
Endoplasmic Reticulum Protein TXNDC5 Interacts with PRDX6 and HSPA9 to Regulate Glutathione Metabolism and Lipid Peroxidation in the Hepatic AML12 Cell Line

 ,
,  , , , and
, , , and
Abstract
:1. Introduction
2. Results
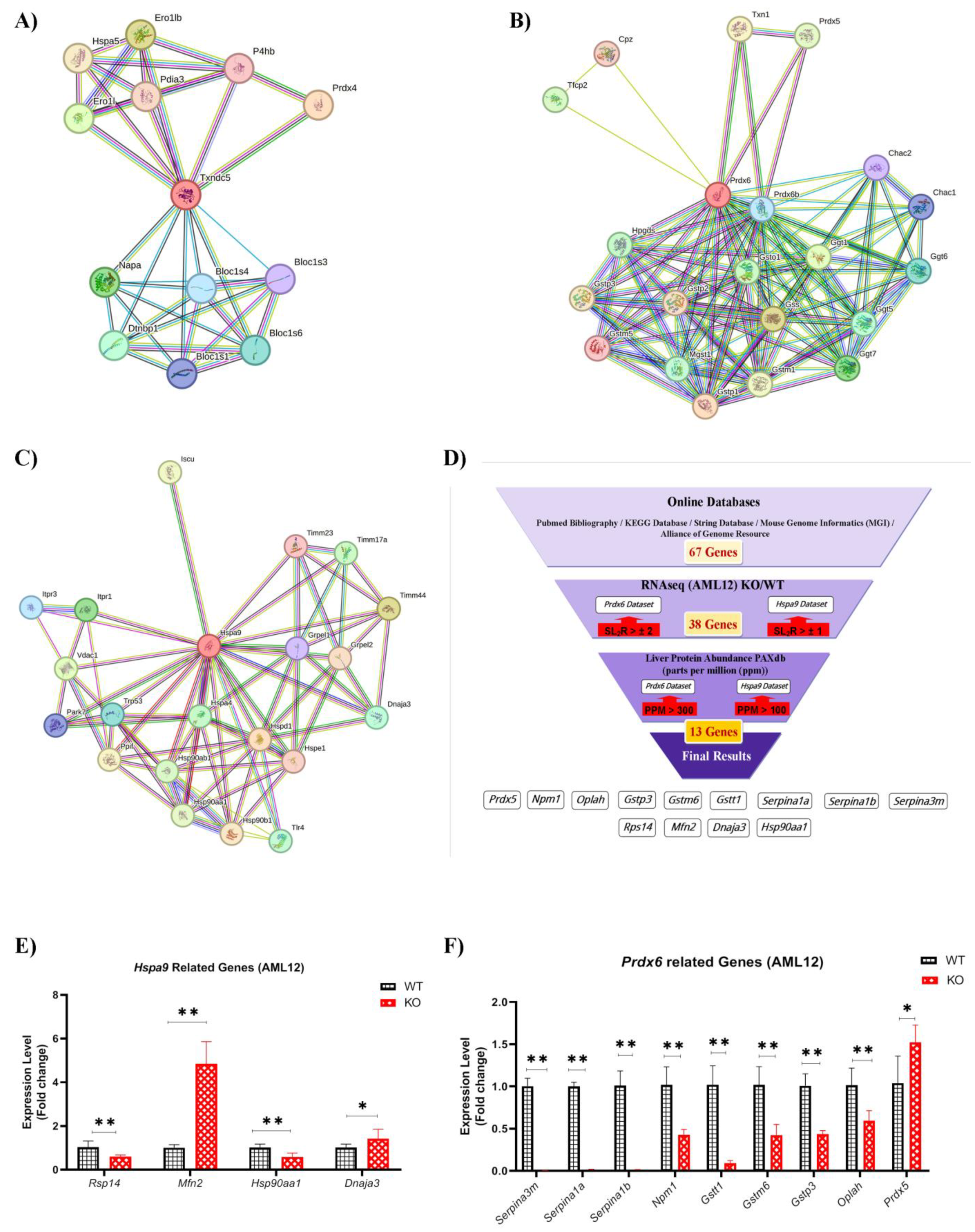
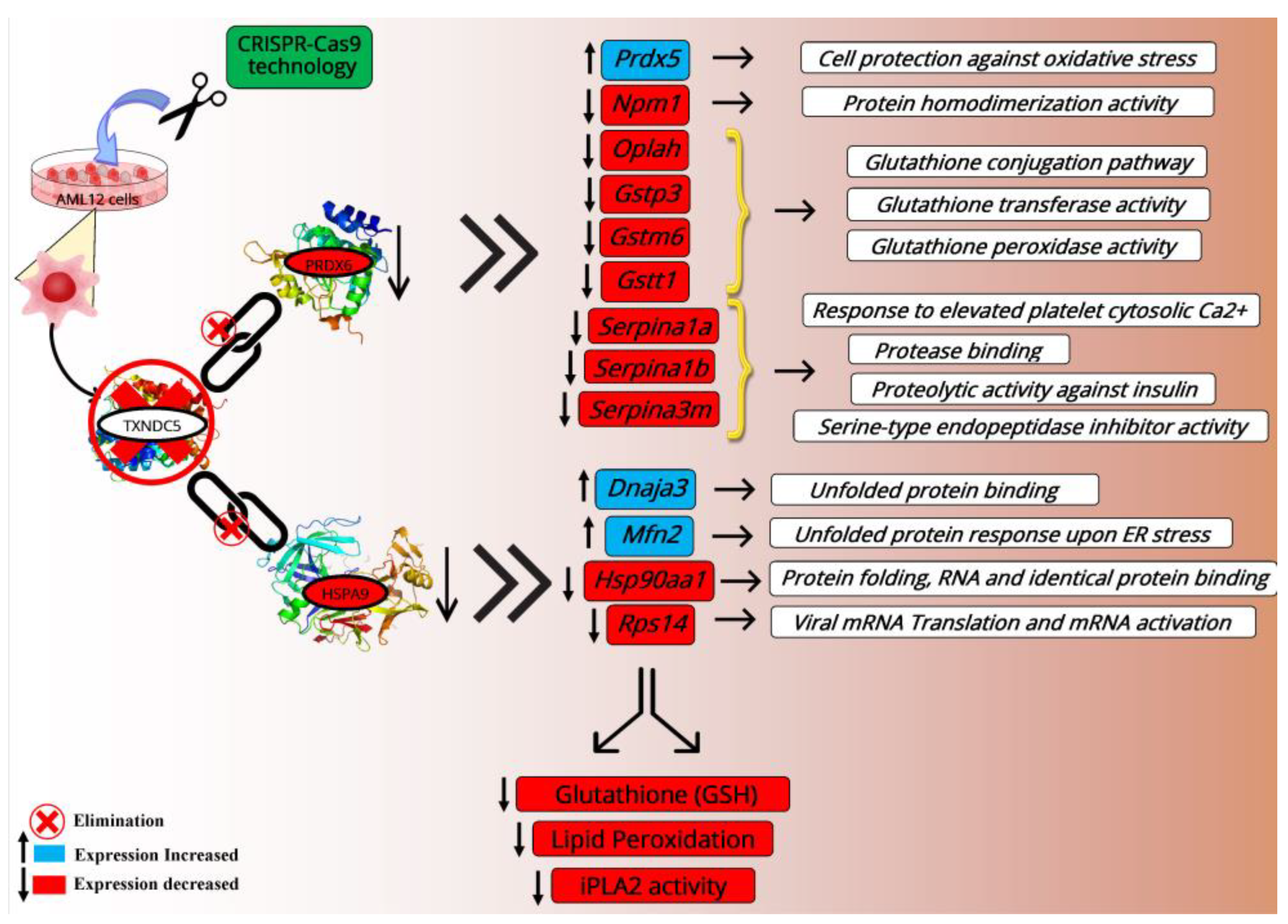
2.1. Identification of PRDX6 and HSPA9 as Novel TXNDC5-Interacting Proteins in the AML12 Cell Line
2.2. HSPA9 mRNA and Protein Levels Are Differentially Influenced by the Absence of TXNDC5
2.3. TXNDC5 Absence Reduces PRDX6 Protein Levels
2.4. Proteasomes Are Not Involved in the Reduction of PRDX6 Protein Induced by the Absence of TXNDC5
2.5. Lipid Peroxidation Was Decreased in the Absence of TXNDC5
2.6. TXNDC5 Deficiency Decreases Reduced Glutathione and IPLA2 Activity
2.7. HSPA9 and PRDX6 Target Genes
2.8. Hepatic Gene Expression
3. Discussion
4. Materials and Methods
4.1. AML12 Cell Culture
4.2. Creation of a Stable TXNDC5 Knockout AML12 Cell Line
4.3. Co-Immunoprecipitation (Co-IP) Assay
4.4. Protein Identification via LC-MS/MS
4.5. Western Blot
4.6. Bioinformatic Analyses
4.7. RNA Extraction
4.8. RNAseq Analyses
4.9. Reverse Transcription and Quantitative Real-Time PCR
4.10. Proteasome Inhibition in AML12 WT and KO Cells
4.11. Determination of Cellular Lipid Peroxidation via MDA Assay
4.12. Total Phospholipase A2 (PLA2) and Calcium-Independent Phospholipase A2 (iPLA2) Activity Assays
4.13. Intracellular Reduced Glutathione (GSH) Concentration Determination
4.14. Statistical Analysis
5. Conclusions
Limitations and Future Research
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Horna-Terrón, E.; Pradilla-Dieste, A.; Sánchez-de-Diego, C.; Osada, J. TXNDC5, a newly discovered disulfide isomerase with a key role in cell physiology and pathology. Int. J. Mol. Sci. 2014, 15, 23501–23518. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Friedman, S.L.; Shulman, G.I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 2021, 184, 2537–2564. [Google Scholar] [CrossRef]
- Ao, N.; Yang, J.; Wang, X.; Du, J. Glucagon-like peptide-1 preserves non-alcoholic fatty liver disease through inhibition of the endoplasmic reticulum stress-associated pathway. Hepatol. Res. 2016, 46, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Gambino, R.; Cassader, M. Recent insights into hepatic lipid metabolism in non-alcoholic fatty liver disease (NAFLD). Prog. Lipid Res. 2009, 48, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Torres, A.; Barceló-Batllori, S.; Martínez-Beamonte, R.; Navarro, M.A.; Surra, J.C.; Arnal, C.; Guillén, N.; Acín, S.; Osada, J. Proteomics and gene expression analyses of squalene-supplemented mice identify microsomal thioredoxin domain-containing protein 5 changes associated with hepatic steatosis. J. Proteom. 2012, 77, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Kaufman, R.J. The role of ER stress in lipid metabolism and lipotoxicity. J. Lipid Res. 2016, 57, 1329–1338. [Google Scholar] [CrossRef]
