

The Plant Parasitic Nematodes Database: A Comprehensive Genomic Data Platform for Plant Parasitic Nematode Research

Abstract
:1. Introduction
2. Results
2.1. Online Platform Construction
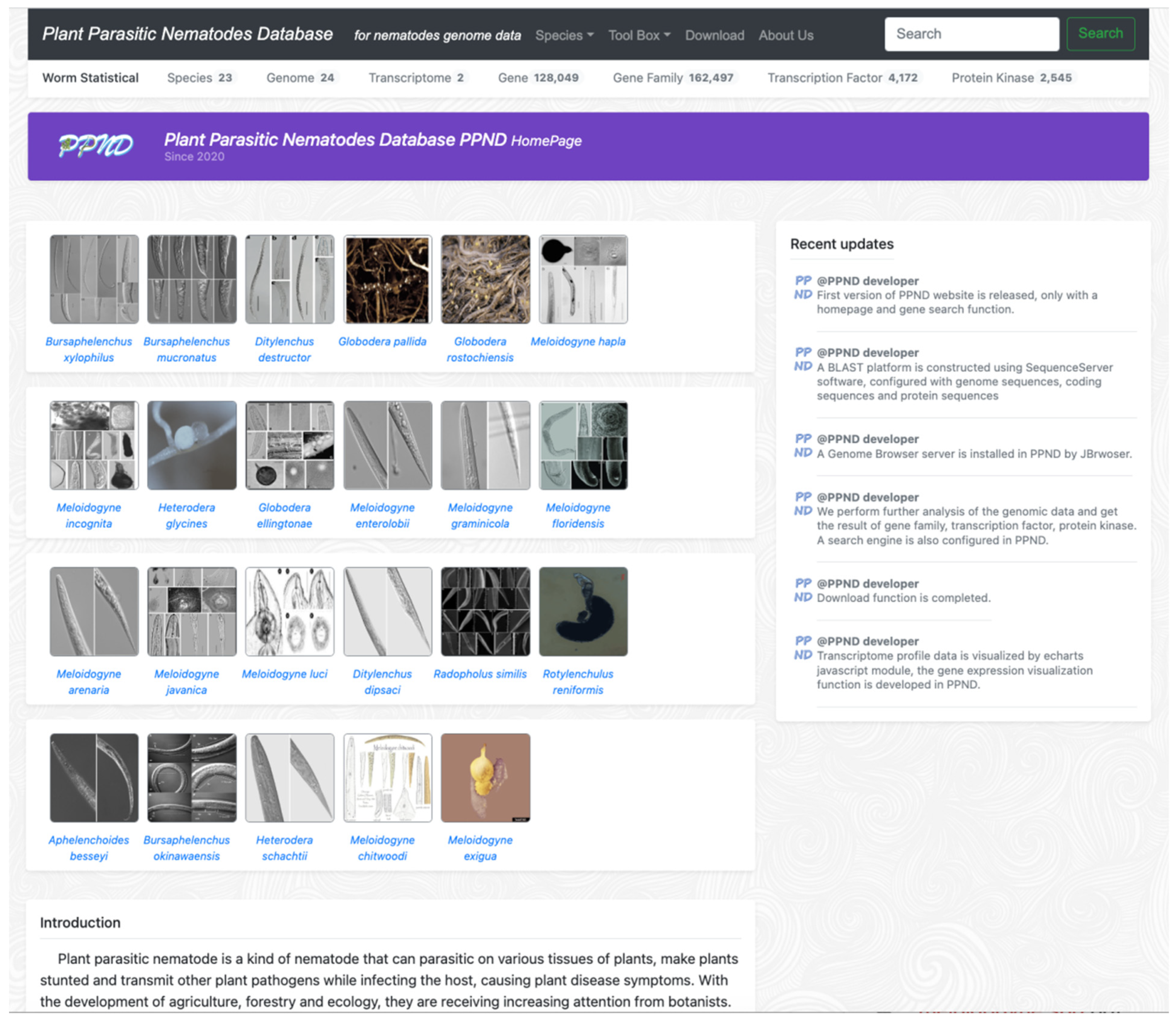
2.2. PPND Homepage
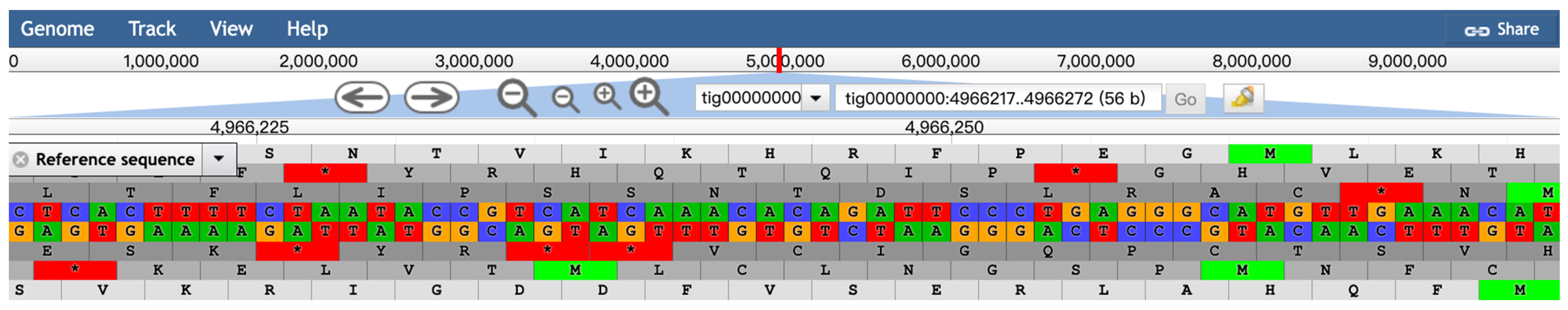
2.3. Genome Browser
2.4. BLAST
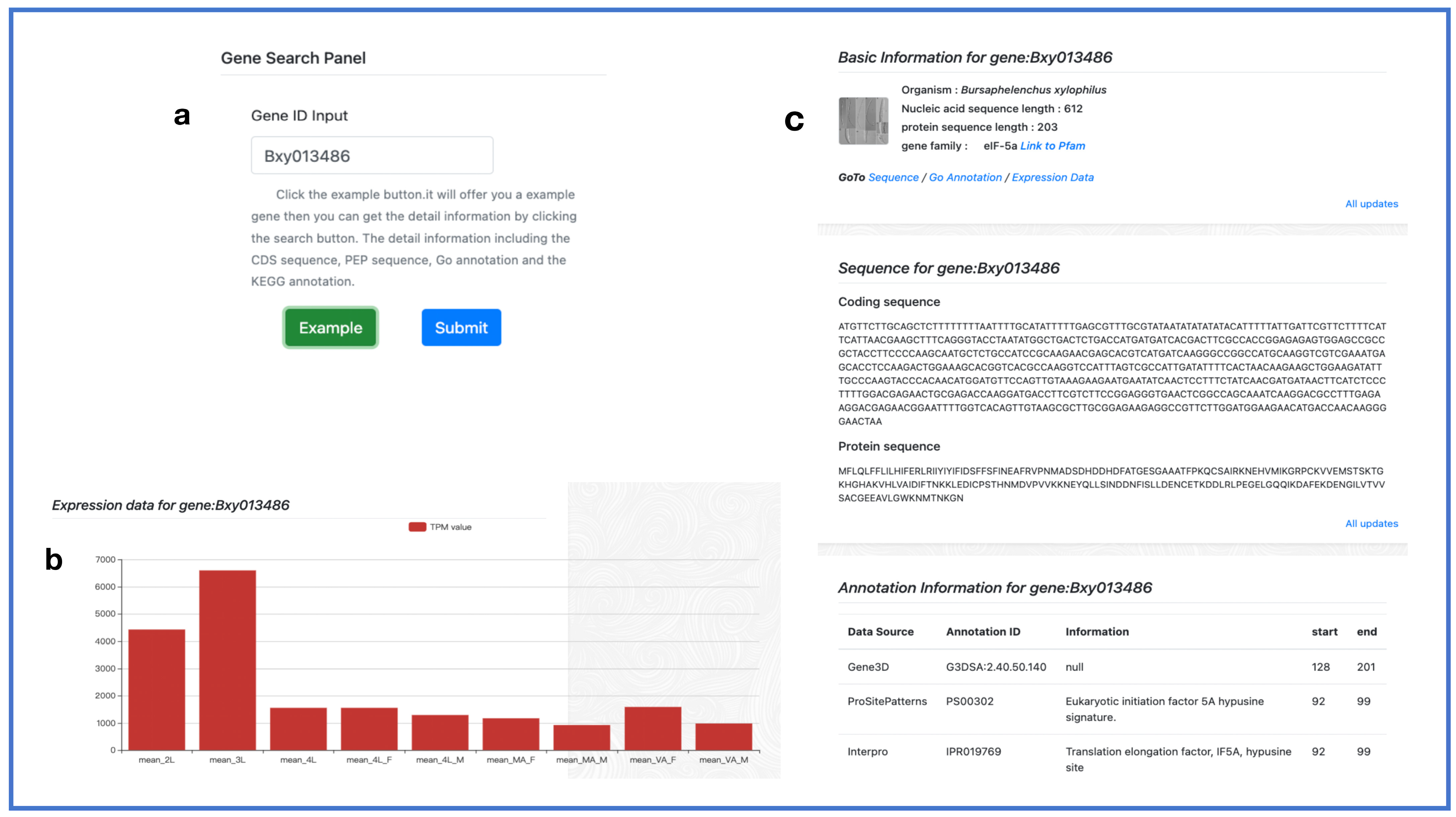
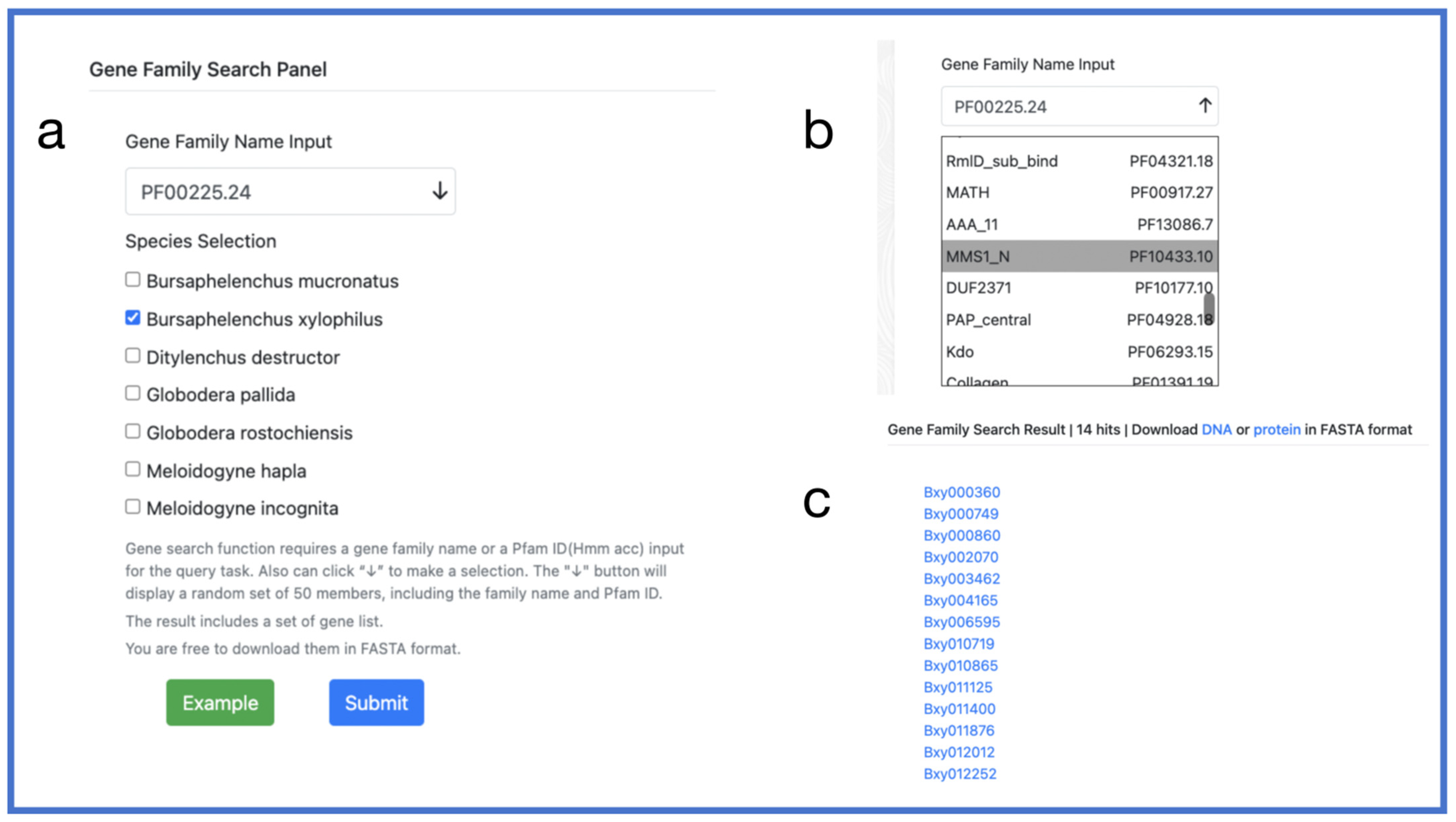
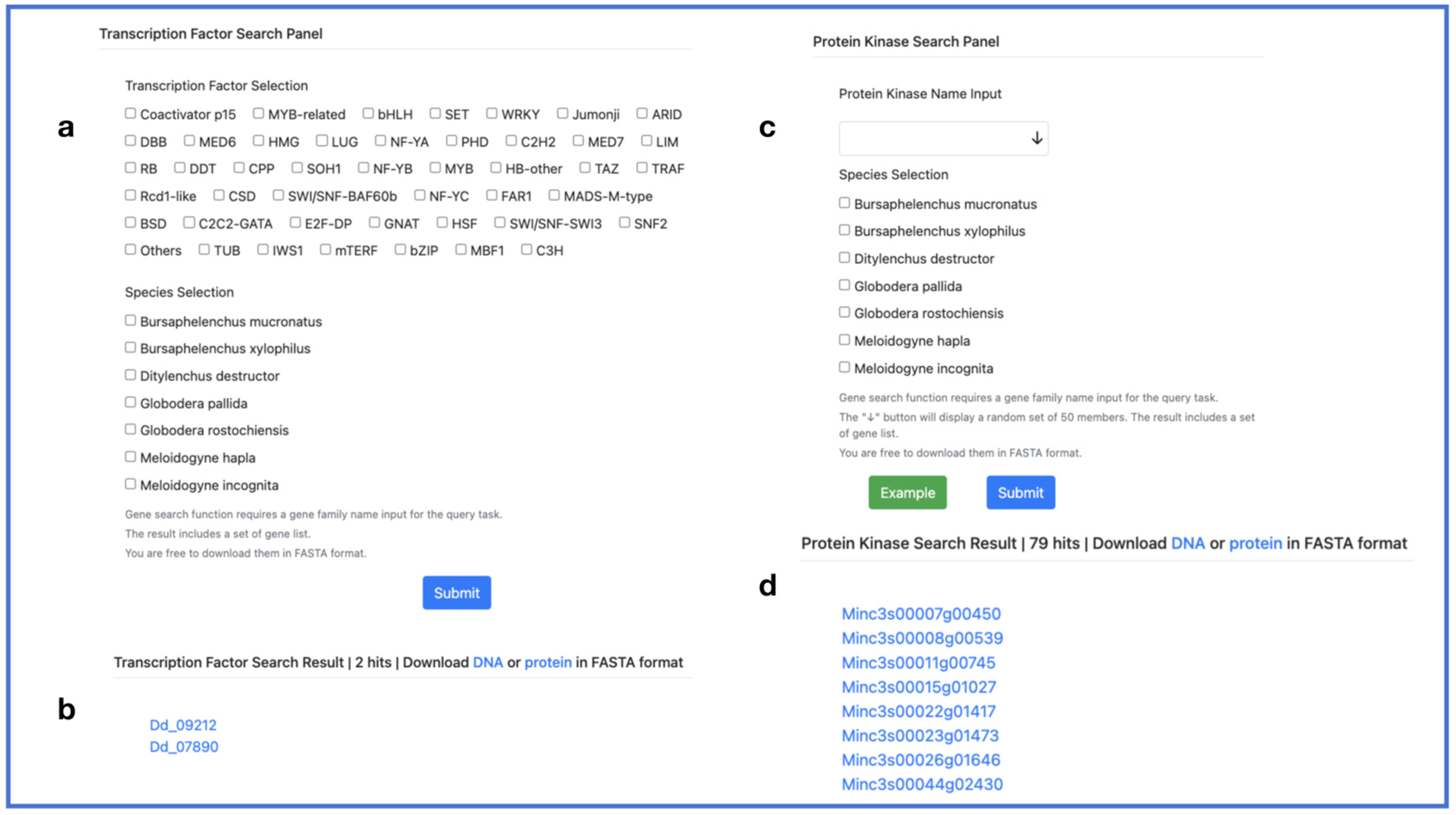
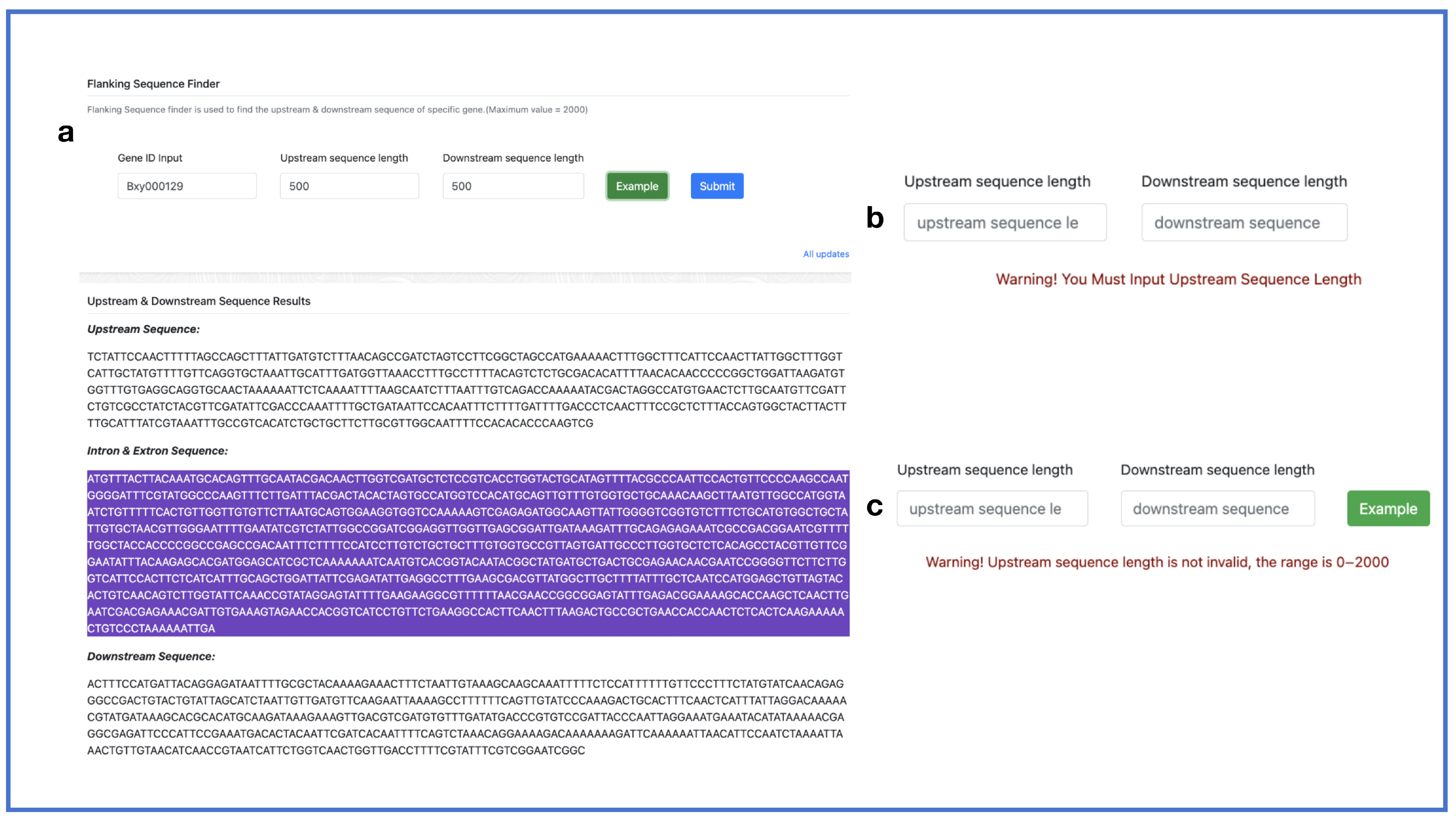
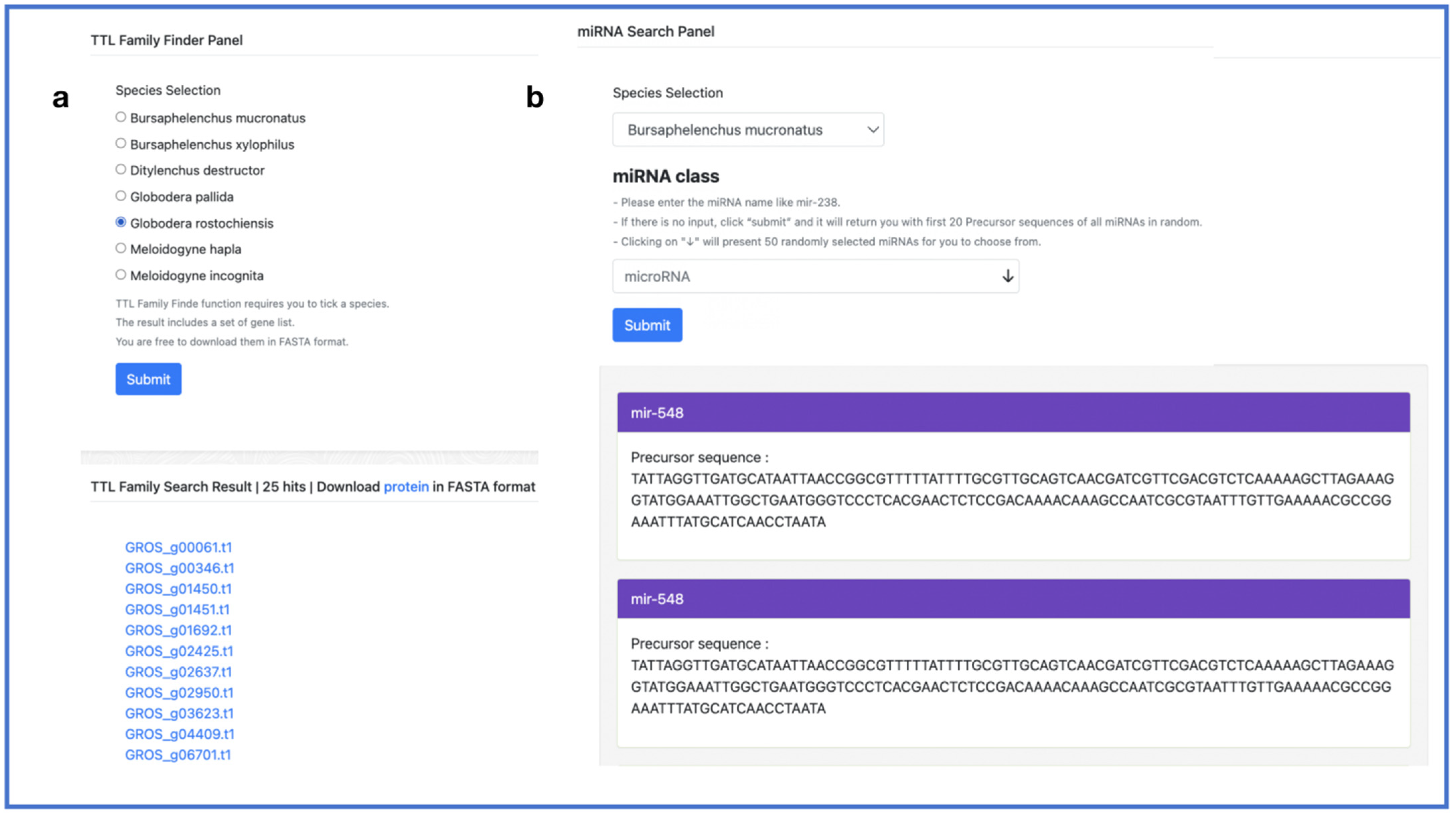
2.5. Search Toolkit
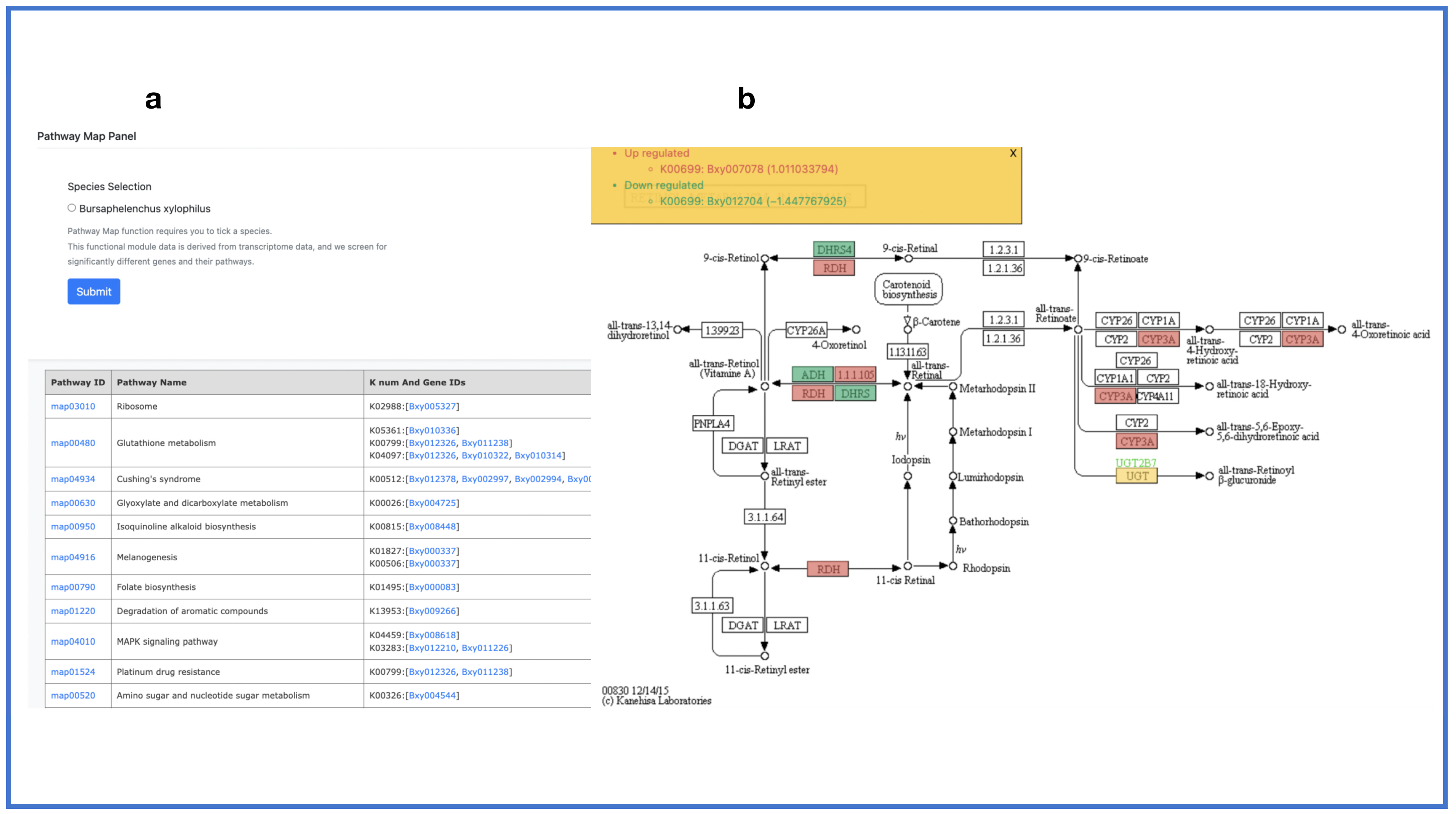
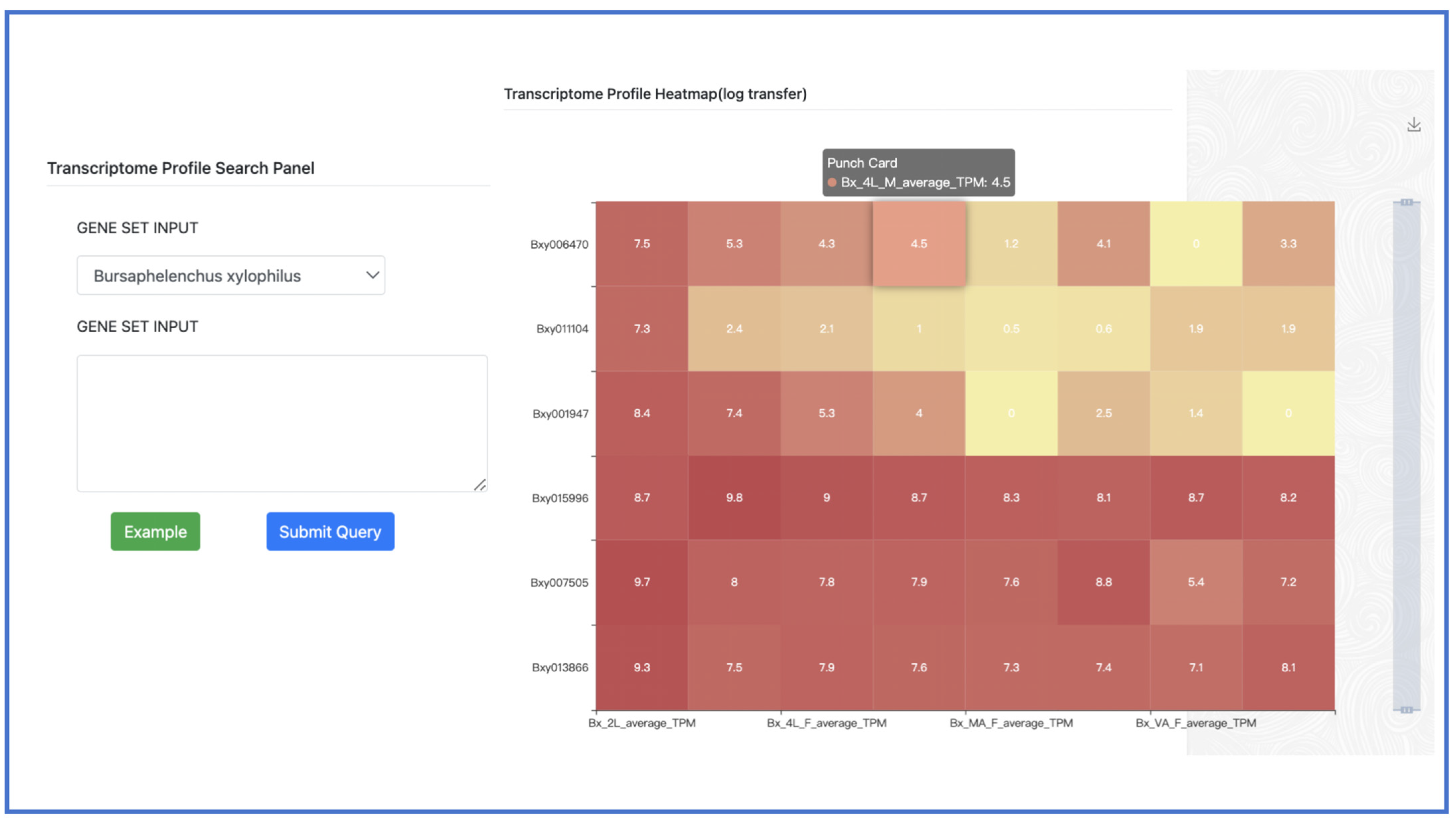
2.6. Transcriptome Profile
2.7. Download and “About Us”
3. Discussion
4. Materials and Methods
4.1. Construction and Content
4.2. Genome Assembly
4.3. Genome Annotation
4.4. Genome Analysis
4.5. Transcriptome Assembly and Annotation
4.6. BLAST and JBrowse Deployment
4.7. Deployment of Other Functionalities
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Decraemer, W.; Hunt, D.J. Structure and classification. In Plant Nematology; Perry, R.N., Moens, M., Eds.; CAB International: Wallingford, UK, 2006; pp. 3–32. [Google Scholar] [CrossRef]
- Nicol, J.M.; Turner, S.J.; Coyne, D.L.; Nijs, L.D.; Hockland, S.; Maafi, Z.T. Current nematode threats to world agriculture. In Genomics and Molecular Genetics of Plant–Nematode Interactions; Jones, J.T., Gheysen, G., Fenoll, C., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 21–44. [Google Scholar] [CrossRef]
- Jones, J.T.; Haegeman, A.; Danchin, E.G.J.; Gaur, H.S.; Jones, M.G.K.; Kikuchi, T.; Manzanilla-López, R.; Palomares-Rius, J.E.; Wesemael, W.M.L.; Perry, R.N. Top 10 plant-parasitic nematodes in molecular plant pathology. Mol. Plant Pathol. 2013, 14, 946–961. [Google Scholar] [CrossRef] [PubMed]
- Karajeh, M.R. Interaction of root-knot nematode (Meloidogyne javanica) and tomato as affected by hydrogen peroxide. J. Plant Prot. Res. 2008, 48, 181–187. [Google Scholar] [CrossRef]
