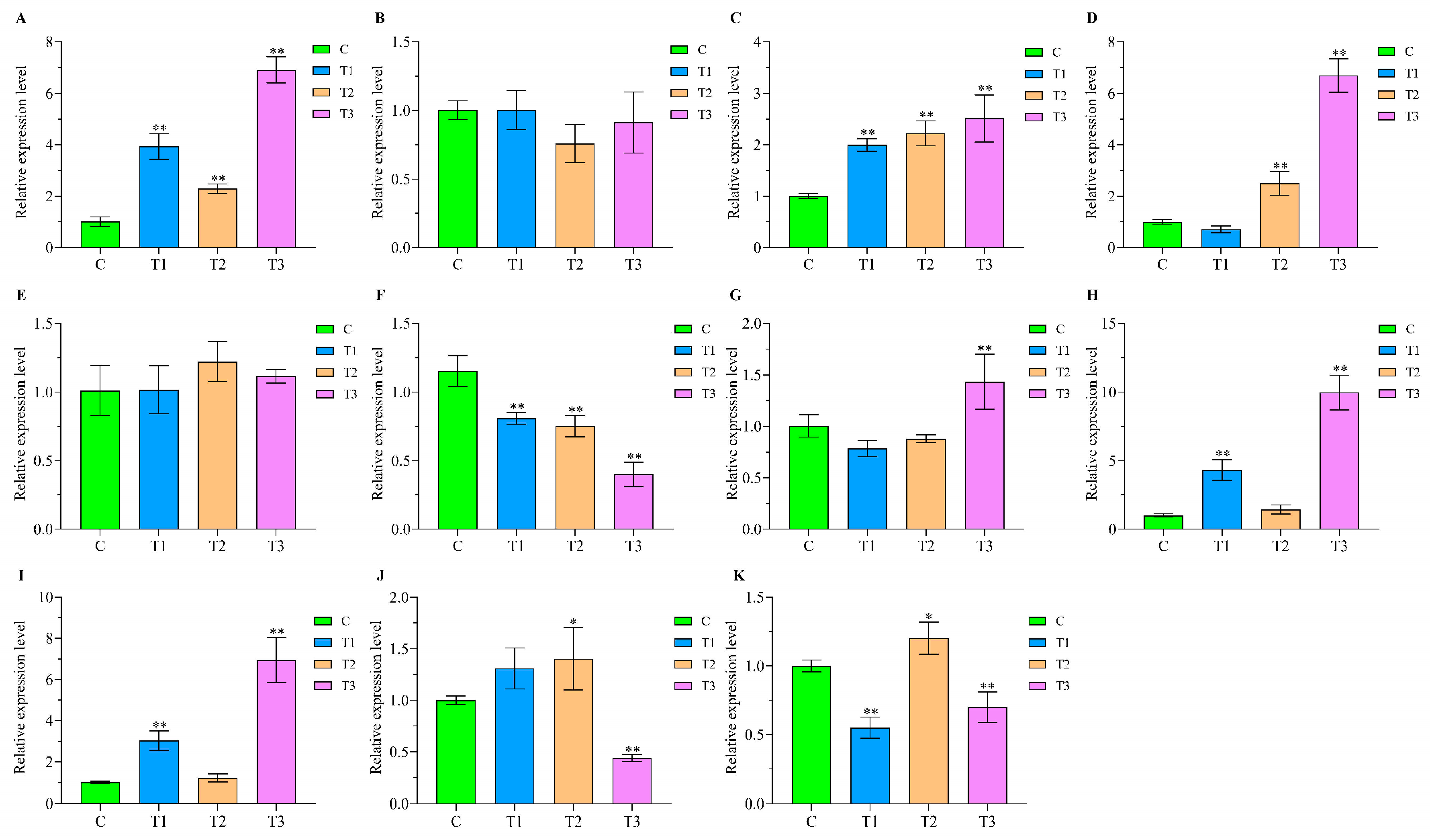
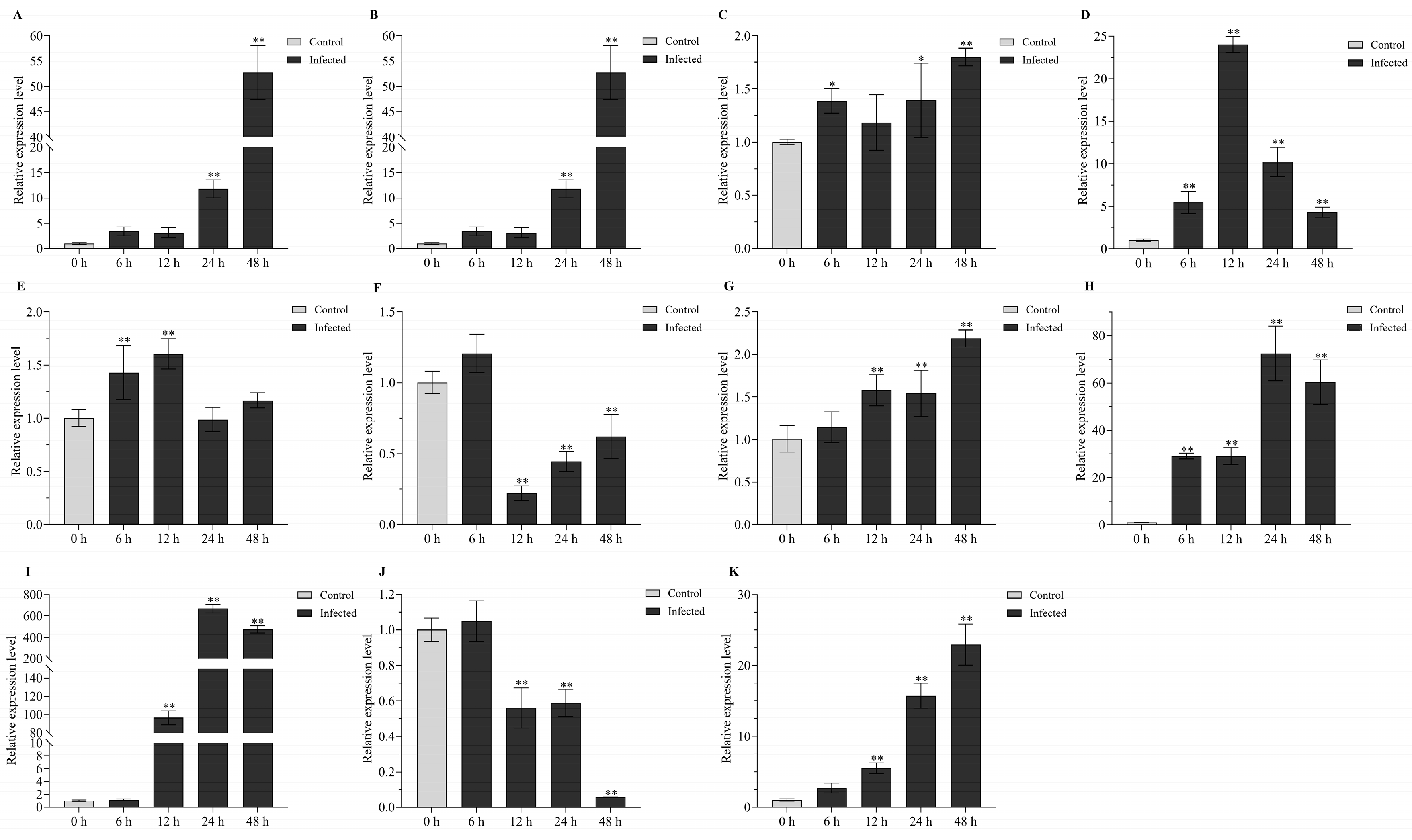
| lncRNA or Gene | Primers | Sequences (5′–3′) | The Length of Product/bp |
|---|---|---|---|
| lnc_MSTRG.17190 | 17190-F | CGAACGCTCACAGGAGACTG | 20 |
| 17190-R | ACAACTCCACAACCTCACCTG | 21 | |
| lnc_MSTRG.28492 | 28492-F | CCATACCCGCGATCTGAAGG | 20 |
| 28492-R | ATCTTCGAGAGCGTCAACCA | 20 | |
| lnc_MSTRG.22156 | 22156-F | GCGCACTTTCTTGACACAGG | 20 |
| 22156-R | CGGCTGGTGCCTAACTAGAG | 20 | |
| linc_MSTRG.12463 | 12463-F | GCAAACCTGAAGGAGTAGGCT | 21 |
| 12463-R | CTAGATAGGCAGGCCTTGGTC | 21 | |
| lnc_MSTRG.7047 | 7047-F | ATAAGTAGCCAGCCGTCGAG | 20 |
| 7047-R | TGGTGCTAGGTTGAATGCTGT | 21 | |
| linc_MSTRG.7983 | 7983-F | TCATTCGATTTCACGCACGC | 20 |
| 7983-R | GCCTCAAGAAGCTGAGAGCA | 20 | |
| lnc_MSTRG.7990 | 7990-F | GCTGTAAAGAGCGCTGCAAG | 20 |
| 7990-R | AGCTGGTGTCTGAACGACAG | 20 | |
| linc_MSTRG.13517 | 13517-F | GCGTAGTTGACGTTGGACTT | 20 |
| 13517-R | GAGGATCACTGCGGCTACG | 19 | |
| lnc_MSTRG.9703 | 9703-F | CACGTCGGCACAATCACAA | 19 |
| 9703-R | ACGACTTTATGAACAGTGGCA | 21 | |
| lnc_MSTRG.4117 | 4117-F | TTCTTCTGGTCCTCCTTGCG | 20 |
