
The Haptoglobin Response after Aneurysmal Subarachnoid Haemorrhage
 , , and
, , and
Abstract
:1. Introduction
2. Results
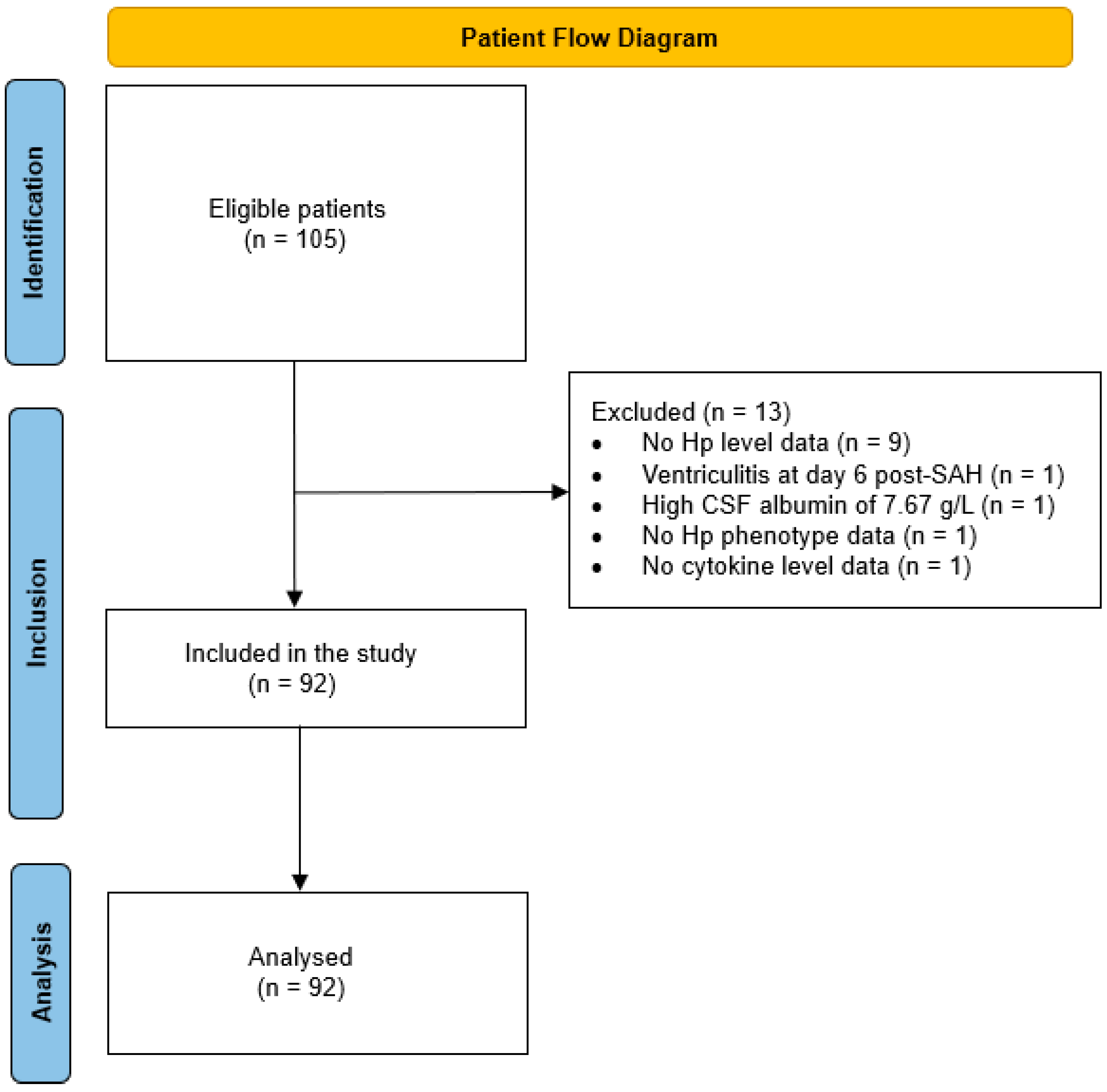
2.1. Demographics and Clinical Characteristics
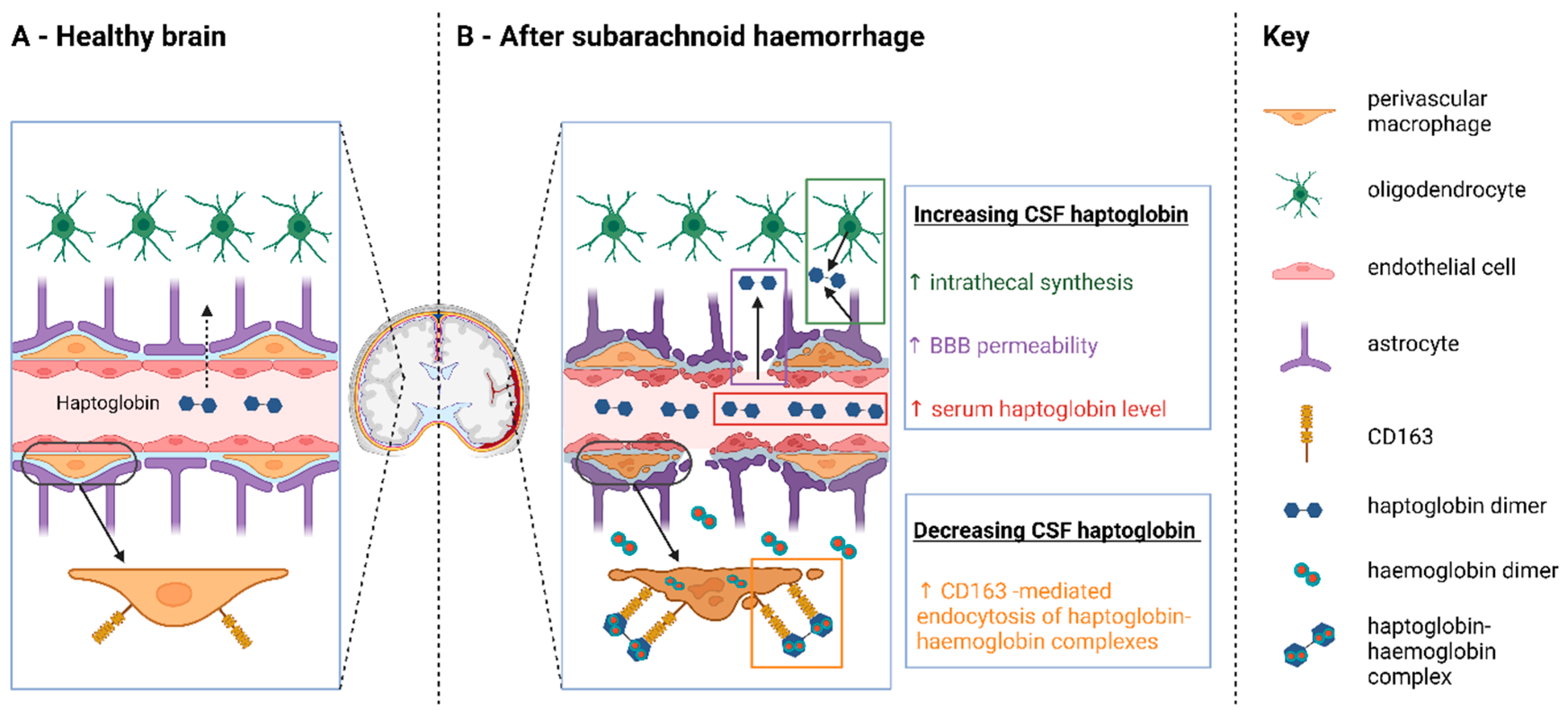
2.2. Evidence for Hp Consumption in the CSF
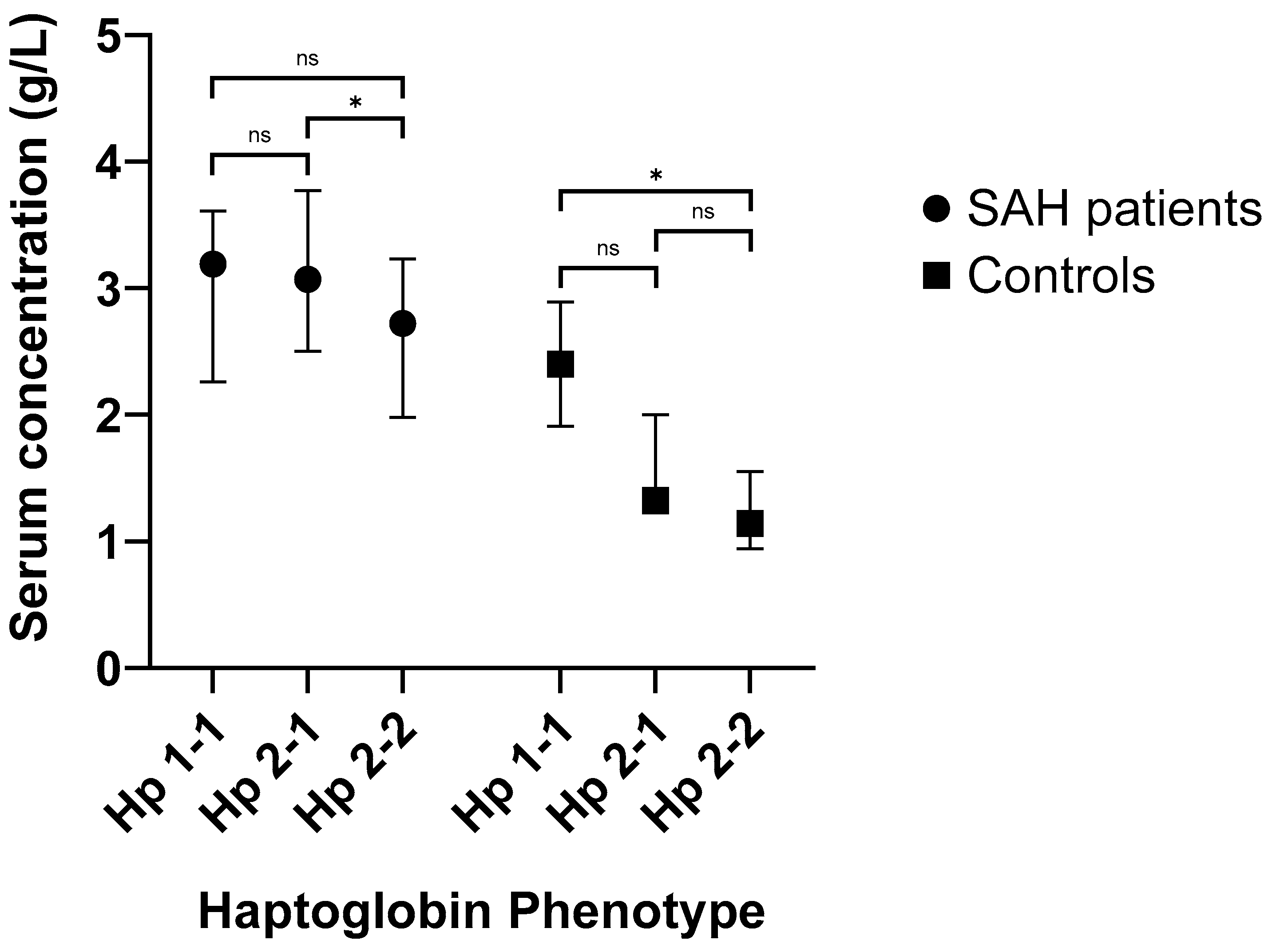
2.3. Serum Hp Increases Following SAH
2.4. Serum Hp Levels Are Associated with CSF Cytokines
2.5. Hp Is Not Associated with Outcome
3. Discussion
3.1. Key Findings
3.2. Implications
4. Materials & Methods
4.1. Study Design and Participants
4.2. CSF and Plasma Sampling
4.3. CSF and Plasma Cytokine Analysis
4.4. CSF/Serum Albumin Quotient
4.5. Haptoglobin and Haemoglobin Analysis
4.6. Clinical Data and Outcome Measures
4.7. Statistical Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Regan, R.F.; Panter, S.S. Neurotoxicity of Hemoglobin in Cortical Cell Culture. Neurosci. Lett. 1993, 153, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Rosen, A.D.; Frumin, N.V. Focal Epileptogenesis after Intracortical Hemoglobin Injection. Exp. Neurol. 1979, 66, 277–284. [Google Scholar] [CrossRef]
- Wu, F.; Liu, Z.; Li, G.; Zhou, L.; Huang, K.; Wu, Z.; Zhan, R.; Shen, J. Inflammation and Oxidative Stress: Potential Targets for Improving Prognosis After Subarachnoid Hemorrhage. Front. Cell. Neurosci. 2021, 15, 363. [Google Scholar] [CrossRef]
- Robicsek, S.A.; Bhattacharya, A.; Rabai, F.; Shukla, K.; Doré, S. Blood-Related Toxicity after Traumatic Brain Injury: Potential Targets for Neuroprotection. Mol. Neurobiol. 2020, 57, 159–178. [Google Scholar] [CrossRef]
- Bulters, D.; Gaastra, B.; Zolnourian, A.; Alexander, S.; Ren, D.; Blackburn, S.L.; Borsody, M.; Doré, S.; Galea, J.; Iihara, K.; et al. Haemoglobin Scavenging in Intracranial Bleeding: Biology and Clinical Implications. Nat. Rev. Neurol. 2018, 14, 416–432. [Google Scholar] [CrossRef]
- Galea, I.; Durnford, A.; Glazier, J.; Mitchell, S.; Kohli, S.; Foulkes, L.; Norman, J.; Darekar, A.; Love, S.; Bulters, D.O.; et al. Iron Deposition in the Brain after Aneurysmal Subarachnoid Hemorrhage. Stroke 2022, 53, 1633–1642. [Google Scholar] [CrossRef]
- Liu, H.; Schwarting, J.; Terpolilli, N.A.; Nehrkorn, K.; Plesnila, N. Scavenging Free Iron Reduces Arteriolar Microvasospasms after Experimental Subarachnoid Hemorrhage. Stroke 2021, 52, 4033–4042. [Google Scholar] [CrossRef]
- Dienel, A.; Kumar, T.P.; Blackburn, S.L.; McBride, D.W. Role of Platelets in the Pathogenesis of Delayed Injury after Subarachnoid Hemorrhage. J. Cereb. Blood Flow Metab. 2021, 41, 2820–2830. [Google Scholar] [CrossRef]
- Fang, Y.; Huang, L.; Wang, X.; Si, X.; Lenahan, C.; Shi, H.; Shao, A.; Tang, J.; Chen, S.; Zhang, J.; et al. A New Perspective on Cerebrospinal Fluid Dynamics after Subarachnoid Hemorrhage: From Normal Physiology to Pathophysiological Changes. J. Cereb. Blood Flow Metab. 2022, 42, 543–558. [Google Scholar] [CrossRef]