- C. elegans Sequencing Consortium. Genome sequence of the nematode C. elegans: A platform for investigating biology. Science 1998, 282, 2012–2018. [Google Scholar] [CrossRef] [PubMed]
- Abad, P.; Gouzy, J.; Aury, J.M.; Castagnone-Sereno, P.; Danchin, E.G.; Deleury, E.; Perfus-Barbeoch, L.; Anthouard, V.; Artiguenave, F.; Blok, V.C.; et al. Genome sequence of the metazoan plant-parasitic nematode Meloidogyne incognita. Nat. Biotechnol. 2008, 26, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Sellers, G.S.; Jeffares, D.C.; Lawson, B.; Prior, T.; Lunt, D.H. Identification of individual root-knot nematodes using low coverage long-read sequencing. PLoS ONE 2021, 16, e0253248. [Google Scholar] [CrossRef]
- Opperman, C.H.; Bird, D.M.; Williamson, V.M.; Rokhsar, D.S.; Burke, M.; Cohn, J.; Cromer, J.; Diener, S.; Gajan, J.; Graham, S.; et al. Sequence and genetic map of Meloidogyne hapla: A compact nematode genome for plant parasitism. Proc. Natl. Acad. Sci. USA 2008, 105, 14802–14807. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, T.; Cotton, J.A.; Dalzell, J.J.; Hasegawa, K.; Kanzaki, N.; McVeigh, P.; Takanashi, T.; Tsai, I.J.; Assefa, S.A.; Cock, P.J.; et al. Genomic insights into the origin of parasitism in the emerging plant pathogen Bursaphelenchus xylophilus. PLoS Pathog. 2011, 7, e1002219. [Google Scholar] [CrossRef]
- Dayi, M.; Sun, S.; Maeda, Y.; Tanaka, R.; Yoshida, A.; Tsai, I.J.; Kikuchi, T. Nearly Complete Genome Assembly of the Pinewood Nematode Bursaphelenchus xylophilus Strain Ka4C1. Microbiol. Resour. Announc. 2020, 9, e01002-20. [Google Scholar] [CrossRef]
- Cotton, J.A.; Lilley, C.J.; Jones, L.M.; Kikuchi, T.; Thorpe, P.; Tsai, I.J.; Beasley, H.; Blok, V.; Cock, P.J.A.; et al. The genome and life-stage specific transcriptomes of Globodera pallida elucidate key aspects of plant parasitism by a cyst nematode. Genome Biol. 2014, 15, R43. [Google Scholar] [CrossRef]
