
Developments in Genetics: Better Management of Ovarian Cancer Patients
 ,
, 
Abstract
1. Introduction
2. Ovarian Cancer Statistics
Ovarian Cancer Risk Factors
3. Histopathologic Classification of Ovarian Cancer
- A.
- Epithelial Tumors
- B.
- Mesenchymal Tumors
- C.
- Sex Cord Stromal Tumors
- D.
- Germ Cell Tumors
- E.
- Miscellaneous and Tumor-Like Lesions
4. Genetic Causes of Ovarian Cancer
4.1. Candidate Genes
4.2. Prognostic Genes
5. Molecular Pathways in Ovarian Cancer
- A.
- The MAPK signaling molecular pathways
- B.
- The MAPK/ERK pathway that acts through the KRAS, MAPK1, NF1, NF2, and BRAF genes (Figure 2).
- Tyrosine-kinase receptor;
- G protein-coupled receptor.
- C.
- The PI3K/AKT/MTOR signaling pathway includes, among others, proteins encoded by the PIK3CA, PTEN, STK11, MTOR, and AKT1 genes It represents one of the most important intracellular signaling pathways involved in various cellular activities, including cell cycle control, proliferation, protein synthesis, transcription, and angiogenesis. In ovarian cancer, the PI3K/MTOR pathway is the most frequently altered [61,62] (Figure 3).
- D.
- The WNT/β-catenin signaling pathway is also involved in the regulation of various cellular processes, including cell proliferation, migration, angiogenesis, carcinogenesis, tumor progression, and treatment resistance (through aberrant activation). The WNT signaling pathway has three main subpathways—a classical/canonical WNT/ β-catenin/T cell factor pathway and two non-canonical pathways WNT/plenary cell polarity (PCP) and WNT/Ca2+ (Figure 4).
- E.
- JAK/STAT Pathway (Figure 5)
- F.
- The TP53 gene is prominently implicated in human cancer, particularly in high-grade serous ovarian cancer (HGSOC), where mutations occur at a frequency of at least 96%. Various TP53 mutations lead to the loss of wild-type functions, either due to the loss of DNA-binding activity or through a dominant-negative effect, wherein the mutated allele inhibits the function of the wild-type counterpart. Intriguingly, certain mutations exhibit gain-of-function properties independent of the wild-type TP53. Such gain-of-function mutations can enhance cell transformation and contribute to resistance against chemotherapy [67]. Additionally, the functional repercussions of TP53 mutations are contingent upon the specific mutation or its type. For instance, frameshift mutations are proposed to manifest distinct phenotypes compared to missense mutations. Notably, certain TP53 missense mutations generate full-length p53 proteins, often characterized by prolonged half-lives and the accumulation of inactive protein [68]. In contrast, frameshift mutations generally do not lead to p53 accumulation, and nonsense mutations typically yield unstable proteins.
- G.
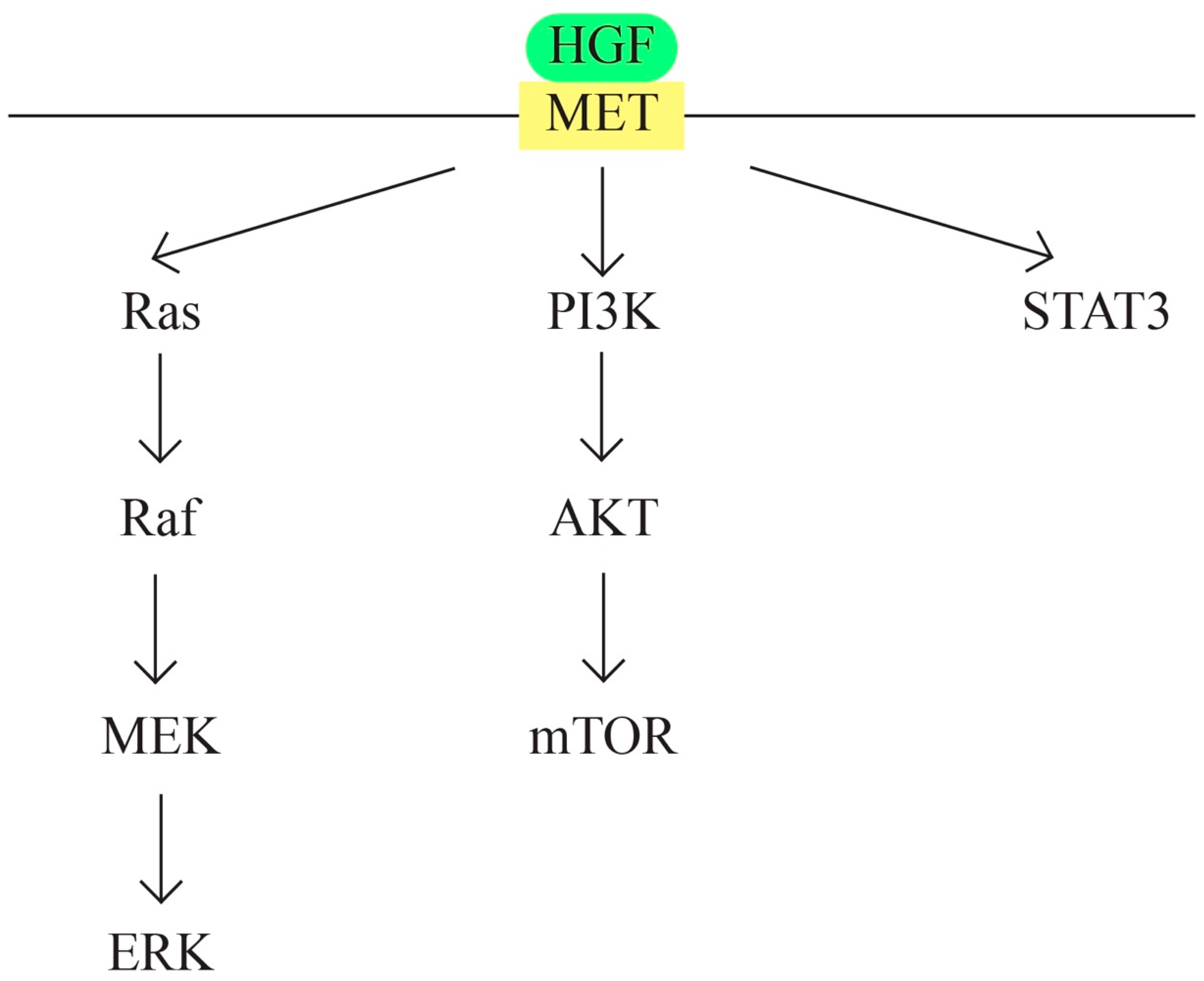
- MET/HGF signaling pathway (Figure 6)
- H.