- Hama, Y.; Morishita, H.; Mizushima, N. Regulation of ER-derived membrane dynamics by the DedA domain-containing proteins VMP1 and TMEM41B. EMBO Rep. 2022, 23, e53894. [Google Scholar] [CrossRef]
- Hwang, C.; Sinskey, A.J.; Lodish, H.F. Oxidized redox state of glutathione in the endoplasmic reticulum. Science 1992, 257, 1496–1502. [Google Scholar] [CrossRef]
- Sánchez-Marco, J.; Martínez-Beamonte, R.; Diego, A.D.; Herrero-Continente, T.; Barranquero, C.; Arnal, C.; Surra, J.; Navarro, M.A.; Osada, J. Thioredoxin Domain Containing 5 Suppression Elicits Serum Amyloid A-Containing High-Density Lipoproteins. Biomedicines 2022, 10, 709. [Google Scholar] [CrossRef]
- Cnop, M.; Foufelle, F.; Velloso, L.A. Endoplasmic reticulum stress, obesity and diabetes. Trends Mol. Med. 2012, 18, 59–68. [Google Scholar] [CrossRef]
- Fu, S.; Yang, L.; Li, P.; Hofmann, O.; Dicker, L.; Hide, W.; Lin, X.; Watkins, S.M.; Ivanov, A.R.; Hotamisligil, G.S. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 2011, 473, 528–531. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Watkins, S.M.; Hotamisligil, G.S. The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metab. 2012, 15, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Tan, F.; Zhu, H.; He, X.; Yu, N.; Zhang, X.; Xu, H.; Pei, H. Role of TXNDC5 in tumorigenesis of colorectal cancer cells: In vivo and in vitro evidence. Int. J. Mol. Med. 2018, 42, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Alberti, A.; Karamessinis, P.; Peroulis, M.; Kypreou, K.; Kavvadas, P.; Pagakis, S.; Politis, P.K.; Charonis, A. ERp46 is reduced by high glucose and regulates insulin content in pancreatic β-cells. Am. J. Physiol.-Endocrinol. Metab. 2009, 297, E812–E821. [Google Scholar] [CrossRef] [PubMed]
- Chawsheen, H.A.; Jiang, H.; Ying, Q.; Ding, N.; Thapa, P.; Wei, Q. The redox regulator sulfiredoxin forms a complex with thioredoxin domain–containing 5 protein in response to ER stress in lung cancer cells. J. Biol. Chem. 2019, 294, 8991–9006. [Google Scholar] [CrossRef] [PubMed]
- Hatahet, F.; Ruddock, L.W. Protein disulfide isomerase: A critical evaluation of its function in disulfide bond formation. Antioxid. Redox Signal. 2009, 11, 2807–2850. [Google Scholar] [CrossRef]
- Benham, A.M. The protein disulfide isomerase family: Key players in health and disease. Antioxid. Redox Signal. 2012, 16, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Duivenvoorden, W.; Hopmans, S.N.; Austin, R.C.; Pinthus, J.H. Endoplasmic reticulum protein ERp46 in prostate adenocarcinoma. Oncol. Lett. 2017, 13, 3624–3630. [Google Scholar] [CrossRef]
- Mo, R.; Peng, J.; Xiao, J.; Ma, J.; Li, W.; Wang, J.; Ruan, Y.; Ma, S.; Hong, Y.; Wang, C. High TXNDC5 expression predicts poor prognosis in renal cell carcinoma. Tumor Biol. 2016, 37, 9797–9806. [Google Scholar] [CrossRef]