- van Steenbrugge, J.J.M.; van den Elsen, S.; Lozano-Torres, J.L.; Putker, V.; Thorpe, P.; Goverse, A.; Sterken, M.G.; Smant, G.; Helder, J.; et al. Comparative genomics among cyst nematodes reveals distinct evolutionary histories among effector families and an irregular distribution of effector-associated promoter motifs. Mol. Ecol. 2023, 6, 1515–1529. [Google Scholar] [CrossRef]
- Eves-van den Akker, S.; Laetsch, D.R.; Thorpe, P.; Lilley, C.J.; Danchin, E.G.J.; Da Rocha, M.; Rancurel, C.; Holroyd, N.E.; Cotton, J.A.; Szitenberg, A.; et al. The genome of the yellow potato cyst nematode, Globodera rostochiensis, reveals insights into the basis of parasitism and virulence. Genome Biol. 2016, 17, 124. [Google Scholar] [CrossRef] [PubMed]
- Masonbrink, R.; Maier, T.R.; Muppirala, U.; Seetharam, A.S.; Lord, E.; Juvale, P.S.; Schmutz, J.; Johnson, N.T.; Korkin, D.; Mitchum, M.G.; et al. The genome of the soybean cyst nematode (Heterodera glycines) reveals complex patterns of duplications involved in the evolution of parasitism genes. BMC Genom. 2019, 20, 119. [Google Scholar] [CrossRef] [PubMed]
- Masonbrink, R.; Maier, T.R.; Hudson, M.; Severin, A.; Baum, T. A chromosomal assembly of the soybean cyst nematode genome. Mol. Ecol. Resour. 2021, 21, 7. [Google Scholar] [CrossRef] [PubMed]
- Phillips, W.S.; Howe, D.K.; Brown, A.M.V.; Den Akker, S.E.-V.; Dettwyler, L.; Peetz, A.B.; Denver, D.R.; Zasada, I.A. The draft genome of Globodera ellingtonae. J. Nematol. 2020, 49, 127–128. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Gao, S.; Wang, S.; Meng, J.; Wickham, J.; Luo, S.; Tan, X.; Yu, H.; Xiang, Y.; Hu, S.; et al. A Reference Genome of Bursaphelenchus mucronatus Provides New Resources for Revealing Its Displacement by Pinewood Nematode. Genes 2020, 11, 570. [Google Scholar] [CrossRef] [PubMed]
- Koutsovoulos, G.D.; Poullet, M.; El Ashry, A.; Kozlowski, D.K.; Sallet, E.; Da Rocha, M.; Martin-Jimenez, C.; Perfus-Barbeoch, L.; Frey, J.-E.; Ahrens, C.; et al. The polyploid genome of the mitotic parthenogenetic root-knot nematode Meloidogyne enterolobii. BioRxiv 2019, 586818. [Google Scholar] [CrossRef]