- Notch pathway (Figure 7)
- I.
- EGFR
- J.
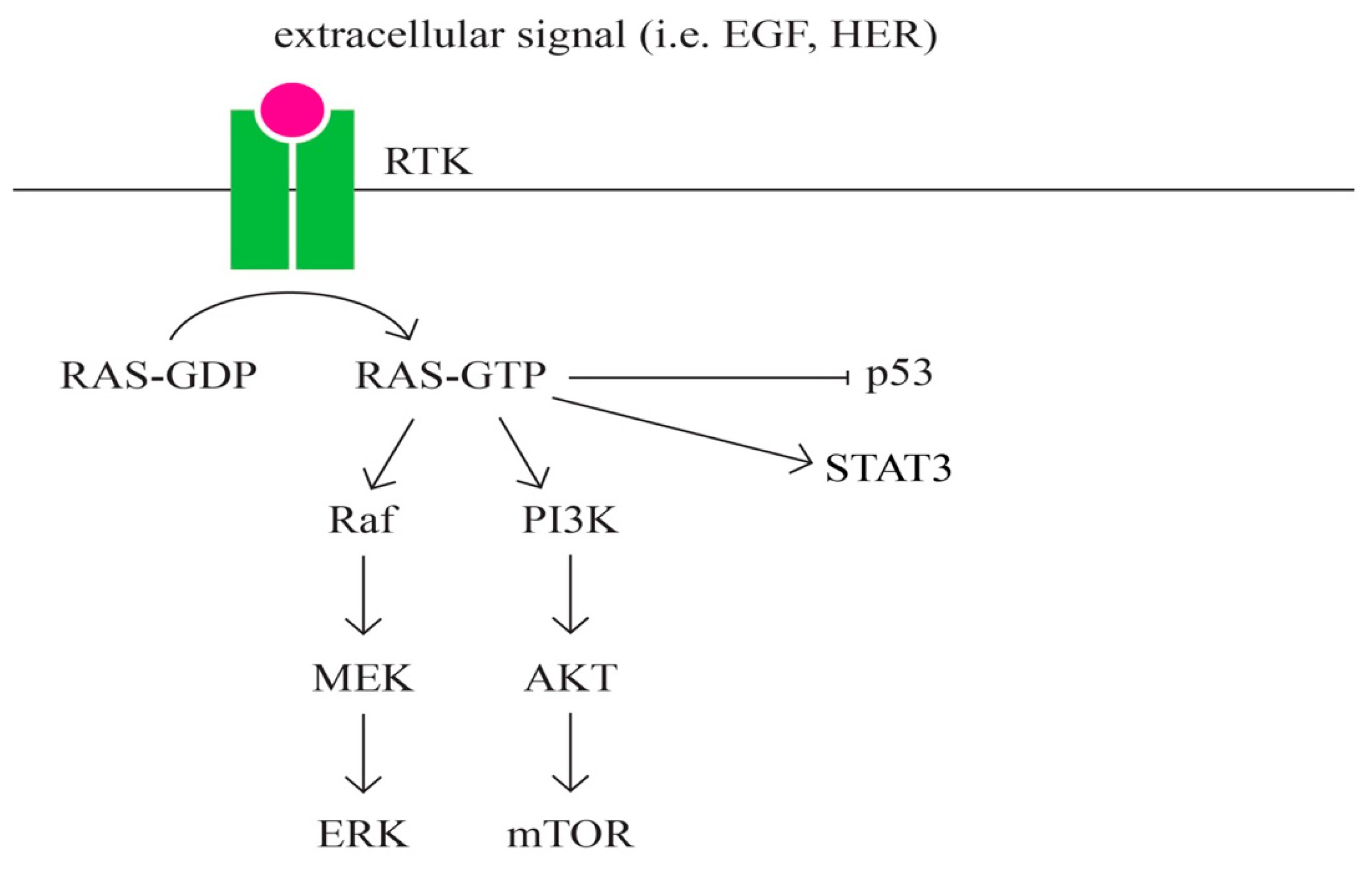
- RAS signaling pathway (Figure 8)
6. Genetic Testing in Ovarian Cancer
- Indications for clinical testing;
- Considerations for testing;
- Scenarios with low likelihood of clinical utility from testing.
- Loss of heterozygosity (LOH), a genomic event in which one allele of a gene is deleted or inactivated, leaving only one functional copy.
- Telomeric allelic imbalance (TAI), which refers to imbalances in the lengths of the telomeres (protective caps at the ends of chromosomes) between two homologous chromosomes. TAI can be indicative of HRD, and it is evaluated in HRD testing.
- Large-scale state transitions (LST), which measure the number of chromosomal breaks, gains, or losses in a cancer genome. A high LST score is associated with HRD.
- Microhomology-mediated end joining (MMEJ), which is a DNA repair mechanism that can be more active in HRD cells. It involves the repair of double-strand DNA breaks by joining DNA ends with short, overlapping sequences.
