
Opportunities in Cancer Therapies: Deciphering the Role of Cancer Stem Cells in Tumour Repopulation

1
UniSA Allied Health & Human Performance, University of South Australia, Adelaide, SA 5001, Australia
2
Faculty of Informatics and Science, University of Oradea, 410087 Oradea, Romania
3
Australian Centre for Quantitative Imaging, School of Medicine, The University of Western Australia, Perth, WA 6009, Australia
4
Faculty of Chemistry & Physics, University of Adelaide, Adelaide, SA 5000, Australia
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2023, 24(24), 17258; https://doi.org/10.3390/ijms242417258
Submission received: 18 November 2023
/
Revised: 6 December 2023
/
Accepted: 6 December 2023
/
Published: 8 December 2023
(This article belongs to the Special Issue Radiobiology and Radiotherapy in Tumour)
{kind=link}
{kind=link}
Abstract
:Tumour repopulation during treatment is a well acknowledged yet still challenging aspect of cancer management. The latest research results show clear evidence towards the existence of cancer stem cells (CSCs) that are responsible for tumour repopulation, dissemination, and distant metastases in most solid cancers. Cancer stem cell quiescence and the loss of asymmetrical division are two powerful mechanisms behind repopulation. Another important aspect in the context of cancer stem cells is cell plasticity, which was shown to be triggered during fractionated radiotherapy, leading to cell dedifferentiation and thus reactivation of stem-like properties. Repopulation during treatment is not limited to radiotherapy, as there is clinical proof for repopulation mechanisms to be activated through other conventional treatment techniques, such as chemotherapy. The dynamic nature of stem-like cancer cells often elicits resistance to treatment by escaping drug-induced cell death. The aims of this scoping review are (1) to describe the main mechanisms used by cancer stem cells to initiate tumour repopulation during therapy; (2) to present clinical evidence for tumour repopulation during radio- and chemotherapy; (3) to illustrate current trends in the identification of CSCs using specific imaging techniques; and (4) to highlight novel technologies that show potential in the eradication of CSCs.
1. Cancer Stem Cell Properties
Cancer stem cells are recognised as an important cellular category that is at the top of the hierarchical scale, presenting similar biological properties and expression profiles to normal stem cells [1]. While the origins of cancer stem cells in solid tumours are not fully elucidated, it is hypothesised that they arise from either differentiated tumour cells or from organ-specific adult stem cells [2].
1.1. Biological Characteristics of CSCs
Generally, cancer stem cells (CSCs) account for a very small percentage of the cells that constitute a malignant solid tumour [3]. The fraction of CSCs within a tumour was shown to be dependent on the neoplasm, but also on the in vivo assays and biomarkers employed for their identification [4]. An interesting observation derived from in vitro studies on CSCs is related to their heterogeneity [5]. Research undertaken on both HPV-positive and HPV-negative head and neck carcinoma cell lines revealed the existence of a diversity of CSCs that differed in their biological properties and response to therapy, explaining the more responsive behaviour of HPV-positive tumours [5]. This finding demonstrates than not only the absolute percentage of CSCs but also their phenotype dictate treatment outcome, showing the complexity that surrounds CSC identification and targeting. The link between oncogenic viruses and CSCs was also investigated in gastric cancers associated with the Epstein–Barr virus [6,7]. Epstein–Barr virus-encoded miRNAS were shown to have a strong impact on the maintenance of stemness through the regulation of epithelial–mesenchymal transition. Recent studies on gastric cancers identified a unique CSC marker (CD44v6/v9) for Epstein–Barr virus-associated carcinomas that might serve as a potential target for this subtype of gastric cancer [7].
Despite their low percentage, the biological properties specific to CSCs are powerful tools that influence the fate of the tumour. Among their biological features, pluripotency is perhaps one of the most notable ones, whereby CSCs are capable of unlimited self-renewal by maintaining an undifferentiated state, also being able to create the entire heterogeneous lineage of the original tumour through differentiation [8]. Stemness is maintained by cellular plasticity—a property that promotes tumour heterogeneity and CSC generation.
1.2. CSC and Hypoxia
A particular property of cancer stem cells is their preference for residing in specific microenvironmental niches, such as hypoxic ones, in order to preserve their functionality and stem-like properties [9]. Studies showed that the hypoxia inducible factor (HIF) plays a key role in the niche mechanism, being implicated in CSC survival and proliferation under hypoxic conditions as well as in the angiogenic switch activation [10]. Knowing the challenge of treatment resistance caused by the presence of hypoxia, tumour control is heavily impacted by the increased expression of HIF found in these hypoxic niches.