- Wang, L.; Song, G.; Chang, X.; Tan, W.; Pan, J.; Zhu, X.; Liu, Z.; Qi, M.; Yu, J.; Han, B. The role of TXNDC5 in castration-resistant prostate cancer—Involvement of androgen receptor signaling pathway. Oncogene 2015, 34, 4735–4745. [Google Scholar] [CrossRef]
- Chawsheen, H.A.; Ying, Q.; Jiang, H.; Wei, Q. A critical role of the thioredoxin domain containing protein 5 (TXNDC5) in redox homeostasis and cancer development. Genes Dis. 2018, 5, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Maiorino, M.; Bosello, V.; Cozza, G.; Roveri, A.; Toppo, S.; Ursini, F. Glutathione peroxidase-4. In Selenium; Springer: Berlin/Heidelberg, Germany, 2011; pp. 181–195. [Google Scholar]
- Janssen-Heininger, Y.; Reynaert, N.L.; van der Vliet, A.; Anathy, V. Endoplasmic reticulum stress and glutathione therapeutics in chronic lung diseases. Redox Biol. 2020, 33, 101516. [Google Scholar] [CrossRef] [PubMed]
- Bidooki, S.H.; Alejo, T.; Sánchez-Marco, J.; Martínez-Beamonte, R.; Abuobeid, R.; Burillo, J.C.; Lasheras, R.; Sebastian, V.; Rodríguez-Yoldi, M.J.; Arruebo, M. Squalene Loaded Nanoparticles Effectively Protect Hepatic AML12 Cell Lines against Oxidative and Endoplasmic Reticulum Stress in a TXNDC5-Dependent Way. Antioxidants 2022, 11, 581. [Google Scholar] [CrossRef] [PubMed]
- Dores-Silva, P.R.; Cauvi, D.M.; Kiraly, V.T.; Borges, J.C.; De Maio, A. Human HSPA9 (mtHsp70, mortalin) interacts with lipid bilayers containing cardiolipin, a major component of the inner mitochondrial membrane. Biochim. Et Biophys. Acta BBA-Biomembr. 2020, 1862, 183436. [Google Scholar] [CrossRef] [PubMed]
- Fujii, J.; Ikeda, Y. Advances in our understanding of peroxiredoxin, a multifunctional, mammalian redox protein. Redox Rep. 2002, 7, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Immenschuh, S.; Baumgart-Vogt, E. Peroxiredoxins, oxidative stress, and cell proliferation. Antioxid. Redox Signal. 2005, 7, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Fujii, S.; Ushioda, R.; Nagata, K. Redox states in the endoplasmic reticulum directly regulate the activity of calcium channel, inositol 1, 4, 5-trisphosphate receptors. Proc. Natl. Acad. Sci. USA 2023, 120, e2216857120. [Google Scholar] [CrossRef]
- Gallagher, B.M.; Phelan, S.A. Investigating transcriptional regulation of Prdx6 in mouse liver cells. Free Radic. Biol. Med. 2007, 42, 1270–1277. [Google Scholar] [CrossRef]
- Yun, H.-M.; Park, K.-R.; Lee, H.P.; Lee, D.H.; Jo, M.; Shin, D.H.; Yoon, D.-Y.; Han, S.B.; Hong, J.T. PRDX6 promotes lung tumor progression via its GPx and iPLA2 activities. Free Radic. Biol. Med. 2014, 69, 367–376. [Google Scholar] [CrossRef]
- Krysiak, K.; Tibbitts, J.F.; Shao, J.; Liu, T.; Ndonwi, M.; Walter, M.J. Reduced levels of Hspa9 attenuate Stat5 activation in mouse B cells. Exp. Hematol. 2015, 43, 319–330.e310. [Google Scholar] [CrossRef]