- Polonovski, M.; Jayle, M. Existence Dans Le Plasma Sanguin d’une Substance Activant l’action Peroxydasique de l’hemoglobine. Comptes Rendus Seances Soc. Biol. Fil. 1938, 29, 457–460. [Google Scholar]
- Chamoun, V.; Zeman, A.; Blennow, K.; Fredman, P.; Wallin, A.; Keir, G.; Giovannoni, G.; Thompson, E.J. Haptoglobins as Markers of Blood-CSF Barrier Dysfunction: The Findings in Normal CSF. J. Neurol. Sci. 2001, 182, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Jia, Y.; Parker Siburt, C.J.; Abraham, B.; Wood, F.; Bonaventura, C.; Henkens, R.; Crumbliss, A.L.; Alayash, A.I. Haptoglobin Alters Oxygenation and Oxidation of Hemoglobin and Decreases Propagation of Peroxide-Induced Oxidative Reactions. Free Radic. Biol. Med. 2012, 53, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Borsody, M.; Burke, A.; Coplin, W.; Miller-Lotan, R.; Levy, A. Haptoglobin and the Development of Cerebral Artery Vasospasm after Subarachnoid Hemorrhage. Neurology 2006, 66, 634–640. [Google Scholar] [CrossRef]
- Cooper, C.E.; Schaer, D.J.; Buehler, P.W.; Wilson, M.T.; Reeder, B.J.; Silkstone, G.; Svistunenko, D.A.; Bulow, L.; Alayash, A.I. Haptoglobin Binding Stabilizes Hemoglobin Ferryl Iron and the Globin Radical on Tyrosine Β145. Antioxid. Redox Signal. 2013, 18, 2264–2273. [Google Scholar] [CrossRef]
- Hugelshofer, M.; Buzzi, R.M.; Schaer, C.A.; Richter, H.; Akeret, K.; Anagnostakou, V.; Mahmoudi, L.; Vaccani, R.; Vallelian, F.; Deuel, J.W.; et al. Haptoglobin Administration into the Subarachnoid Space Prevents Hemoglobin-Induced Cerebral Vasospasm. J. Clin. Investig. 2019, 129, 5219–5235. [Google Scholar] [CrossRef] [PubMed]
- Durnford, A.; Dunbar, J.; Galea, J.; Bulters, D.; Nicoll, J.A.R.; Boche, D.; Galea, I. Haemoglobin Scavenging after Subarachnoid Haemorrhage. Acta Neurochir. Suppl. 2015, 120, 51–54. [Google Scholar] [CrossRef]
- Hostettler, I.C.; Morton, M.J.; Ambler, G.; Kazmi, N.; Gaunt, T.; Wilson, D.; Shakeshaft, C.; Jäger, H.R.; Cohen, H.; Yousry, T.A.; et al. Haptoglobin Genotype and Outcome after Spontaneous Intracerebral Haemorrhage. J. Neurol. Neurosurg. Psychiatry 2020, 91, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Kristiansen, M.; Graversen, J.H.; Jacobsen, C.; Sonne, O.; Hoffman, H.J.; Law, S.K.A.; Moestrup, S.K. Identification of the Haemoglobin Scavenger Receptor. Nature 2001, 409, 198–201. [Google Scholar] [CrossRef]
- Galea, J.; Cruickshank, G.; Teeling, J.L.; Boche, D.; Garland, P.; Perry, V.H.; Galea, I. The Intrathecal CD163-Haptoglobin-Hemoglobin Scavenging System in Subarachnoid Hemorrhage. J. Neurochem. 2012, 121, 785–792. [Google Scholar] [CrossRef]
- Philippidis, P.; Mason, J.C.; Evans, B.J.; Nadra, I.; Taylor, K.M.; Haskard, D.O.; Landis, R.C. Hemoglobin Scavenger Receptor CD163 Mediates Interleukin-10 Release and Heme Oxygenase-1 Synthesis. Circ. Res. 2004, 94, 119–126. [Google Scholar] [CrossRef]