- Depletion of RAD51 foci, which is a reduction in the formation protein involved in homologous recombination repair.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviation
- HGSOC—high-grade serous ovarian carcinoma
- LGSOC—low-grade ovarian serous carcinoma
- EnOC—endometrioid ovarian carcinoma
- OCCC—ovarian clear cell carcinoma
- EOT—Epithelial ovarian tumor
- OC—Ovarian Cancer
- AGCT—adult granulosa cell tumor
- EMT—epithelial to mesenchymal
- DBD—DNA-binding domain
- GoF—gain-of-function
- LoF—loss-of-function
- HRGs—heregulins
- PV—pathogenic variant
- LPV—likely pathogenic variant
- CPG—cytosine–phosphate–guanine
- PARP—poly (ADP-ribose) polymerase
- PARPI—poly (ADP-ribose) polymerase inhibitors
- NGS—next-generation sequencing
- FFPE—formalin-fixed paraffin-embedded
- HR—homologous recombination
- TCGA—The Cancer Genome Atlas
- LOH—loss of heterozygosity
- VAF—variant allele frequency
- HRD—Homologous Recombination Deficiency
- TAI—Telomeric allelic imbalance
- LST—Large-scale state transitions
- MMEJ—Microhomology-mediated end joining
References
- Available online: https://gco.iarc.fr/ (accessed on 20 August 2023).
- Available online: https://gco.iarc.fr/tomorrow/en/dataviz/trends?types=0_1&sexes=2&mode=population&group_populations=0&multiple_populations=1&multiple_cancers=0&cancers=25&populations=900 (accessed on 20 August 2023).
- Available online: https://seer.cancer.gov/statfacts/html/ovary.html (accessed on 20 August 2023).
- America Cancer Society. Key Statistics for Ovarian Cancer. Available online: https://www.cancer.org/cancer/types/ovarian-cancer/about/key-statistics.html#:~:text=Ovarian%20cancer%20ranks%20fifth%20in,is%20about%201%20in%20108, (accessed on 20 August 2023).
- McLemore, M.R.; Miaskowski, C.; Aouizerat, B.E.; Dodd, M.J. Epidemiologic and Genetic Factors Associated with Ovarian Cancer. Cancer Nurs. 2009, 32, 281. [Google Scholar] [CrossRef] [PubMed]
- Momenimovahed, Z.; Tiznobaik, A.; Taheri, S.; Salehiniya, H. Ovarian cancer in the world: Epidemiology and risk factors. Int. J. Women’s Health 2019, 11, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Ramus, S.J.; Gayther, S.A. The Contribution of BRCA1 and BRCA2 to Ovarian Cancer. Mol. Oncology 2009, 3, 138–150. [Google Scholar] [CrossRef]
- Toss, A.; Tomasello, C.; Razzaboni, E.; Contu, G.; Grandi, G.; Cagnacci, A.; Schilder, R.J.; Cortesi, L. Hereditary Ovarian Cancer: Not Only BRCA 1 and 2 Genes; BioMed Research International: New York, NY, USA, 2015. [Google Scholar] [CrossRef]
- Adhikari, L.; Hassell, L.A. WHO Classification. Pathology Outlines.com Website. Available online: https://www.pathologyoutlines.com/topic/ovarytumorwhoclassif.html (accessed on 29 August 2023).
- Guo, T.; Dong, X.; Xie, S.; Zhang, L.; Zeng, P.; Zhang, L. Cellular Mechanism of Gene Mutations and Potential Therapeutic Targets in Ovarian Cancer. Cancer Manag. Res. 2021, 13, 3081–3100. [Google Scholar] [CrossRef] [PubMed]
- Radu, M.R.; Prădatu, A.; Duică, F.; Micu, R.; Creţoiu, S.M.; Suciu, N.; Creţoiu, D.; Varlas, V.N.; Rădoi, V.E. Ovarian Cancer: Biomarkers and Targeted Therapy. Biomedicines 2021, 9, 693. [Google Scholar] [CrossRef]
- Meyer, L.A.; Broaddus, R.R.; Lu, K.H. Endometrial cancer and Lynch syndrome: Clinical and pathologic considerations. Cancer Control. 2009, 16, 14–22. [Google Scholar] [CrossRef] [PubMed]
- dos Anjos, L.G.; da Cunha, I.W.; Baracat, E.C.; Carvalho, K.C. Genetic and Epigenetic Features in Uterine Smooth Muscle Tumors: An Update. Clin. Oncol. 2019, 4, 1637. [Google Scholar]
- Bertsch, E.; Qiang, W.; Zhang, Q.; Druschitz, S.; Liu, Y.; Mittal, K.; Kong, B.; Kurita, T.; Wei, J. MED12 and HMGA2 mutations: Two independent genetic events in uterine leiomyoma and leiomyosarcoma. Mod. Pathol. 2014, 27, 1144–1153. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Minh Tran, T.; Shen Choo, Y.; Alexiadis, M.; Fuller, P.J.; Chu, S. Genetics and Mutational Landscape of Ovarian Sex Cord-Stromal Tumors; IntechOpen: London, UK, 2021. [Google Scholar] [CrossRef]
- Llano, E.; Todeschini, A.L.; Felipe-Medina, N.; Corte-Torres, M.D.; Condezo, Y.B.; Sanchez-Martin, M.; López-Tamargo, S.; Astudillo, A.; Puente, X.S.; Pendas, A.M.; et al. The Oncogenic FOXL2 C134W Mutation Is a Key Driver of Granulosa Cell Tumors. Cancer Res. 2023, 83, 239–250. [Google Scholar] [CrossRef]
- Available online: https://www.mycancergenome.org/content/disease/germ-cell-tumor/ (accessed on 29 August 2023).