- Guo, W.; Yan, L.; Yang, L.; Liu, X.; E, Q.; Gao, P.; Ye, X.; Liu, W.; Zuo, J. Targeting GRP75 improves HSP90 inhibitor efficacy by enhancing p53-mediated apoptosis in hepatocellular carcinoma. PLoS ONE 2014, 9, e85766. [Google Scholar] [CrossRef] [PubMed]
- Bassot, A.; Prip-Buus, C.; Alves, A.; Berdeaux, O.; Perrier, J.; Lenoir, V.; Ji-Cao, J.; Berger, M.-A.; Loizon, E.; Cabaret, S. Loss and gain of function of Grp75 or mitofusin 2 distinctly alter cholesterol metabolism, but all promote triglyceride accumulation in hepatocytes. Biochim. Et Biophys. Acta BBA-Mol. Cell Biol. Lipids 2021, 1866, 159030. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Cao, X.; Qin, X.; Liu, H.; Chen, S.; Zhong, S.; Li, Y. Proteomic analysis of the molecular mechanism of curcumin/β-cyclodextrin polymer inclusion complex inhibiting HepG2 cells growth. J. Food Biochem. 2020, 44, e13119. [Google Scholar] [CrossRef]
- López-Grueso, M.J.; Lagal, D.J.; García-Jiménez, Á.F.; Tarradas, R.M.; Carmona-Hidalgo, B.; Peinado, J.; Requejo-Aguilar, R.; Bárcena, J.A.; Padilla, C.A. Knockout of PRDX6 induces mitochondrial dysfunction and cell cycle arrest at G2/M in HepG2 hepatocarcinoma cells. Redox Biol. 2020, 37, 101737. [Google Scholar] [CrossRef] [PubMed]
- Azimi, A.; Kaufman, K.L.; Ali, M.; Arthur, J.; Kossard, S.; Fernandez-Penas, P. Differential proteomic analysis of actinic keratosis, Bowen’s disease and cutaneous squamous cell carcinoma by label-free LC–MS/MS. J. Dermatol. Sci. 2018, 91, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Yafune, A.; Kawai, M.; Itahashi, M.; Kimura, M.; Nakane, F.; Mitsumori, K.; Shibutani, M. Global DNA methylation screening of liver in piperonyl butoxide-treated mice in a two-stage hepatocarcinogenesis model. Toxicol. Lett. 2013, 222, 295–302. [Google Scholar] [CrossRef]
- Charlton, H.K.; Webster, J.; Kruger, S.; Simpson, F.; Richards, A.A.; Whitehead, J.P. ERp46 binds to AdipoR1, but not AdipoR2, and modulates adiponectin signalling. Biochem. Biophys. Res. Commun. 2010, 392, 234–239. [Google Scholar] [CrossRef]
- Heiker, J.T.; Kosel, D.; Beck-Sickinger, A.G. Molecular Mechanisms of Signal Transduction via Adiponectin and Adiponectin Receptors. Biol. Chem. 2010, 391. [Google Scholar] [CrossRef]
- Lu, W.; Lee, N.; Kaul, S.; Lan, F.; Poon, R.; Wadhwa, R.; Luk, J. Mortalin–p53 interaction in cancer cells is stress dependent and constitutes a selective target for cancer therapy. Cell Death Differ. 2011, 18, 1046–1056. [Google Scholar] [CrossRef]
- Ornatsky, O.; Connor, M.; Hood, D. Expression of stress proteins and mitochondrial chaperonins in chronically stimulated skeletal muscle. Biochem. J. 1995, 311, 119–123. [Google Scholar] [CrossRef]