- Falk, J.E.; Dresel, E.I.B.; Rimington, C.; Gibson, K.D.; Neuberger, A.; Scott, J.J.; Goldberg, A.; Rimington, C.C.R.; Overhoff, J.; Hiller, A.; et al. Zone Electrophoresis in Starch Gels: Group Variations in the Serum Proteins of Normal Human Adults. Biochem. J. 1955, 61, 629. [Google Scholar] [CrossRef]
- Smithies, O.; Walker, N.F. Notation for Serum-Protein Groups and the Genes Controlling Their Inheritance. Nature 1956, 178, 694–695. [Google Scholar] [CrossRef] [PubMed]
- Gaastra, B.; Glazier, J.; Bulters, D.; Galea, I. Haptoglobin Genotype and Outcome after Subarachnoid Haemorrhage: New Insights from a Meta-Analysis. Oxidative Med. Cell. Longev. 2017, 2017, 6747940. [Google Scholar] [CrossRef] [PubMed]
- Gaastra, B.; Ren, D.; Alexander, S.; Bennett, E.R.; Bielawski, D.M.; Blackburn, S.L.; Borsody, M.K.; Doré, S.; Galea, J.; Garland, P.; et al. Haptoglobin Genotype and Aneurysmal Subarachnoid Hemorrhage. Neurology 2019, 92, e2150–e2164. [Google Scholar] [CrossRef] [PubMed]
- Andersen, C.B.F.; Stødkilde, K.; Sæderup, K.L.; Kuhlee, A.; Raunser, S.; Graversen, J.H.; Moestrup, S.K. Haptoglobin. Antioxid. Redox Signal. 2017, 26, 814–831. [Google Scholar] [CrossRef]
- Galea, I.; Bandyopadhyay, S.; Bulters, D.; Humar, R.; Hugelshofer, M.; Schaer, D.J.; Abdulazim, A.; Alalade, A.F.; Alexander, S.A.; Amaro, S.; et al. Haptoglobin Treatment for Aneurysmal Subarachnoid Hemorrhage: Review and Expert Consensus on Clinical Translation. Stroke 2023, 54, 1930–1942. [Google Scholar] [CrossRef]
- Kumar, D.M.; Thota, B.; Shinde, S.V.; Prasanna, K.V.; Hegde, A.S.; Arivazhagan, A.; Chandramouli, B.A.; Santosh, V.; Somasundaram, K. Proteomic Identification of Haptoglobin A2 as a Glioblastoma Serum Biomarker: Implications in Cancer Cell Migration and Tumor Growth. J. Proteome Res. 2010, 9, 5557–5567. [Google Scholar] [CrossRef]
- Wilcockson, D.C.; Campbell, S.J.; Anthony, D.C.; Perry, V.H. The Systemic and Local Acute Phase Response Following Acute Brain Injury. J. Cereb. Blood Flow Metab. 2002, 22, 318–326. [Google Scholar] [CrossRef]
- Williams, R.; Speyer, B.E.; Billing, B.H. Serum Haptoglobin in Liver Disease. Gut 1961, 2, 297. [Google Scholar] [CrossRef]
- van Heugten, C.; Rasquin, S.; Winkens, I.; Beusmans, G.; Verhey, F. Checklist for Cognitive and Emotional Consequences Following Stroke (CLCE-24): Development, Usability and Quality of the Self-Report Version. Clin. Neurol. Neurosurg. 2007, 109, 257–262. [Google Scholar] [CrossRef]
- Wilson, J.T.L.; Pettigrew, L.E.L.; Teasdale, G.M. Structured Interviews for the Glasgow Outcome Scale and the Extended Glasgow Outcome Scale: Guidelines for Their Use. J. Neurotrauma 1998, 15, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Van Swieten, J.C.; Koudstaal, P.J.; Visser, M.C.; Schouten, H.; Van Gijn, J. Interobserver Agreement for the Assessment of Handicap in Stroke Patients. Stroke 1988, 19, 604–607. [Google Scholar] [CrossRef] [PubMed]