- Varghese, A.; Lele, S. Rare Ovarian Tumors. In Ovarian Cancer; Lele, S., Ed.; Exon Publications: Brisbane, Australia, 2022; Volume 1. [Google Scholar] [CrossRef]
- Mostert, M.; Rosenberg, C.; Stoop, H.; Schuyer, M.; Timmer, A.; Oosterhuis, W.; Looijenga, L. Comparative Genomic and In Situ Hybridization of Germ Cell Tumors of the Infantile Testis. Lab. Investig. 2000, 80, 1055–1064. [Google Scholar] [CrossRef]
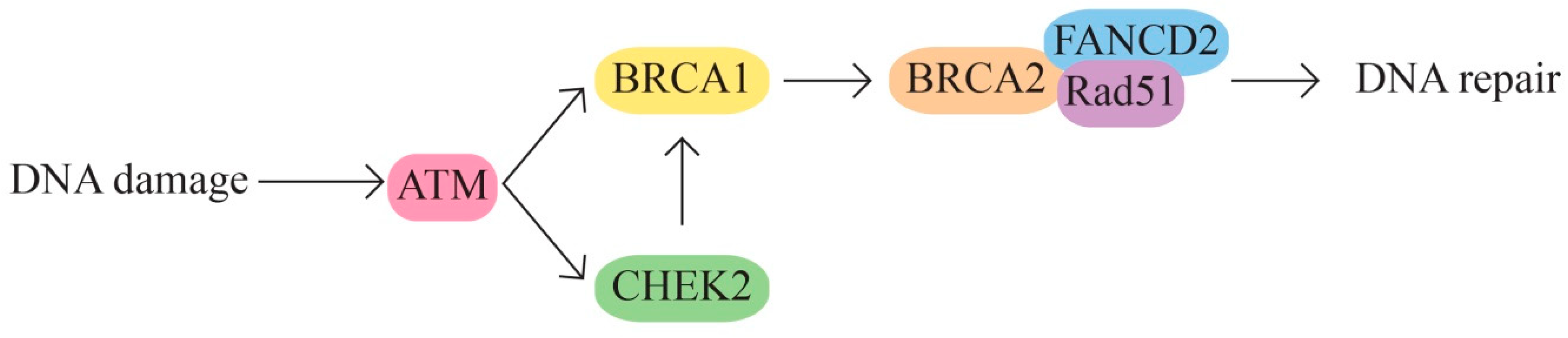
- Moynahan, M.E.; Pierce, A.J.; Jasin, M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol. Cell. 2001, 7, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Venkitaraman, A.R. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell 2002, 108, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Gorodetska, I.; Kozeretska, I.; Dubrovska, A. BRCA Genes: The Role in Genome Stability, Cancer Stemness and Therapy Resistance. J. Cancer 2019, 10, 2109–2127. [Google Scholar] [CrossRef] [PubMed]
- Karami, F.; Mehdipour, P. A comprehensive focus on global spectrum of BRCA1 and BRCA2 mutations in breast cancer. Biomed. Res. Int. 2013, 2013, 928562. [Google Scholar] [CrossRef]
- Albertsen, H.; Plaetke, R.; Ballard, L.; Fujimoto, E.; Connolly, J.; Lawrence, E.; Rodriguez, P.; Robertson, M.; Bradley, P.; Milner, B.; et al. Genetic mapping of the BRCA1 region on chromosome 17q21. Am. J. Hum. Genet. 1994, 54, 516–525. [Google Scholar] [PubMed]
- Fackenthal, J.D.; Olopade, O.I. Breast cancer risk associated with BRCA1 and BRCA2 in diverse populations. Nat. Rev. Cancer. 2007, 7, 937–948. [Google Scholar] [CrossRef]
- Godet, I.; Gilkes, D.M. BRCA1 and BRCA2 mutations and treatment strategies for breast cancer. Integr. Cancer Sci. Ther. 2017, 4. [Google Scholar] [CrossRef] [PubMed]
- Le, H.P.; Heyer, W.-D.; Liu, J. Guardians of the Genome: BRCA2 and Its Partners. Genes 2021, 12, 1229. [Google Scholar] [CrossRef]
- Xie, C.; Luo, J.; He, Y.B.; Jiang, L.; Zhong, L.; Shi, Y. BRCA2 gene mutation in cancer. Medicine 2022, 101, e31705. [Google Scholar] [CrossRef] [PubMed]
- Breast Cancer Linkage Consortium. Cancer risks in BRCA2 mutation carriers. J. Natl. Cancer Inst. 1999, 91, 1310–1316. [Google Scholar] [CrossRef] [PubMed]
- Leung, M.; Rosen, D.; Fields, S.; Cesano, A.; Budman, D.R. Poly(ADP-ribose) polymerase-1 inhibition: Preclinical and clinical development of synthetic lethality. Mol. Med. 2011, 17, 854–862. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhou, J.; Zhang, K.; Chen, H.; Luo, M.; Lu, Y.; Sun, Y.; Chen, Y. Molecular Mechanisms of PALB2 Function and Its Role in Breast Cancer Management. Front. Oncol. 2020, 10, 301. [Google Scholar] [CrossRef]
- Oliver, A.W.; Swift, S.; Lord, C.J.; Ashworth, A.; Pearl, L.H. Structural basis for recruitment of BRCA2 by PALB2. EMBO Rep. 2009, 10, 990–996, Erratum in EMBO Rep. 2017, 18, 1264. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-Y.; Singh, T.R.; Nassar, N.; Zhang, F.; Freund, M.; Hanenberg, H.; Meetei, A.R.; Andreassen, P.R. Breast cancer-associated missense mutants of the PALB2 WD40 domain, which directly binds RAD51C, RAD51 and BRCA2, disrupt DNA repair. Oncogene 2014, 33, 4803–4812. [Google Scholar] [CrossRef] [PubMed]
- Bleuyard, J.Y.; Buisson, R.; Masson, J.Y.; Esashi, F. ChAM, a novel motif that mediates PALB2 intrinsic chromatin binding and facilitates DNA repair. EMBO Rep. 2012, 13, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Dray, E.; Etchin, J.; Wiese, C.; Saro, D.; Williams, G.J.; Hammel, M.; Yu, X.; E Galkin, V.; Liu, D.; Tsai, M.-S.; et al. Enhancement of RAD51 recombinase activity by the tumor suppressor PALB2. Nat. Struct. Mol. Biol. 2010, 17, 1255–1259. [Google Scholar] [CrossRef] [PubMed]
- Rahman, N.; Seal, S.; Thompson, D.; Kelly, P.; Renwick, A.; Elliott, A.; Reid, S.; Spanova, K.; Barfoot, R.; Chagtai, T.; et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat. Genet. 2007, 39, 165–167. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.; Dorsman, J.C.; Ameziane, N.; de Vries, Y.; A Rooimans, M.; Sheng, Q.; Pals, G.; Errami, A.; Gluckman, E.; Llera, J.; et al. Fanconi anemia is associated with a defect in the BRCA2 partner PALB2. Nat. Genet. 2007, 39, 159–161. [Google Scholar] [CrossRef]
- Reid, S.; Schindler, D.; Hanenberg, H.; Barker, K.; Hanks, S.; Kalb, R.; Neveling, K.; Kelly, P.; Seal, S.; Freund, M.; et al. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat. Genet. 2007, 39, 162–164. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, B.; Hengel, S.R.; Grundy, M.K.; Bernstein, K.A. RAD51 Gene Family Structure and Function. Annu. Rev. Genet. 2020, 54, 25–46. [Google Scholar] [CrossRef] [PubMed]
- Thacker, J. The RAD51 gene family, genetic instability and cancer. Cancer Lett. 2005, 219, 125–135. [Google Scholar] [CrossRef] [PubMed]
- RAD51. RAD51 Recombinase [Homo sapiens (Human)]. Available online: https://www.ncbi.nlm.nih.gov/gene/5888 (accessed on 25 August 2023).