- Somvanshi, V.S.; Tathode, M.; Shukla, R.N.; Rao, U. Nematode genome announcement: A draft genome for rice root-knot nematode, Meloidogyne graminicola. J. Nematol. 2018, 50, 111–116. [Google Scholar] [CrossRef]
- Phan, N.T.; Orjuela, J.; Danchin, E.G.J.; Klopp, C.; Perfus-Barbeoch, L.; Kozlowski, D.K.; Koutsovoulos, G.D.; Lopez-Roques, C.; Bouchez, O.; Zahm, M.; et al. Genome structure and content of the rice root-knot nematode (Meloidogyne graminicola). Ecol. Evol. 2020, 20, 11006–11021. [Google Scholar] [CrossRef]
- Lunt, D.H.; Kumar, S.; Koutsovoulos, G.; Blaxter, M.L. The complex hybrid origins of the root knot nematodes revealed through comparative genomics. PeerJ 2014, 2, e356. [Google Scholar] [CrossRef]
- Sato, K.; Kadota, Y.; Gan, P.; Bino, T.; Uehara, T.; Yamaguchi, K.; Ichihashi, Y.; Maki, N.; Iwahori, H.; Suzuki, T.; et al. High-quality genome sequence of the root-knot nematode Meloidogyne arenaria genotype A2-O. Genome Announc. 2018, 6, e00519-18. [Google Scholar] [CrossRef]
- Blanc-Mathieu, R.; Perfus-Barbeoch, L.; Aury, J.-M.; Da Rocha, M.; Gouzy, J.; Sallet, E.; Martin-Jimenez, C.; Bailly-Bechet, M.; Castagnone-Sereno, P.; Flot, J.-F.; et al. Hybridization and polyploidy enable genomic plasticity without sex in the most devastating plant-parasitic nematodes. PLoS Genet. 2017, 13, e1006777. [Google Scholar] [CrossRef] [PubMed]
- Susič, N.; Koutsovoulos, G.D.; Riccio, C.; Danchin, E.G.J.; Blaxter, M.L.; Luntl, D.H.; Strajnar, P.; Širca, S.; Urek, G.; Stare, B.G. Genome sequence of the root-knot nematode Meloidogyne luci. J. Nematol. 2020, 52, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Peng, D.; Chen, L.; Liu, H.; Chen, F.; Xu, M.; Ju, S.; Ruan, L.; Sun, M. The Ditylenchus destructor genome provides new insights into the evolution of plant parasitic nematodes. Proc. R. Soc. B 2016, 283, 20160942. [Google Scholar] [CrossRef] [PubMed]