- Pace, A.; Mitchell, S.; Casselden, E.; Zolnourian, A.; Glazier, J.; Foulkes, L.; Bulters, D.; Galea, I. A Subarachnoid Haemorrhage-Specific Outcome Tool. Brain 2018, 141, 1111–1121. [Google Scholar] [CrossRef] [PubMed]
- Blau, J.N.; Harris, H.; Robson, E.B. Haptoglobins in Cerebrospinal Fluid. Clin. Chim. Acta 1963, 8, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Felgenhauer, K. Protein Size and Cerebrospinal Fluid Composition. Wien. Klin. Wochenschr. 1974, 52, 1158–1164. [Google Scholar] [CrossRef] [PubMed]
- Louagie, H.; Delanghe, J.; Desombere, I.; De Buyzere, M.; Hauser, P.; Leroux-Roels, G. Haptoglobin Polymorphism and the Immune Response after Hepatitis B Vaccination. Vaccine 1993, 11, 1188–1190. [Google Scholar] [CrossRef] [PubMed]
- Márquez, L.; Shen, C.; Cleynen, I.; De Hertogh, G.; Van Steen, K.; Machiels, K.; Perrier, C.; Ballet, V.; Organe, S.; Ferrante, M.; et al. Effects of Haptoglobin Polymorphisms and Deficiency on Susceptibility to Inflammatory Bowel Disease and on Severity of Murine Colitis. Gut 2012, 61, 528–534. [Google Scholar] [CrossRef]
- Bjornsson, E.; Helgason, H.; Halldorsson, G.; Helgadottir, A.; Gylfason, A.; Kehr, B.; Jonasdottir, A.; Jonasdottir, A.; Sigurdsson, A.; Oddsson, A.; et al. A Rare Splice Donor Mutation in the Haptoglobin Gene Associates with Blood Lipid Levels and Coronary Artery Disease. Hum. Mol. Genet. 2017, 26, 2364–2376. [Google Scholar] [CrossRef]
- Soejima, M.; Sagata, N.; Komatsu, N.; Sasada, T.; Kawaguchi, A.; Itoh, K.; Koda, Y. Genetic Factors Associated with Serum Haptoglobin Level in a Japanese Population. Clin. Chim. Acta 2014, 433, 54–57. [Google Scholar] [CrossRef]
- Park, K.U.; Song, J.; Kim, J.Q. Haptoglobin Genotypic Distribution (Including Hp0 Allele) and Associated Serum Haptoglobin Concentrations in Koreans. J. Clin. Pathol. 2004, 57, 1094–1095. [Google Scholar] [CrossRef]
- Kasvosve, I.; Gomo, Z.A.R.; Gangaidzo, I.T.; Mvundura, E.; Saungweme, T.; Moyo, V.M.; Khumalo, H.; Boelaert, J.R.; Gordeuk, V.R.; Delanghe, J.R. Reference Range of Serum Haptoglobin Is Haptoglobin Phenotype-Dependent in Blacks. Clin. Chim. Acta 2000, 296, 163–170. [Google Scholar] [CrossRef]
- Fowkes, F.J.I.; Imrie, H.; Migot-Nabias, F.; Michon, P.; Justice, A.; Deloron, P.; Luty, A.J.F.; Day, K.P. Association of Haptoglobin Levels with Age, Parasite Density, and Haptoglobin Genotype in a Malaria-Endemic Area of Gabon. Am. J. Trop. Med. Hyg. 2006, 74, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Imrie, H.; Fowkes, F.J.I.; Michon, P.; Tavul, L.; Hume, J.C.C.; Piper, K.P.; Reeder, J.C.; Day, K.P. Haptoglobin Levels Are Associated with Haptoglobin Genotype and Alpha+-Thalassemia in a Malaria-Endemic Area. Am. J. Trop. Med. Hyg. 2006, 74, 965–971. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.H.; Tseng, M.Y.; Ro, L.S.; Lyu, R.K.; Tai, Y.H.; Chang, H.S.; Wu, Y.R.; Huang, C.C.; Hsu, W.C.; Kuo, H.C.; et al. Analyses of Haptoglobin Level in the Cerebrospinal Fluid and Serum of Patients with Neuromyelitis Optica and Multiple Sclerosis. Clin. Chim. Acta 2013, 417, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Asleh, R.; Marsh, S.; Shilkrut, M.; Binah, O.; Guetta, J.; Lejbkowicz, F.; Enav, B.; Shehadeh, N.; Kanter, Y.; Lache, O.; et al. Genetically Determined Heterogeneity in Hemoglobin Scavenging and Susceptibility to Diabetic Cardiovascular Disease. Circ. Res. 2003, 92, 1193–1200. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A Paravascular Pathway Facilitates CSF Flow through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid β. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef]
- Jones, H.C.; Keep, R.F.; Drewes, L.R. CNS Fluid and Solute Movement: Physiology, Modelling and Imaging. Fluids Barriers CNS 2020, 17, 12. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Lee, H.; Yu, M.; Feng, T.; Logan, J.; Nedergaard, M.; Benveniste, H. Brain-Wide Pathway for Waste Clearance Captured by Contrast-Enhanced MRI. J. Clin. Investig. 2013, 123, 1299–1309. [Google Scholar] [CrossRef]
- Carare, R.O.; Bernardes-Silva, M.; Newman, T.A.; Page, A.M.; Nicoll, J.A.R.; Perry, V.H.; Weller, R.O. Solutes, But Not Cells, Drain from the Brain Parenchyma along Basement Membranes of Capillaries and Arteries: Significance for Cerebral Amyloid Angiopathy and Neuroimmunology. Neuropathol. Appl. Neurobiol. 2008, 34, 131–144. [Google Scholar] [CrossRef]
- Morris, A.W.J.; Sharp, M.M.G.; Albargothy, N.J.; Fernandes, R.; Hawkes, C.A.; Verma, A.; Weller, R.O.; Carare, R.O. Vascular Basement Membranes as Pathways for the Passage of Fluid into and out of the Brain. Acta Neuropathol. 2016, 131, 725. [Google Scholar] [CrossRef]
- Arbel-Ornath, M.; Hudry, E.; Eikermann-Haerter, K.; Hou, S.; Gregory, J.L.; Zhao, L.; Betensky, R.A.; Frosch, M.P.; Greenberg, S.M.; Bacskai, B.J. Interstitial Fluid Drainage Is Impaired in Ischemic Stroke and Alzheimer’s Disease Mouse Models. Acta Neuropathol. 2013, 126, 353–364. [Google Scholar] [CrossRef]
- Mehta, N.H.; Sherbansky, J.; Kamer, A.R.; Carare, R.O.; Butler, T.; Rusinek, H.; Chiang, G.C.; Li, Y.; Strauss, S.; Saint-Louis, L.A.; et al. The Brain-Nose Interface: A Potential Cerebrospinal Fluid Clearance Site in Humans. Front. Physiol. 2021, 12, 769948. [Google Scholar] [CrossRef]
- Gaberel, T.; Gakuba, C.; Goulay, R.; De Lizarrondo, S.M.; Hanouz, J.L.; Emery, E.; Touze, E.; Vivien, D.; Gauberti, M. Impaired Glymphatic Perfusion after Strokes Revealed by Contrast-Enhanced MRI: A New Target for Fibrinolysis? Stroke 2014, 45, 3092–3096. [Google Scholar] [CrossRef] [PubMed]
- Pizzo, M.E.; Wolak, D.J.; Kumar, N.N.; Brunette, E.; Brunnquell, C.L.; Hannocks, M.J.; Abbott, N.J.; Meyerand, M.E.; Sorokin, L.; Stanimirovic, D.B.; et al. Intrathecal Antibody Distribution in the Rat Brain: Surface Diffusion, Perivascular Transport and Osmotic Enhancement of Delivery. J. Physiol. 2018, 596, 445–475. [Google Scholar] [CrossRef] [PubMed]