- Noureddine, B. Constitutional Oncogenetics, 1st ed; Wiley: Hoboken, NJ, USA, 2021. [Google Scholar]
- Schmidt, M.K.; Hogervorst, F.; van Hien, R.; Cornelissen, S.; Broeks, A.; Adank, M.A.; Meijers, H.; Waisfisz, Q.; Hollestelle, A.; Schutte, M.; et al. Age- and Tumor Subtype-Specific Breast Cancer Risk Estimates for CHEK2*1100delC Carriers. J. Clin. Oncol. 2016, 34, 2750–2760. [Google Scholar] [CrossRef] [PubMed]
- Apostolou, P.; Papasotiriou, I. Current perspectives on CHEK2 mutations in breast cancer. Breast Cancer (Dove Med. Press) 2017, 9, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cao, L.; Nguyen, D.; Lu, H. TP53 mutations in epithelial ovarian cancer. Transl. Cancer Res. 2016, 5, 650–663. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.-J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef]
- Takahashi, K.; Takenaka, M.; Okamoto, A.; Bowtell, D.D.L.; Kohno, T. Treatment Strategies for ARID1A-Deficient Ovarian Clear Cell Carcinoma. Cancers 2021, 13, 1769. [Google Scholar] [CrossRef] [PubMed]
- Mullen, J.; Kato, S.; Sicklick, J.K.; Kurzrock, R. Targeting ARID1A mutations in cancer. Cancer Treat. Rev. 2021, 100, 102287. [Google Scholar] [CrossRef]
- Mandal, J.; Mandal, P.; Wang, T.-L.; Shih, I.-M. Treating ARID1A mutated cancers by harnessing synthetic lethality and DNA damage response. J. Biomed. Sci. 2022, 29, 71. [Google Scholar] [CrossRef]
- Prokofyeva, D.S.; Mingajeva, E.T.; Bogdanova, N.V.; Faiskhanova, R.R.; Sakaeva, D.D.; Dörk, T.; Khusnutdinova, E.K. The search for new candidate genes involved in ovarian cancer pathogenesis by exome sequencing. Russ. J. Genet. 2016, 52, 1105–1109. [Google Scholar] [CrossRef]
- Rinki, S.; Anup, S. Identification of common candidate genes and pathways for progression of ovarian, cervical and endometrial cancers. Meta Gene 2020, 23, 100634. [Google Scholar] [CrossRef]
- Nero, C.; Ciccarone, F.; Pietragalla, A.; Scambia, G. PTEN and Gynecological Cancers. Cancers 2019, 11, 1458. [Google Scholar] [CrossRef]
- Mayr, D.; Hirschmann, A.; Löhrs, U.; Diebold, J. KRAS and BRAF mutations in ovarian tumors: A comprehensive study of invasive carcinomas, borderline tumors and extraovarian implants. Gynecol. Oncol. 2006, 103, 883–887. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.T.H.; Fuller, P.J.; Chu, S. Impact of FOXL2 mutations on signaling in ovarian granulosa cell tumors. Int. J. Biochem. Cell Biol. 2016, 72, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Bethany, B.; Frimer, M.; Cheng, K.; John, V.S. The role of CCNE1 amplification in refractory ovarian and endometrial cancer. Gynecol. Oncol. 2020, 159, 216. [Google Scholar] [CrossRef]
- Li, H.; Li, M.; Tang, C.; Xu, L. Screening and prognostic value of potential biomarkers for ovarian cancer. Ann. Transl. Med. 2021, 9, 1007. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Hua, F.; Li, X.; Zhang, J.; Li, S.; Wang, W.; Zhou, J.; Wang, W.; Liao, P.; Yan, Y.; et al. Loss of Optineurin Drives Cancer Immune Evasion via Palmitoylation-Dependent IFNGR1 Lysosomal Sorting and Degradation. Cancer Discov. 2021, 11, 1826–1843. [Google Scholar] [CrossRef] [PubMed]
- LaFleur, M.W.; Nguyen, T.H.; Coxe, M.A.; Miller, B.C.; Yates, K.B.; Gillis, J.E.; Sen, D.R.; Gaudiano, E.F.; Al Abosy, R.; Freeman, G.J.; et al. PTPN2 regulates the generation of exhausted CD8+ T cell subpopulations and restrains tumor immunity. Nat. Immunol. 2019, 20, 1335–1347. [Google Scholar] [CrossRef]
- Zhuge, J.; Wang, X.; Li, J.; Wang, T.; Wang, H.; Yang, M.; Dong, W.; Gao, Y. Construction of the model for predicting prognosis by key genes regulating EGFR-TKI resistance. Front. Genet. 2022, 13, 968376. [Google Scholar] [CrossRef] [PubMed]
- Ji, M.; Zhao, Z.; Li, Y.; Xu, P.; Shi, J.; Li, Z.; Wang, K.; Huang, X.; Ji, J.; Liu, W.; et al. FBXO16-mediated hnRNPL ubiquitination and degradation plays a tumor suppressor role in ovarian cancer. Cell Death Dis. 2021, 12, 758. [Google Scholar] [CrossRef]
- Huang, R.Y.; Huang, K.; Chen, K.; Hsiao, S.; Tan, T.Z.; Wu, C.; Hsu, C.; Chang, W.; Pan, C.; Sheu, B.; et al. Immune-Hot tumor features associated with recurrence in early-stage ovarian clear cell carcinoma. Int. J. Cancer 2023, 152, 2174–2185. [Google Scholar] [CrossRef]
- Mabuchi, S.; Sasano, T.; Komura, N. Targeting Myeloid-Derived Suppressor Cells in Ovarian Cancer. Cells 2021, 10, 329. [Google Scholar] [CrossRef] [PubMed]