- Mimee, B.; Lord, E.; Véronneau, P.Y.; Masonbrink, R.; Yu, Q.; Eves-van den Akker, S. The draft genome of Ditylenchus dipsaci. J. Nematol. 2019, 51, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Burke, M.; Scholl, E.H.; Bird, D.M.; Schaff, J.E.; Colman, S.D.; Crowell, R.; Diener, S.; Gordon, O.; Graham, S.; Wang, X.; et al. The plant parasite Pratylenchus coffeae carries a minimal nematode genome. Nematology 2015, 17, 621–637. [Google Scholar] [CrossRef]
- Mathew, R.; Opperman, C.H. The genome of the migratory nematode, Radopholus similis, reveals signatures of close association to the sedentary cyst nematodes. PLoS ONE 2019, 14, e0224391. [Google Scholar] [CrossRef]
- Wram, C.L.; Hesse, C.N.; Wasala, S.K.; Howe, D.K.; Peetz, A.B.; Denver, D.R.; Humphreys-Pereira, D.; Zasada, I.A. Genome announcement: The draft genomes of two Radopholus similis populations from Costa Rica. J. Nematol. 2019, 51, 1–4. [Google Scholar] [CrossRef]
- Showmaker, K.C.; Sanders, W.S.; den Akker, S.E.-V.; Martin, B.E.; Platt, R.N.; Stokes, J.V.; Hsu, C.-Y.; Bartlett, B.D.; Peterson, D.G.; Wubben, M.J. A genomic resource for the sedentary semi-endoparasitic reniform nematode, Rotylenchulus reniformis Linford & Oliveira. J. Nematol. 2019, 51, 1–2. [Google Scholar] [CrossRef]
- Siddique, S.; Radakovic, Z.S.; Hiltl, C.; Pellegrin, C.; Baum, T.J.; Beasley, H.; Bent, A.F.; Chitambo, O.; Chopra, D.; Danchin, E.G.J.; et al. The genome and lifestage-specific transcriptomes of a plant-parasitic nematode and its host reveal susceptibility genes involved in trans-kingdom synthesis of vitamin B5. Nat. Commun. 2022, 13, 6190. [Google Scholar] [CrossRef]
- Humphreys-Pereira, D.A.; Elling, A.A. Mitochondrial genomes of Meloidogyne chitwoodi and M. incognita (Nematoda: Tylenchina): Comparative analysis, gene order and phylogenetic relationships with other nematodes. Mol. Biochem. Parasitol. 2014, 194, 20–32. [Google Scholar] [CrossRef]