- Xu, J.; Xiao, H.H.; Sartorelli, A.C. Attenuation of the induced differentiation of HL-60 leukemia cells by mitochondrial chaperone HSP70. Oncol. Res. 1999, 11, 429–435. [Google Scholar] [PubMed]
- Pizzatti, L.; Sá, L.A.; de Souza, J.M.; Bisch, P.M.; Abdelhay, E. Altered protein profile in chronic myeloid leukemia chronic phase identified by a comparative proteomic study. Biochim. Et Biophys. Acta BBA-Proteins Proteom. 2006, 1764, 929–942. [Google Scholar] [CrossRef] [PubMed]
- Takano, S.; Wadhwa, R.; Yoshii, Y.; Nose, T.; Kaul, S.C.; Mitsui, Y. Elevated levels of mortalin expression in human brain tumors. Exp. Cell Res. 1997, 237, 38–45. [Google Scholar] [CrossRef]
- Rozenberg, P.; Kocsis, J.; Saar, M.; Prohászka, Z.; Füst, G.; Fishelson, Z. Elevated levels of mitochondrial mortalin and cytosolic HSP70 in blood as risk factors in patients with colorectal cancer. Int. J. Cancer 2013, 133, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Wadhwa, R.; Takano, S.; Kaur, K.; Deocaris, C.C.; Pereira-Smith, O.M.; Reddel, R.R.; Kaul, S.C. Upregulation of mortalin/mthsp70/Grp75 contributes to human carcinogenesis. Int. J. Cancer 2006, 118, 2973–2980. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Luk, J.M.; Lee, N.P.; Peng, J.; Leng, X.; Guan, X.-Y.; Lau, G.K.; Beretta, L.; Fan, S.-T. Association of mortalin (HSPA9) with liver cancer metastasis and prediction for early tumor recurrence. Mol. Cell. Proteom. 2008, 7, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Wadhwa, R.; Takano, S.; Taira, K.; Kaul, S.C. Reduction in mortalin level by its antisense expression causes senescence-like growth arrest in human immortalized cells. J. Gene Med. A Cross-Discip. J. Res. Sci. Gene Transf. Its Clin. Appl. 2004, 6, 439–444. [Google Scholar] [CrossRef]
- Hofmann, B.; Hecht, H.-J.; Flohé, L. Peroxiredoxins. Biol. Chem. 2002, 383. [Google Scholar] [CrossRef]
- Rhee, S.G.; Chae, H.Z.; Kim, K. Peroxiredoxins: A historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic. Biol. Med. 2005, 38, 1543–1552. [Google Scholar] [CrossRef]
- Fisher, A.B.; Dodia, C.; Manevich, Y.; Chen, J.-W.; Feinstein, S.I. Phospholipid hydroperoxides are substrates for non-selenium glutathione peroxidase. J. Biol. Chem. 1999, 274, 21326–21334. [Google Scholar] [CrossRef]
- Sparling, N.E.; Phelan, S.A. Identification of multiple transcripts for antioxidant protein 2 (Aop2): Differential regulation by oxidative stress and growth factors. Redox Rep. 2003, 8, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Phelan, S.A.; Forsman-Semb, K.; Taylor, E.F.; Petros, C.; Brown, A.; Lerner, C.P.; Paigen, B. Mice with targeted mutation of peroxiredoxin 6 develop normally but are susceptible to oxidative stress. J. Biol. Chem. 2003, 278, 25179–25190. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.-S.; Dodia, C.; Chen, X.; Hennigan, B.B.; Jain, M.; Feinstein, S.I.; Fisher, A.B. Cloning and expression of rat lung acidic Ca2+-independent PLA2 and its organ distribution. Am. J. Physiol.-Lung Cell. Mol. Physiol. 1998, 274, L750–L761. [Google Scholar] [CrossRef] [PubMed]