- Campbell, S.J.; Hughes, P.M.; Iredale, J.P.; Wilcockson, D.C.; Waters, S.; Docagne, F.; Perry, V.H.; Anthony, D.C. CINC-1 Is an Acute-Phase Protein Induced by Focal Brain Injury Causing Leukocyte Mobilization and Liver Injury. FASEB J. 2003, 17, 1168–1170. [Google Scholar] [CrossRef]
- Campbell, S.J.; Perry, V.H.; Pitossi, F.J.; Butchart, A.G.; Chertoff, M.; Waters, S.; Dempster, R.; Anthony, D.C. Central Nervous System Injury Triggers Hepatic CC and CXC Chemokine Expression That Is Associated with Leukocyte Mobilization and Recruitment to Both the Central Nervous System and the Liver. Am. J. Pathol. 2005, 166, 1487–1497. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Stoorvogel, W. Extracellular Vesicles: Exosomes, Microvesicles, and Friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Couch, Y.; Akbar, N.; Roodselaar, J.; Evans, M.C.; Gardiner, C.; Sargent, I.; Romero, I.A.; Bristow, A.; Buchan, A.M.; Haughey, N.; et al. Circulating Endothelial Cell-Derived Extracellular Vesicles Mediate the Acute Phase Response and Sickness Behaviour Associated with CNS Inflammation. Sci. Rep. 2017, 7, 9574. [Google Scholar] [CrossRef] [PubMed]
- Gusdon, A.M.; Savarraj, J.P.J.; Shihabeddin, E.; Paz, A.; Assing, A.; Ko, S.B.; McCullough, L.D.; Choi, H.A. Time Course of Peripheral Leukocytosis and Clinical Outcomes after Aneurysmal Subarachnoid Hemorrhage. Front. Neurol. 2021, 12, 694996. [Google Scholar] [CrossRef] [PubMed]
- Rappl, P.; Brüne, B.; Schmid, T. Role of Tristetraprolin in the Resolution of Inflammation. Biology 2021, 10, 66. [Google Scholar] [CrossRef]
- Zolnourian, A.; Galea, I.; Bulters, D. Neuroprotective Role of the Nrf2 Pathway in Subarachnoid Haemorrhage and Its Therapeutic Potential. Oxidative Med. Cell. Longev. 2019, 2019, 6218239. [Google Scholar] [CrossRef]
- Reiber, H. Dynamics of Brain-Derived Proteins in Cerebrospinal Fluid. Clin. Chim. Acta 2001, 310, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Reiber, H. Proteins in Cerebrospinal Fluid and Blood: Barriers, CSF Flow Rate and Source-Related Dynamics. Restor. Neurol. Neurosci. 2003, 21, 79–96. [Google Scholar]
- Messina, R.; de Gennaro, L.; De Robertis, M.; Pop, R.; Chibbaro, S.; Severac, F.; Blagia, M.; Balducci, M.T.; Bozzi, M.T.; Signorelli, F. Cerebrospinal Fluid Lactate and Glucose Levels as Predictors of Symptomatic Delayed Cerebral Ischemia in Patients with Aneurysmal Subarachnoid Hemorrhage. World Neurosurg. 2023, 170, e596–e602. [Google Scholar] [CrossRef] [PubMed]
- Zolnourian, A.H.; Franklin, S.; Galea, I.; Bulters, D.O. Study Protocol for SFX-01 after Subarachnoid Haemorrhage (SAS): A Multicentre Randomised Double-Blinded, Placebo Controlled Trial. BMJ Open 2020, 10, e028514. [Google Scholar] [CrossRef] [PubMed]
- Meso Scale Discovery Assay Kits. Available online: https://www.mesoscale.com/en/products_and_services/assay_kits (accessed on 21 September 2023).