- Phan, N.T.; Besnard, G.; Ouazahrou, R.; Sánchez, W.S.; Gil, L.; Manzi, S.; Bellafiore, S. Genome sequence of the coffee root-knot nematode. J. Nematol. 2021, 53, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Xie, J.; Han, Z.; Yang, F.; Yu, W.; Peng, Y.; Qing, X. Complete genome sequencing of nematode Aphelenchoides besseyi, an economically important pest causing rice white-tip disease. Phytopathol. Res. 2023, 5, 5. [Google Scholar] [CrossRef]
- Shinya, R.; Sun, S.; Dayi, M.; Tsai, I.J.; Miyama, A.; Chen, A.F.; Hasegawa, K.; Antoshechkin, I.; Kikuchi, T.; Sternberg, P.W. Possible stochastic sex determination in Bursaphelenchus nematodes. Nat. Commun. 2022, 13, 2574. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, T.; Eves-van den Akker, S.; Jones, J.T. Genome evolution of plant-parasitic nematodes. Annu. Rev. Phytopathol. 2017, 55, 333–354. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.W.; Antoshechkin, I.; Bieri, T.; Blasiar, D.; Chan, J.; Chen, W.J.; De La Cruz, N.; Davis, P.; Duesbury, M.; Fang, R.; et al. WormBase: A comprehensive resource for nematode research. Nucleic Acids Res. 2010, 38, D463–D467. [Google Scholar] [CrossRef]
- NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2016, 44, D7–D19. [Google Scholar] [CrossRef] [PubMed]
- Raggett, D.; Le Hors, A.; Jacobs, I. HTML 4.01 Specification. W3C Proposed Recommendation. 1999. Available online: http://www.w3.org/TR/html40 (accessed on 23 November 2023).
- Duckett, J. HTML & CSS: Design and Build Websites; John Wiley & Sons: Hoboken, NJ, USA, 2011. [Google Scholar]
- Jensen, S.H.; Møller, A.; Thiemann, P. Type analysis for JavaScript. In Static Analysis. SAS 2009. Lecture Notes in Computer Science; Palsberg, J., Su, Z., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; Volume 5673, pp. 238–255. [Google Scholar] [CrossRef]
- Hesterberg, T. Bootstrap. Wiley Interdisciplinary Reviews. Comput. Stat. 2011, 3, 497–516. [Google Scholar] [CrossRef]