- Frank, S.; Munz, B.; Werner, S. The human homologue of a bovine non-selenium glutathione peroxidase is a novel keratinocyte growth factor-regulated gene. Oncogene 1997, 14, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Munz, B.; FRANK, S.; HÜBNER, G.; OLSEN, E.; WERNER, S. A novel type of glutathione peroxidase: Expression and regulation during wound repair. Biochem. J. 1997, 326, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Avram, H.; Aaron, C.; Alexander, V. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar]
- Chen, L.; Madura, K. Increased proteasome activity, ubiquitin-conjugating enzymes, and eEF1A translation factor detected in breast cancer tissue. Cancer Res. 2005, 65, 5599–5606. [Google Scholar] [CrossRef]
- Arlt, A.; Bauer, I.; Schafmayer, C.; Tepel, J.; Müerköster, S.S.; Brosch, M.; Röder, C.; Kalthoff, H.; Hampe, J.; Moyer, M. Increased proteasome subunit protein expression and proteasome activity in colon cancer relate to an enhanced activation of nuclear factor E2-related factor 2 (Nrf2). Oncogene 2009, 28, 3983–3996. [Google Scholar] [CrossRef]
- Quero, J.; Cabello, S.; Fuertes, T.; Mármol, I.; Laplaza, R.; Polo, V.; Gimeno, M.C.; Rodriguez-Yoldi, M.J.; Cerrada, E. Proteasome versus thioredoxin reductase competition as possible biological targets in antitumor mixed thiolate-dithiocarbamate gold (III) complexes. Inorg. Chem. 2018, 57, 10832–10845. [Google Scholar] [CrossRef]
- Manevich, Y.; Sweitzer, T.; Pak, J.H.; Feinstein, S.I.; Muzykantov, V.; Fisher, A.B. 1-Cys peroxiredoxin overexpression protects cells against phospholipid peroxidation-mediated membrane damage. Proc. Natl. Acad. Sci. USA 2002, 99, 11599–11604. [Google Scholar] [CrossRef]
- Pak, J.H.; Manevich, Y.; Kim, H.S.; Feinstein, S.I.; Fisher, A.B. An antisense oligonucleotide to 1-cys peroxiredoxin causes lipid peroxidation and apoptosis in lung epithelial cells. J. Biol. Chem. 2002, 277, 49927–49934. [Google Scholar] [CrossRef] [PubMed]
- Sharapov, M.G.; Novoselov, V.I.; Gudkov, S.V. Radioprotective role of peroxiredoxin 6. Antioxidants 2019, 8, 15. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Chen, X.-B.; Hong, Y.-C.; Zhu, H.; He, Q.-J.; Yang, B.; Ying, M.-D.; Cao, J. Identification of PRDX6 as a regulator of ferroptosis. Acta Pharmacol. Sin. 2019, 40, 1334–1342. [Google Scholar] [CrossRef] [PubMed]
- Goswami, A.V.; Chittoor, B.; D’Silva, P. Understanding the functional interplay between mammalian mitochondrial Hsp70 chaperone machine components. J. Biol. Chem. 2010, 285, 19472–19482. [Google Scholar] [CrossRef] [PubMed]