- Rah, B.; A Rather, R.; Bhat, G.R.; Baba, A.B.; Mushtaq, I.; Farooq, M.; Yousuf, T.; Dar, S.B.; Parveen, S.; Hassan, R.; et al. JAK/STAT Signaling: Molecular Targets, Therapeutic Opportunities, and Limitations of Targeted Inhibitions in Solid Malignancies. Front. Pharmacol. 2022, 13, 821344. [Google Scholar] [CrossRef] [PubMed]
- Gao, A.H.; Hu, Y.R.; Zhu, W.P. IFN-γ inhibits ovarian cancer progression via SOCS1/JAK/STAT signaling pathway. Clin. Transl. Oncol. 2021, 24, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Groner, B.; von Manstein, V. Jak Stat signaling and cancer: Opportunities, benefits and side effects of targeted inhibition. Mol. Cell. Endocrinol. 2017, 451, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Poli, V.; Camporeale, A. STAT3-Mediated Metabolic Reprograming in Cellular Transformation and Implications for Drug Resistance. Front. Oncol. 2015, 5, 121. [Google Scholar] [CrossRef]
- Muller, P.A.J.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef]
- Hashimoto, T.; Tokuchi, Y.; Hayashi, M.; Kobayashi, Y.; Nishida, K.; Hayashi, S.; Ishikawa, Y.; Tsuchiya, S.; Nakagawa, K.; Hayashi, J.; et al. p53 null mutations undetected by immunohistochemical staining predict a poor outcome with early-stage non-small cell lung carcinomas. Cancer Res. 1999, 59, 5572–5577. [Google Scholar]
- Vang, R.; Shih, I.M.; Kurman, R.J. Ovarian low-grade and high-grade serous carcinoma: Pathogenesis, clinicopathologic and molecular biologic features, and diagnostic problems. Adv. Anat. Pathol. 2009, 16, 267–282. [Google Scholar] [CrossRef]
- Lengyel, E. Ovarian cancer development and metastasis. Am. J. Pathol. 2010, 177, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Comoglio, P.M. Pathway specificity for Met signalling. Nat. Cell Biol. 2001, 3, E161–E162. [Google Scholar] [CrossRef] [PubMed]
- Birchmeier, C.; Birchmeier, W.; Gherardi, E.; Vande Woude, G.F. Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol. 2003, 4, 915–925. [Google Scholar] [CrossRef]
- Harvey, P.; Clark, I.M.; Jaurand, M.-C.; Warn, R.M.; Edwards, D.R. Hepatocyte growth factor/scatter factor enhances the invasion of mesothelioma cell lines and the expression of matrix metalloproteinases. Br. J. Cancer 2000, 83, 1147–1153. [Google Scholar] [CrossRef]
- Mhawech-Fauceglia, P.; Afkhami, M.; Pejovic, T. MET/HGF Signaling Pathway in Ovarian Carcinoma: Clinical Implications and Future Direction. Pathol. Res. Int. 2012, 2012, 960327. [Google Scholar] [CrossRef] [PubMed]
- Maggiora, P.; Lorenzato, A.; Fracchioli, S.; Costa, B.; Castagnaro, M.; Arisio, R.; Katsaros, D.; Massobrio, M.; Comoglio, P.M.; Di Renzo, M.F. The RON and MET oncogenes are co-expressed in human ovarian carcinomas and cooperate in activating invasiveness. Exp. Cell Res. 2003, 288, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.Y.; Pon, Y.L.; Wong, A.S.T. Synergistic effects of epidermal growth factor and hepatocyte growth factor on human ovarian cancer cell invasion and migration: Role of extracellular signal-regulated kinase 1/2 and p38 mitogen-activated protein kinase. Endocrinology 2007, 148, 5195–5208. [Google Scholar] [CrossRef] [PubMed]
- Perez-Fidalgo, J.A.; Ortega, B.; Simon, S.; Samartzis, E.P.; Boussios, S. NOTCH signalling in ovarian cancer angiogenesis. Ann. Transl. Med. 2020, 8, 1705. [Google Scholar] [CrossRef] [PubMed]
- Chiaramonte, R.; Colombo, M.; Bulfamante, G.; Falleni, M.; Tosi, D.; Garavelli, S.; De Simone, D.; Vigolo, E.; Todoerti, K.; Neri, A.; et al. Notch pathway promotes ovarian cancer growth and migration via CXCR4/SDF1α chemokine system. Int. J. Biochem. Cell Biol. 2015, 66, 134–140. [Google Scholar] [CrossRef]
- Bray, S.J. Notch signalling: A simple pathway becomes complex. Nat. Rev. Mol. Cell Biol. 2006, 7, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Hopfer, O.; Zwahlen, D.; Fey, M.F.; Aebi, S. The Notch pathway in ovarian carcinomas and adenomas. Br. J. Cancer 2005, 93, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Joazeiro, C.; Fang, N.; Wang, H.-Y.; Elly, C.; Altman, Y.; Fang, D.; Hunter, T.; Liu, Y.-C. Recognition and Ubiquitination of Notch by Itch, a Hect-type E3 Ubiquitin Ligase. J. Biol. Chem. 2000, 275, 35734–35737. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, J.J.; Henkel, T.; Salmon, P.; Robey, E.; Peterson, M.G.; Hayward, S.D. Truncated mammalian Notch 1 activates CBFl/RBPJk-repressed genes by a mechanism resembling that of Epstein–Barr virus EBNA2. Mol. Cell Biol. 1996, 16, 952–959. [Google Scholar] [CrossRef] [PubMed]