- Grinberg, M. Flask Web Development: Developing Web Applications with Python; O’Reilly Media, Inc.: Sebastopol, CA, USA, 2018. [Google Scholar]
- Reese, W. Nginx: The High-Performance Web Server and Reverse Proxy. Linux J. 2008. Available online: https://dl.acm.org/doi/fullHtml/10.5555/1412202.1412204 (accessed on 23 November 2023).
- Percival, H.J.W. Test-Driven Development with Python: Obey the Testing Goat: Using Django, Selenium, and JavaScript; O’Reilly Media, Inc.: Sebastopol, CA, USA, 2014. [Google Scholar]
- Jacob, J.; Vanholme, B.; Haegeman, A.; Gheysen, G. Four transthyretin-like genes of the migratory plant-parasitic nematode Radopholus similis: Members of an extensive nematode-specific family. Gene 2007, 402, 9–19. [Google Scholar] [CrossRef]
- Hu, L.; Liu, S. Genome-wide Analysis of the MADS-box Gene Family in Cucumber. Genome 2012, 55, 245–256. [Google Scholar] [CrossRef]
- Li, B.; Ruotti, V.; Stewart, R.M.; Thomson, J.A.; Dewey, C.N. RNA-Seq Gene Expression Estimation with Read Mapping Uncertainty. Bioinformatics 2009, 26, 493–500. [Google Scholar] [CrossRef]
- Marcais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.L.; Chen, Y.; Xie, S.-Q.; Chen, K.-N.; Wang, Y.; Han, Y.; Luo, F.; Xie, Z. MECAT: Fast mapping, error correction, and de novo assembly for single-molecule sequencing reads. Nat. Methods 2017, 14, 1072–1074. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.S.; Peluso, P.; Sedlazeck, F.J.; Nattestad, M.; Concepcion, G.T.; Clum, A.; Dunn, C.; O’Malley, R.; Figueroa-Balderas, R.; Morales-Cruz, A.; et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 2016, 13, 1050–1054. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E.; et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, R.M.; Seppey, M.; Simão, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Smit, A.F.A. Repeat-Masker Open-3.0. 2004. Available online: http://www.repeatmasker.org (accessed on 23 November 2023).