- Dhennin-Duthille, I.; Nyga, R.; Yahiaoui, S.; Gouilleux-Gruart, V.; Régnier, A.; Lassoued, K.; Gouilleux, F. The tumor suppressor hTid1 inhibits STAT5b activity via functional interaction. J. Biol. Chem. 2011, 286, 5034–5042. [Google Scholar] [CrossRef] [PubMed]
- Gooljarsingh, L.T.; Fernandes, C.; Yan, K.; Zhang, H.; Grooms, M.; Johanson, K.; Sinnamon, R.H.; Kirkpatrick, R.B.; Kerrigan, J.; Lewis, T. A biochemical rationale for the anticancer effects of Hsp90 inhibitors: Slow, tight binding inhibition by geldanamycin and its analogues. Proc. Natl. Acad. Sci. USA 2006, 103, 7625–7630. [Google Scholar] [CrossRef]
- Pascale, R.M.; Simile, M.M.; Calvisi, D.F.; Frau, M.; Muroni, M.R.; Seddaiu, M.A.; Daino, L.; Muntoni, M.D.; De Miglio, M.R.; Thorgeirsson, S.S. Role of HSP90, CDC37, and CRM1 as modulators of P16INK4A activity in rat liver carcinogenesis and human liver cancer. Hepatology 2005, 42, 1310–1319. [Google Scholar] [CrossRef]
- Muñoz, J.P.; Ivanova, S.; Sánchez-Wandelmer, J.; Martínez-Cristóbal, P.; Noguera, E.; Sancho, A.; Díaz-Ramos, A.; Hernández-Alvarez, M.I.; Sebastián, D.; Mauvezin, C. Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J. 2013, 32, 2348–2361. [Google Scholar] [CrossRef]
- Liu, T.; Krysiak, K.; Shirai, C.L.; Kim, S.; Shao, J.; Ndonwi, M.; Walter, M.J. Knockdown of HSPA9 induces TP53-dependent apoptosis in human hematopoietic progenitor cells. PLoS ONE 2017, 12, e0170470. [Google Scholar] [CrossRef]
- Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019, 28, 1947–1951. [Google Scholar] [CrossRef]
- Almaghlouth, I.; Mohamed, J.; Al-Amoudi, M.; Al-Ahaidib, L.; Al-Odaib, A.; Alkuraya, F. 5-Oxoprolinase deficiency: Report of the first human OPLAH mutation. Clin. Genet. 2012, 82, 193–196. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Shanmugam, A.; Markand, S.; Zorrilla, E.; Ganapathy, V.; Smith, S.B. Sigma 1 receptor regulates the oxidative stress response in primary retinal Müller glial cells via NRF2 signaling and system xc−, the Na+-independent glutamate–cystine exchanger. Free Radic. Biol. Med. 2015, 86, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Parl, F.F. Glutathione S-transferase genotypes and cancer risk. Cancer Lett. 2005, 221, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, H.; Boesch-Saadatmandi, C.; Regos, I.; Treutter, D.; Wolffram, S.; Rimbach, G. Effects of quercetin and catechin on hepatic glutathione-S transferase (GST), NAD (P) H quinone oxidoreductase 1 (NQO1), and antioxidant enzyme activity levels in rats. Nutr. Cancer 2009, 61, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Karatas, E.; Raymond, A.-A.; Leon, C.; Dupuy, J.-W.; Di-Tommaso, S.; Senant, N.; Collardeau-Frachon, S.; Ruiz, M.; Lachaux, A.; Saltel, F. Hepatocyte proteomes reveal the role of protein disulfide isomerase 4 in alpha 1-antitrypsin deficiency. JHEP Rep. 2021, 3, 100297. [Google Scholar] [CrossRef]