- Hov, K.R.; Berg, J.P.; Frihagen, F.; Ræder, J.; Hall, R.; Wyller, T.B.; Watne, L.O. Blood-Cerebrospinal Fluid Barrier Integrity in Delirium Determined by Q-Albumin. Dement. Geriatr. Cogn. Disord. 2016, 41, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Musaeus, C.S.; Gleerup, H.S.; Høgh, P.; Waldemar, G.; Hasselbalch, S.G.; Simonsen, A.H. Cerebrospinal Fluid/Plasma Albumin Ratio as a Biomarker for Blood–brain Barrier Impairment Across Neurodegenerative Dementias. J. Alzheimer’s Dis. 2020, 75, 429–436. [Google Scholar] [CrossRef]
- Garland, P.; Morton, M.J.; Haskins, W.; Zolnourian, A.; Durnford, A.; Gaastra, B.; Toombs, J.; Heslegrave, A.J.; More, J.; Okemefuna, A.I.; et al. Haemoglobin Causes Neuronal Damage in Vivo Which Is Preventable by Haptoglobin. Brain Commun. 2020, 2, fcz053. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hp 1-1 (n = 19) | Hp 2-1 (n = 46) | Hp 2-2 (n = 27) | p-Value | ||
|---|---|---|---|---|---|
| Age [median, IQR] | 56 (48–62) | 55 (49–59) | 55 (51–68) | 0.707 b | |
| Sex (n, %) | Female | 14 (73.7) | 33 (71.7) | 22 (81.5) | 0.663 a |
| Male | 5 (26.3) | 13 (28.3) | 6 (18.5) | ||
| Race (n, %) | White | 19 (100.0) | 46 (100.0) | 24 (88.9) | 0.082 a |
| Black | 0 (0.0) | 0 (0.0) | 1 (3.7) | ||
| Asian | 0 (0.0) | 0 (0.0) | 2 (7.4) | ||
| Premorbid hypertension | Yes | 6 (31.6) | 11 (23.9) | 9 (33.3) | 0.592 a |
| No | 13 (68.4) | 35 (76.1) | 18 (66.7) | ||
| WFNS Grade (n, %) | I | 8 (42.1) | 20 (43.5) | 13 (44.4) | 0.985 a |
| II | 3 (15.8) | 9 (19.6) | 4 (14.8) | ||
| III | 2 (10.5) | 2 (4.4) | 3 (11.1) | ||
| IV | 5 (26.3) | 12 (26.1) | 7 (25.9) | ||
| V | 1 (5.3) | 3 (6.5) | 1 (3.7) | ||
| Blood volume (n = 91) (cm3) [median, IQR] | 11.8 (4.2–29.0) | 21.2 (8.0–39.6) | 24.3 (10.1–32.4) | 0.367 b | |
| Intracerebral haemorrhage present (n, %) | Yes | 1 (5.3) | 6 (13.0) | 7 (25.9) | 0.179 a |
| No | 17 (89.5) | 40 (87.0) | 20 (74.1) | ||
| Missing | 1 (5.3) | 0 (0.0) | 0 (0.0) | NA | |
| Intraventricular haemorrhage present (n, %) | Yes | 14 (73.7) | 32 (69.6) | 23 (85.2) | 0.340 a |
| No | 4 (21.0) | 14 (30.4) | 4 (14.8) | ||
| Missing | 1 (5.3) | 0 (0.0) | 0 (0.0) | NA | |
| EVD inserted (n, %) | Yes | 9 (47.4) | 16 (34.8) | 7 (25.9) | 0.326 a |
| No | 10 (52.6) | 30 (65.2) | 20 (74.1) | ||
| Aneurysm location (n, %) | Anterior Cerebral | 5 (26.3) | 21 (45.7) | 10 (37.0) | 0.391 a |
| Internal Carotid | 6 (31.6) | 4 (8.7) | 7 (25.9) | ||
| Middle Cerebral | 5 (26.3) | 14 (30.4) | 6 (22.2) | ||
| Vertebrobasilar | 3 (15.8) | 6 (13.0) | 3 (11.1) | ||
| Non-aneurysmal | 0 (0.0) | 1 (2.2) | 1 (3.7) | ||
| Securing of the aneurysm (n, %) | Clipping | 4 (21.1) | 11 (23.9) | 6 (22.2) | 1.000 a |
| Coiling | 15 (78.9) | 34 (73.9) | 20 (74.1) | ||
| Not applicable | 0 (0.0) | 1 (2.2) | 1 (3.7) | ||
| Cytokines (pg/mL) | Variables | β (95% CI) | p Value |
|---|---|---|---|
| IFN-gamma | Serum Hp level | 11.591 (0.055–23.128) | 0.049 |
| Hp 2-1 (relative to Hp 1-1) | 22.577 (−17.964–63.119) | 0.271 | |
| Hp 2-2 (relative to Hp 1-1) | 50.113 (7.011–93.215) | 0.023 | |
| Interaction with Hp level and Hp 2-1 | −9.729 (−22,420–2962) | 0.113 | |
| Interaction with Hp level and Hp 2-2 | −15.987 (−30.198–−1.776) | 0.028 | |
| IL-1beta | Serum Hp level | 1.413 (0.116–2.710) | 0.033 |
| Hp 2-1 (relative to Hp 1-1) | 1.589 (−2.968–6.146) | 0.489 | |
| Hp 2-2 (relative to Hp 1-1) | 5.470 (0.625–10.315) | 0.027 | |
| Interaction with Hp level and Hp 2-1 | −0.598 (−2.025–0.828) | 0.406 | |
| Interaction with Hp level and Hp 2-2 | −1.725 (−3.322–−0.127) | 0.035 | |
| IL-10 | Serum Hp level | 1.271 (−3.917–6.458) | 0.627 |
| Hp 2-1 (relative to Hp 1-1) | 2.120 (−16.110–20.350) | 0.817 | |
| Hp 2-2 (relative to Hp 1-1) | 18.501 (−0.880–37.883) | 0.061 | |
| Interaction with Hp level and Hp 2-1 | −0.723 (−6.430–4.984) | 0.801 | |
| Interaction with Hp level and Hp 2-2 | −5.227 (−11.617–1.164) | 0.107 | |
| IL-12p70 | Serum Hp level | 9.796 (2.787–16.806) | 0.007 |
| Hp 2-1 (relative to Hp 1-1) | 17.235 (−7.397–41.867) | 0.167 | |
| Hp 2-2 (relative to Hp 1-1) | 28.436 (2.248–54.624) | 0.034 | |
| Interaction with Hp level and Hp 2-1 | −7.208 (−14.918–0.503) | 0.066 | |
| Interaction with Hp level and Hp 2-2 | −9.518 (−18.152–−0.884) | 0.031 | |
| IL-13 | Serum Hp level | 9.833 (−2.544–22.210) | 0.118 |
| Hp 2-1 (relative to Hp 1-1) | −5.668 (−49.164–37.829) | 0.796 | |
| Hp 2-2 (relative to Hp 1-1) | 49.495 (3.252–95.739) | 0.036 | |