- Edgar, R.C.; Myers, E.W. PILER: Identification and classification of genomic repeats. Bioinformatics 2005, 21, i152–i158. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, H. Ltr_finder: An efficient tool for the prediction of full-length ltr retrotransposons. Nucl. Acids Res. 2007, 35, W265–W268. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Birney, E.; Clamp, M.; Durbin, R. GeneWise and Genomewise. Genome Res. 2004, 14, 988–995. [Google Scholar] [CrossRef]
- Holt, C.; Yandell, M. MAKER2: An annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinform. 2011, 12, 491. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, Y.; Li, S.; Hu, N.; He, Y.; Pong, R.; Lin, D.; Lu, L.; Law, M. Comparison of Next-Generation Sequencing Systems. J. Biomed. Biotechnol. 2012, 2012, 251364. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Attwood, T.K.; Babbitt, P.C.; Bateman, A.; Bork, P.; Bridge, A.J.; Chang, H.-Y.; Dosztányi, Z.; El-Gebali, S.; Fraser, M.; et al. InterPro in 2017—Beyond Protein Family and Domain Annotations. Nucleic Acids Res. 2017, 45, D190–D199. [Google Scholar] [CrossRef] [PubMed]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smar, A.; et al. The Pfam Protein Families Database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER Web Server: Interactive Sequence Similarity Searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [PubMed]
- The Gene Ontology Consortium. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y. KEGG Mapper for Inferring Cellular Functions from Protein Sequences. Protein Sci. 2020, 29, 28–35. [Google Scholar] [CrossRef]
- Zheng, Y.; Jiao, C.; Sun, H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.; Banf, M.; Dai, X.; Martin, G.B.; Giovannoni, J.J.; et al. iTAK: A Program for Genome-wide Prediction and Classification of Plant Transcription Factors, Transcriptional Regulators, and Protein Kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2013, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Priyam, A.; Woodcroft, B.J.; Rai, V.; Moghul, I.; Munagala, A.; Ter, F.; Chowdhary, H.; Pieniak, I.; Maynard, L.J.; Gibbins, M.A.; et al. SequenceServer: A Modern Graphical User Interface for Custom BLAST Databases. Mol. Biol. Evol. 2019, 36, 2922–2924. [Google Scholar] [CrossRef]
- Buels, R.; Yao, E.; Diesh, C.M.; Hayes, R.D.; Munoz-Torres, M.; Helt, G.; Goodstein, D.M.; Elsik, C.G.; Lewis, S.E.; Stein, L.; et al. JBrowse: A Dynamic Web Platform for Genome Visualization and Analysis. Genome Biol. 2016, 17, 66. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Type | Count |
|---|---|
| Nuclear genome | 25 |
| Coding sequence | 128,049 |
| Protein | 128,049 |
| Protein kinase | 2545 |
| Transcription factors | 4172 |
| Annotation items | 779,406 |
| Species | Total Assembly | Assembly Level | Number of Scaffolds | Scaffold N50 | Number of Contigs | Contig N50 | Public Time | Cite |
|---|---|---|---|---|---|---|---|---|
| Aphelenchoides besseyi | 47.4 Mb | Scaffold | 39 | 17.8 Mb | 112 | 1.1 Mb | 2022-08 | Ji, H. et al., 2023 [35] |
| Bursaphelenchus mucronatus (Bmu) | 80.4 Mb | Contig | - | - | 227 | 1.8 Mb | - | - |
| Bursaphelenchus mucronatus 2020 | 73 Mb | Chromosome | 72 | 11.5 Mb | 181 | 1.6 Mb | 2022-09 | Wu, S. et al., 2020 [18] |
| Bursaphelenchus okinawaensis | 70 Mb | Scaffold | 7 | 11.6 Mb | 11 | 9.8 Mb | 2021-08 | Shinya, R. et al., 2022 [36] |
| Bursaphelenchus xylophilus (Bxy) | 77.1 Mb | Scaffold | 11 | 12.6 Mb | 52 | 5.7 Mb | - | - |
| Bursaphelenchus xylophilus 2020 | 78.3 Mb | Scaffold | 11 | 12.8 Mb | 54 | 5.9 Mb | 2020-10 | Dayi, M. et al., 2020 [10] |
| Ditylenchus destructor | 139.4 Mb | Contig | - | - | 1236 | 782 kb | 2016-03 | Zheng, J. et al., 2016 [25] |
| Ditylenchus dipsaci | 227.2 Mb | Scaffold | 1394 | 287.4 kb | 1631 | 246.9 kb | 2019-02 | Mimee, B. et al., 2019 [26] |
| Globodera ellingtonae | 105.1 Mb | Scaffold | 2246 | 327.2 kb | 13,948 | 13.2 kb | 2016-12 | Phillips, W.S. et al., 2020 [16] |
| Globodera pallida | 112.3 Mb | Scaffold | 163 | 2.9 Mb | 1466 | 662.1 kb | 2021-10 | van Steenbrugge, J.J.W. et al., 2023 [12] |
| Globodera rostochiensis | 92.2 Mb | Scaffold | 88 | 3.3 Mb | 282 | 1 Mb | 2021-05 | van Steenbrugge, J.J.W. et al., 2023 [12] |
| Heterodera glycines | 156.3 Mb | Chromosome | 9 | 17.9 Mb | 2121 | 138.3 kb | 2021-07 | Masonbrink, R. et al., 2021 [15] |
| Heterodera schachtii | 174.3 Mb | Scaffold | 395 | 1.3 Mb | 1682 | 301.4 kb | 2022-05 | Siddique, S. et al., 2022 [31] |
| Meloidogyne arenaria | 281.7 Mb | Contig | - | - | 1430 | 434.7 kb | 2021-03 | - |
| Meloidogyne chitwoodi | 47.5 Mb | Contig | - | - | 30 | 2.5 Mb | 2020-11 | Sellers, G.S. et al., 2021 [7] |
| Meloidogyne enterolobii | 240.1 Mb | Scaffold | 4437 | 143.5 kb | 4451 | 143.3 kb | 2020-08 | Sellers, G.S. et al., 2021 [7] |
| Meloidogyne exigua | 42.1 Mb | Contig | - | - | 206 | 1.9 Mb | 2021-01 | Phan, N.T. et al., 2021 [33] |
| Meloidogyne floridensis | 74.6 Mb | Scaffold | 8887 | 13.3 kb | 13,362 | 8.1 kb | 2018-10 | Sellers, G.S. et al., 2021 [7] |
| Meloidogyne graminicola | 41.5 Mb | Scaffold | 283 | 294.9 kb | 286 | 294.9 kb | 2020-09 | Phan, N.T. et al., 2020 [33] |
| Meloidogyne hapla | 53 Mb | Contig | - | - | 3450 | 37.6 kb | 2008-09 | Opperman, C.H. et al., 2008 [8] |
| Meloidogyne incognita | 193.2 Mb | Contig | - | - | 374 | 974.8 kb | 2020-07 | Sellers, G.S. et al., 2021 [7] |
| Meloidogyne javanica | 149.9 Mb | Scaffold | 34,316 | 14.1 kb | 38,690 | 11.9 kb | 2018-10 | Blanc-Mathieu, R. et al., 2017 [23] |
| Meloidogyne luci | 209.2 Mb | Contig | - | - | 327 | 1.7 Mb | 2019-12 | Susič, N. et al., 2020 [24] |
| Radopholus similis | 50.5 Mb | Scaffold | 5192 | 27.8 kb | 5339 | 26.5 kb | 2020-06 | Wram, C.L. et al., 2019 [29] |
| Rotylenchulus reniformis | 310.8 Mb | Scaffold | 100,524 | 22.7 kb | 129,027 | 6 kb | 2015-06 | Showmaker, K.C. et al., 2019 [30] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhuge, J.; Zhou, X.; Zhou, L.; Hu, J.; Guo, K. The Plant Parasitic Nematodes Database: A Comprehensive Genomic Data Platform for Plant Parasitic Nematode Research. Int. J. Mol. Sci. 2023, 24, 16841. https://doi.org/10.3390/ijms242316841
Zhuge J, Zhou X, Zhou L, Hu J, Guo K. The Plant Parasitic Nematodes Database: A Comprehensive Genomic Data Platform for Plant Parasitic Nematode Research. International Journal of Molecular Sciences. 2023; 24(23):16841. https://doi.org/10.3390/ijms242316841
Chicago/Turabian StyleZhuge, Junhao, Xiang Zhou, Lifeng Zhou, Jiafu Hu, and Kai Guo. 2023. "The Plant Parasitic Nematodes Database: A Comprehensive Genomic Data Platform for Plant Parasitic Nematode Research" International Journal of Molecular Sciences 24, no. 23: 16841. https://doi.org/10.3390/ijms242316841