- Francavilla, R.; Castellaneta, S.P.; Hadzic, N.; Chambers, S.M.; Portmann, B.; Tung, J.; Cheeseman, P.; Rela, M.; Heaton, N.D.; Mieli-Vergani, G. Prognosis of alpha-1-antitrypsin deficiency-related liver disease in the era of paediatric liver transplantion. J. Hepatol. 2000, 32, 986–992. [Google Scholar] [CrossRef]
- Herrera-Marcos, L.V.; Martínez-Beamonte, R.; Macías-Herranz, M.; Arnal, C.; Barranquero, C.; Puente-Lanzarote, J.J.; Gascón, S.; Herrero-Continente, T.; Gonzalo-Romeo, G.; Alastrué-Vera, V. Hepatic galectin-3 is associated with lipid droplet area in non-alcoholic steatohepatitis in a new swine model. Sci. Rep. 2022, 12, 1–17. [Google Scholar] [CrossRef]
- Abuobeid, R.; Herrera-Marcos, L.; Navarro, M.A.; Arnal, C.; Martínez-Beamonte, R.; Surra, J.; Osada, J. Dietary erythrodiol modifies hepatic transcriptome in mice in a sex and dose-dependent way. Int. J. Mol. Sci. 2020, 21, 7331. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Gene ID (NCBI) | SL2R RNAseq (AML12) | Regulation (AML12) KO/WT |
|---|---|---|---|
| Dnaja3 [31] | 83945 | 1.2331 | UP |
| Hsp90aa1 [32] | 15519 | −1.2332 | DOWN |
| Mfn2 [33] | 170731 | 1.6794 | UP |
| Rps14 [31] | 20044 | −1.6093 | DOWN |
| Gene Symbol | Gene ID (NCBI) | SL2R RNAseq (AML12) | Regulation (AML12) KO/WT |
|---|---|---|---|
| Gstm6 | 14867 | −2.1758 | DOWN |
| Gstp3 | 225884 | −2.6291 | DOWN |
| Gstt1 | 14871 | −3.3955 | DOWN |
| Npm1 [34] | 18148 | −2.3854 | DOWN |
| Oplah | 75475 | −2.5423 | DOWN |
| Prdx5 [35] | 54683 | −2.0351 | DOWN |
| Serpina1a [36] | 20700 | −3.8105 | DOWN |
| Serpina1b [36] | 20701 | −2.5978 | DOWN |
| Serpina3m [37] | 20717 | −6.7645 | DOWN |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bidooki, S.H.; Sánchez-Marco, J.; Martínez-Beamonte, R.; Herrero-Continente, T.; Navarro, M.A.; Rodríguez-Yoldi, M.J.; Osada, J. Endoplasmic Reticulum Protein TXNDC5 Interacts with PRDX6 and HSPA9 to Regulate Glutathione Metabolism and Lipid Peroxidation in the Hepatic AML12 Cell Line. Int. J. Mol. Sci. 2023, 24, 17131. https://doi.org/10.3390/ijms242417131
Bidooki SH, Sánchez-Marco J, Martínez-Beamonte R, Herrero-Continente T, Navarro MA, Rodríguez-Yoldi MJ, Osada J. Endoplasmic Reticulum Protein TXNDC5 Interacts with PRDX6 and HSPA9 to Regulate Glutathione Metabolism and Lipid Peroxidation in the Hepatic AML12 Cell Line. International Journal of Molecular Sciences. 2023; 24(24):17131. https://doi.org/10.3390/ijms242417131
Chicago/Turabian StyleBidooki, Seyed Hesamoddin, Javier Sánchez-Marco, Roberto Martínez-Beamonte, Tania Herrero-Continente, María A. Navarro, María J. Rodríguez-Yoldi, and Jesús Osada. 2023. "Endoplasmic Reticulum Protein TXNDC5 Interacts with PRDX6 and HSPA9 to Regulate Glutathione Metabolism and Lipid Peroxidation in the Hepatic AML12 Cell Line" International Journal of Molecular Sciences 24, no. 24: 17131. https://doi.org/10.3390/ijms242417131