| 4117-R | TCAGCCTCGTGACCTTGAAC | 20 | |
| lnc_MSTRG.17785 | 17785-F | CGTCTCCTCTCACTGCTCCA | 20 |
| 17785-R | CTGAGCTCCTCCACCACGTC | 20 | |
| β-actin | β-F | ACAACGGATCCGGTATGTGC | 20 |
| β-R | CTCTGGGCTTCATCACCTACG | 21 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, W.; Chen, Y.; Wang, Y.; Chen, S.; Xu, X.-w. Genome-Wide Identification and Involvement in Response to Biotic and Abiotic Stresses of lncRNAs in Turbot (Scophthalmus maximus). Int. J. Mol. Sci. 2023, 24, 15870. https://doi.org/10.3390/ijms242115870
Zheng W, Chen Y, Wang Y, Chen S, Xu X-w. Genome-Wide Identification and Involvement in Response to Biotic and Abiotic Stresses of lncRNAs in Turbot (Scophthalmus maximus). International Journal of Molecular Sciences. 2023; 24(21):15870. https://doi.org/10.3390/ijms242115870
Chicago/Turabian StyleZheng, Weiwei, Yadong Chen, Yaning Wang, Songlin Chen, and Xi-wen Xu. 2023. "Genome-Wide Identification and Involvement in Response to Biotic and Abiotic Stresses of lncRNAs in Turbot (Scophthalmus maximus)" International Journal of Molecular Sciences 24, no. 21: 15870. https://doi.org/10.3390/ijms242115870