| Interaction with Hp level and Hp 2-1 | −0.481 (−14.097–13.135) | 0.944 | |
| Interaction with Hp level and Hp 2-2 | −18.042 (−33.289–−2.795) | 0.021 | |
| IL-2 | Serum Hp level | 1.214 (0.064–2.365) | 0.039 |
| Hp 2-1 (relative to Hp 1-1) | 1.584 (−2.460–5.627) | 0.438 | |
| Hp 2-2 (relative to Hp 1-1) | 5.498 (1.199–9.797) | 0.013 | |
| Interaction with Hp level and Hp 2-1 | −0.654 (−1.919–0.612) | 0.307 | |
| Interaction with Hp level and Hp 2-2 | −1.800 (−3.217–−0.382) | 0.014 | |
| IL-4 | Serum Hp level | 8.570 (2.683–14.458) | 0.005 |
| Hp 2-1 (relative to Hp 1-1) | 14.054 (−6.634–34.743) | 0.180 | |
| Hp 2-2 (relative to Hp 1-1) | 26.247 (4.252–48.242) | 0.020 | |
| Interaction with Hp level and Hp 2-1 | −6.243 (−12.719–0.234) | 0.059 | |
| Interaction with Hp level and Hp 2-2 | −8.675 (−15.927–−1.423) | 0.020 | |
| IL-6 | Serum Hp level | 3057.935 (938.419–5177.451) | 0.005 |
| Hp 2-1 (relative to Hp 1-1) | 5570.533 (−1877.866–13,018.93) | 0.140 | |
| Hp 2-2 (relative to Hp 1-1) | 9021.475 (1102.616–16,940.33) | 0.026 | |
| Interaction with Hp level and Hp 2-1 | −2391.482 (−4723.056–−59.908) | 0.045 | |
| Interaction with Hp level and Hp 2-2 | −2988.535 (−559−9.454–−377.617) | 0.025 | |
| IL-8 | Serum Hp level | 408.761 (−567.051–1384.573) | 0.407 |
| Hp 2-1 (relative to Hp 1-1) | 230.141 (−3199.058–3659.339) | 0.894 | |
| Hp 2-2 (relative to Hp 1-1) | 2266.3 (−1379.495–5912.095) | 0.219 | |
| Interaction with Hp level and Hp 2-1 | 84.080 (−989.363–1157.523) | 0.876 | |
| Interaction with Hp level and Hp 2-2 | −491.748 (−1693.799–710.303) | 0.418 | |
| TNF-alpha | Serum Hp level | 2.023 (−0.899–4.945) | 0.172 |
| Hp 2-1 (relative to Hp 1-1) | 1.579 (−8.690–11.848) | 0.760 | |
| Hp 2-2 (relative to Hp 1-1) | 14.599 (3.681–25.517) | 0.009 | |
| Interaction with Hp level and Hp 2-1 | −0.507 (−3.722–2.707) | 0.754 | |
| Interaction with Hp level and Hp 2-2 | −4.730 (−8.305–−1.155) | 0.011 | |
| Principal Component 1 | Serum Hp level | 1.850 (0.580–3.120) | 0.005 |
| Hp 2-1 (relative to Hp 1-1) | 2.460 (−2.002–6.921) | 0.276 | |
| Hp 2-2 (relative to Hp 1-1) | 7.520 (2.776–12.264) | 0.002 | |
| Interaction with Hp level and Hp 2-1 | −1.041 (−2.438–0.355) | 0.142 | |
| Interaction with Hp level and Hp 2-2 | −2.428 (−3.992–−0.864) | 0.003 | |
| Principal Component 2 | Serum Hp level | −0.482 (−1.339–0.375) | 0.266 |
| Hp 2-1 (relative to Hp 1-1) | −1.546 (−4.557–1.464) | 0.309 | |
| Hp 2-2 (relative to Hp 1-1) | −0.011 (−3.212–3.190) | 0.995 | |
| Interaction with Hp level and Hp 2-1 | 0.702 (−0.240–1.645) | 0.142 | |
| Interaction with Hp level and Hp 2-2 | 0.070 (−0.986–1.125) | 0.896 | |
| Principal Component 3 | Serum Hp level | −0.078 (−0.724–0.569) | 0.812 |
| Hp 2-1 (relative to Hp 1-1) | 0.028 (−2.244–2.300) | 0.980 | |
| Hp 2-2 (relative to Hp 1-1) | 0.847 (−1.569–3.263) | 0.487 | |
| Interaction with Hp level and Hp 2-1 | 0.040 (−0.672–0.751) | 0.912 | |
| Interaction with Hp level and Hp 2-2 | −0.170 (−0.967–0.626) | 0.671 |
| Variable | Component 1 | Component 2 | Component 3 | Unexplained |
|---|---|---|---|---|
| CSF IFN-gamma | 0.2979 | −0.1914 | −0.1327 | 0.4555 |
| CSF IL-1beta | 0.3856 | 0.1303 | 0.3081 | 0.1071 |
| CSF IL-10 | 0.1334 | 0.0327 | 0.8927 | 0.05044 |
| CSF IL-12p70 | 0.3444 | −0.3718 | −0.064 | 0.1134 |
| CSF IL-13 | 0.3426 | 0.2788 | −0.2138 | 0.1967 |
| CSF IL-2 | 0.3455 | 0.1562 | −0.1825 | 0.3075 |
| CSF IL-4 | 0.3479 | −0.4017 | −0.0369 | 0.05734 |
| CSF IL-6 | 0.331 | −0.3883 | 0.0043 | 0.1386 |
| CSF IL-8 | 0.2652 | 0.4453 | −0.0242 | 0.2419 |
| CSF TNF-alpha | 0.2975 | 0.441 | −0.0744 | 0.1519 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bandyopadhyay, S.; Garland, P.; Gaastra, B.; Zolnourian, A.; Bulters, D.; Galea, I. The Haptoglobin Response after Aneurysmal Subarachnoid Haemorrhage. Int. J. Mol. Sci. 2023, 24, 16922. https://doi.org/10.3390/ijms242316922
Bandyopadhyay S, Garland P, Gaastra B, Zolnourian A, Bulters D, Galea I. The Haptoglobin Response after Aneurysmal Subarachnoid Haemorrhage. International Journal of Molecular Sciences. 2023; 24(23):16922. https://doi.org/10.3390/ijms242316922
Chicago/Turabian StyleBandyopadhyay, Soham, Patrick Garland, Ben Gaastra, Ardalan Zolnourian, Diederik Bulters, and Ian Galea. 2023. "The Haptoglobin Response after Aneurysmal Subarachnoid Haemorrhage" International Journal of Molecular Sciences 24, no. 23: 16922. https://doi.org/10.3390/ijms242316922