- Ansieau, S.; Strobl, L.J.; Leutz, A. Activation of the Notch-regulated transcription factor CBF1/RBP-Jκ through the 13SE1A oncoprotein. Genes Dev. 2001, 15, 380–385. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Blanco, R.; Gerhardt, H. VEGF and Notch in Tip and Stalk Cell Selection. Cold Spring Harb. Perspect. Med. 2012, 3, a006569. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.-J.; Pena, G.N.A.; Pradeep, S.; Cho, M.S.; Coleman, R.L.; Sood, A.K. Anti-vascular therapies in ovarian cancer: Moving beyond anti-VEGF approaches. Cancer Metastasis Rev. 2015, 34, 19–40. [Google Scholar] [CrossRef] [PubMed]
- Siwak, D.R.; Carey, M.; Hennessy, B.T.; Nguyen, C.T.; Murray, M.J.M.; Nolden, L.; Mills, G.B. Targeting the Epidermal Growth Factor Receptor in Epithelial Ovarian Cancer: Current Knowledge and Future Challenges. J. Oncol. 2010, 2010, 568938. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Berezov, A.; Wang, Q.; Zhang, G.; Drebin, J.; Murali, R.; Greene, M.I. ErbB receptors: From oncogenes to targeted cancer therapies. J. Clin. Invest. 2007, 117, 2051–2058. [Google Scholar] [CrossRef] [PubMed]
- Oda, K.; Matsuoka, Y.; Funahashi, A.; Kitano, H. A comprehensive pathway map of epidermal growth factor receptor signaling. Mol. Syst. Biol. 2005, 1, 2005.0010. [Google Scholar] [CrossRef]
- Castellano, E.; Molina-Arcas, M.; Krygowska, A.A.; East, P.; Warne, P.; Nicol, A.; Downward, J. RAS signalling through PI3-Kinase controls cell migration via modulation of Reelin expression. Nat. Commun. 2016, 7, 11245. [Google Scholar] [CrossRef] [PubMed]
- Therachiyil, L.; Anand, A.; Azmi, A.; Bhat, A.; Korashy, H.M.; Uddin, S. Role of RAS signaling in ovarian cancer. F1000Research 2022, 11, 1253. [Google Scholar] [CrossRef]
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.W.; Agarwal, R.; Mills, G.B. Ras-superfamily GTP-ases in ovarian cancer. Cancer Treat. Res. 2009, 149, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Heald, B.; Marquard, J.; Funchain, P. Strategies for clinical implementation of screening for hereditary cancer syndromes. Semin. Oncol. 2016, 43, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Robson, M.E.; Bradbury, A.R.; Arun, B.; Domchek, S.M.; Ford, J.M.; Hampel, H.L.; Lipkin, S.M.; Syngal, S.; Wollins, D.S.; Lindor, N.M. American Society of Clinical Oncology Policy Statement Update: Genetic and Genomic Testing for Cancer Susceptibility. J. Clin. Oncol. 2015, 33, 3660–3667. [Google Scholar] [CrossRef]
- Easton, D.F.; Pharoah, P.D.; Antoniou, A.C.; Tischkowitz, M.; Tavtigian, S.V.; Nathanson, K.L.; Devilee, P.; Meindl, A.; Couch, F.J.; Southey, M.; et al. Gene-panel sequencing and the prediction of breast-cancer risk. N. Engl. J. Med. 2015, 372, 2243–2257. [Google Scholar] [CrossRef] [PubMed]
- Alvarado, M.; Tiller, G.E.; Chung, J.; Haque, R. Prevalence of mutations in a diverse cohort of 3162 women tested via the same multigene cancer panel in a managed care health plan. J. Community Genet. 2020, 11, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Corredor, J.; Woodson, A.H.; Barrera, A.G.; Arun, B. Multigene panel testing results in patients with multiple breast cancer primaries. Breast J. 2020, 26, 1337–1342. [Google Scholar] [CrossRef] [PubMed]
- Daly, M.B.; Pal, T.; Berry, M.P.; Buys, S.S.; Dickson, P.; Domchek, S.M.; Elkhanany, A.; Friedman, S.; Goggins, M.; Hutton, M.L.; et al. Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc Netw. 2021, 19, 77–102. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.K.; Alvarez, R.D.; Backes, F.J.; Bakkum-Gamez, J.N.; Barroilhet, L.; Behbakht, K.; Berchuck, A.; Chen, L.-M.; Chitiyo, V.C.; Cristea, M.; et al. NCCN Guidelines® Insights: Ovarian Cancer, Version 3.2022: Featured Updates to the NCCN Guidelines. J. Natl. Compr. Canc Netw. 2022, 20, 972–980. [Google Scholar] [CrossRef] [PubMed]
- Rahman, N. Mainstreaming genetic testing of cancer predisposition genes. Clin Med 2014, 14, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.H.; Wood, M.E.; Daniels, M.; Burke, C.; Ford, J.; Kauff, N.D.; Kohlmann, W.; Lindor, N.M.; Mulvey, T.M.; Robinson, L.; et al. American Society of Clinical Oncology Expert Statement: Collection and use of a cancer family history for oncology providers. J. Clin. Oncol. 2014, 32, 833–840. [Google Scholar] [CrossRef] [PubMed]
- Society of Gynecologic Oncology. SGO Clinical Practice Statement: Genetic Testing for Ovarian Cancer. 2014. Available online: https://www.sgo.org/resources/genetic-testing-for-gynecologic-cancer/ (accessed on 6 September 2023).
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Pan, Z.; Xie, X. BRCA mutations in the manifestation and treatment of ovarian cancer. Oncotarget 2017, 8, 97657–97670. [Google Scholar] [CrossRef] [PubMed]
- Rahman, N. Realizing the promise of cancer predisposition genes. Nature 2014, 505, 302–308, Erratum in Nature 2014, 510, 176. [Google Scholar] [CrossRef]
- Grancher, A.; Michel, P.; Di Fiore, F.; Sefrioui, D. Colorectal cancer chemoprevention: Is aspirin still in the game? Cancer Biol. Ther. 2022, 23, 446–461. [Google Scholar] [CrossRef] [PubMed]
- Levy, S.E.; Boone, B.E. Next-Generation Sequencing Strategies. Cold Spring Harb. Perspect. Med. 2019, 9, a025791. [Google Scholar] [CrossRef] [PubMed]
- Fostira, F.; Papadimitriou, M.; Papadimitriou, C. Current practices on genetic testing in ovarian cancer. Ann. Transl. Med. 2020, 8, 1703. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, B.T.J.; Timms, K.M.; Carey, M.S.; Gutin, A.; Meyer, L.A.; Flake, D.D., 2nd; Abkevich, V.; Potter, J.; Pruss, D.; Glenn, P.; et al. Somatic mutations in BRCA1 and BRCA2 could expand the number of patients that benefit from poly (ADP ribose) polymerase inhibitors in ovarian cancer. J. Clin. Oncol. 2010, 28, 3570–3576. [Google Scholar] [CrossRef] [PubMed]
- Pennington, K.P.; Walsh, T.; Harrell, M.I.; Lee, M.K.; Pennil, C.C.; Rendi, M.H.; Thornton, A.; Norquist, B.M.; Casadei, S.; Nord, A.S.; et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin. Cancer Res. 2014, 20, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.; Thompson, E.R.; Nannya, Y.; Yamamoto, G.; Pillai, R.; Ogawa, S.; Bailey, D.K.; Campbell, I.G. Genome-wide, high-resolution detection of copy number, loss of heterozygosity, and genotypes from formalin-fixed, paraffin-embedded tumor tissue using microarrays. Cancer Res. 2007, 67, 2544–2551. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, P.; Jonkers, J. Molecular pathways: How can BRCA-mutated tumors become resistant to PARP inhibitors? Clin. Cancer Res. 2014, 20, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef] [PubMed]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Dicks, E.; Ramus, S.J.; Tyrer, J.P.; Intermaggio, M.P.; Hayward, J.; Edlund, C.K.; Conti, D.; Harrington, P.; Fraser, L.; et al. Contribution of Germline Mutations in the RAD51B, RAD51C, and RAD51D Genes to Ovarian Cancer in the Population. J. Clin. Oncol. 2015, 33, 2901–2907. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subtype | Frequency of Genetic Alterations [10] | |||
|---|---|---|---|---|
| TP53 | BRCA 1/2 | PIK3CA | KRAS | |
| HGSOC | 96% | 22–40% | 2.9% | 5.9% |
| LGSOC | 8.3 | 10% | 12.5% | 54% |
| EnOC | 5–54.5% | 11.1% | 31.4% | 10.3% |
| OCCC | 10% | 4.5% | 51% | 15% |
| Mucinous | 56.8% | 0 | 13.5% | 57.1–64.9% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maioru, O.-V.; Radoi, V.-E.; Coman, M.-C.; Hotinceanu, I.-A.; Dan, A.; Eftenoiu, A.-E.; Burtavel, L.-M.; Bohiltea, L.-C.; Severin, E.-M. Developments in Genetics: Better Management of Ovarian Cancer Patients. Int. J. Mol. Sci. 2023, 24, 15987. https://doi.org/10.3390/ijms242115987
Maioru O-V, Radoi V-E, Coman M-C, Hotinceanu I-A, Dan A, Eftenoiu A-E, Burtavel L-M, Bohiltea L-C, Severin E-M. Developments in Genetics: Better Management of Ovarian Cancer Patients. International Journal of Molecular Sciences. 2023; 24(21):15987. https://doi.org/10.3390/ijms242115987
Chicago/Turabian StyleMaioru, Ovidiu-Virgil, Viorica-Elena Radoi, Madalin-Codrut Coman, Iulian-Andrei Hotinceanu, Andra Dan, Anca-Elena Eftenoiu, Livia-Mălina Burtavel, Laurentiu-Camil Bohiltea, and Emilia-Maria Severin. 2023. "Developments in Genetics: Better Management of Ovarian Cancer Patients" International Journal of Molecular Sciences 24, no. 21: 15987. https://doi.org/10.3390/ijms242115987
APA StyleMaioru, O.-V., Radoi, V.-E., Coman, M.-C., Hotinceanu, I.-A., Dan, A., Eftenoiu, A.-E., Burtavel, L.-M., Bohiltea, L.-C., & Severin, E.-M. (2023). Developments in Genetics: Better Management of Ovarian Cancer Patients. International Journal of Molecular Sciences, 24(21), 15987. https://doi.org/10.3390/ijms242115987