1.3. CSC Repopulation and Resistance to Therapy
CSCs are also characterised by quiescence, a state that fosters prolongation of their life span, preservation of cellular functions, and protection from therapeutic stress [11]. CSC quiescence was shown to induce treatment resistance and tumour recurrence while also promoting metastases. Another mechanism adopted by CSCs is cellular senescence, which was shown to be responsible for genetic reprogramming and the activation of stemness, contributing to tumour progression and distant spread [11].
All the above-mentioned CSC properties have important clinical implications, as stem-like cancer cells have been shown to be more resistant to treatment than their non-stem counterparts, often leading to treatment failure [12]. One of the reasons for poor treatment outcome is credited to tumour repopulation during therapy. Today it is known that repopulation during treatment is not limited to radiotherapy, as there is clinical proof of repopulation mechanisms being activated through other conventional treatment techniques, such as chemotherapy. The dynamic nature of stem-like cancer cells often elicits resistance to treatment by escaping drug-induced cell death.
The mechanisms behind repopulation are complex and most are attributed to the presence of CSCs [13,14,15]. According to previous research, CSCs adopt various repopulation mechanisms in response to the effect of chemo/radiotherapy. Among them, cell recruitment, shortening of cell cycle duration, and the loss of asymmetrical division are the most commonly investigated and agreed upon mechanisms [13,14,15,16]. Quiescence or dormancy, as previously mentioned, is a state outside the cell cycle where CSCs reside to prolong their lifespan and preserve functionality. However, a situation might arise, such as cellular eradication, which triggers the re-entry of CSCs into the cell cycle through the process of cell recruitment. Once in the mitotic cycle, stem cells can further activate repopulation mechanisms by shortening the length of their passage through the cycle, thus promoting faster cell turnover. Perhaps the most efficient repopulation mechanism is the symmetrical division of CSCs (i.e., the creation of two cancer stem cells) which is acquired through the loss of asymmetrical division (i.e., the creation of a stem and a differentiated cell) [15,17].
The goals of this scoping review are to gather evidence towards the role played by cancer stem cells in tumour repopulation during cancer treatment, and to identify techniques to either counteract these mechanisms or to overcome repopulation by direct targeting of cancer stem cells.
2. Repopulation during Radiotherapy
2.1. Cancer Stem Cells and Their Role in Repopulation during Radiotherapy
Tumour repopulation after radiotherapy remains a significant challenge in cancer therapy. Despite advancements in photon radiation therapy techniques, such as intensity modulated radiotherapy (IMRT) or volumetric modulated arc radiotherapy (VMAT), the recurrence of tumours post-treatment remains an issue for many cancers, including head and neck, breast, colorectal, prostate, and hepatocellular cancers [18,19,20,21].
As discussed elsewhere in this paper, once again, clonogenic stem cells have emerged in the literature as primary drivers in the process of tumour repopulation that ultimately lead to treatment failure [22,23]. Because of CSCs’ propensity for renewal, differentiation into various cell types, as well as recruitment of differentiated cells and treatment resistance, they drive tumour repopulation during and post radiation therapy. In other words, the ongoing division of clonogenic cancer stem cells has been shown to be the cause of tumour regrowth in, for example, intestinal, breast, and brain tumours [24].
Similarly to chemotherapy, CSCs are often more resistant to radiation therapy compared to the bulk of tumour cells [25]. This CSC resistance is attributed to various mechanisms, including enhanced DNA repair capacity, quiescence, higher expression of drug efflux pumps, resting in tumour hypoxic regions, etc., which collectively contribute to their survival and capacity to initiate tumour repopulation and regrowth [26]. This phenomenon is a significant obstacle to achieving durable therapy responses in cancer patients.
In addition to radiation resistance, another key characteristic of CSCs is their ability to be recruited from the quiescent state following the irradiation damage and re-enter a standard cell cycle, thus initiating cell proliferation and differentiation, leading to the regrowth of the tumour. Research has shown that about one third of CSCs in glioma and breast cancer cell lines remain quiescent but become active in the cell cycle following radiation treatment [27,28]. The work of Bao et al. [29], conducted in animal models using primary human gliomas, showed that CD133 (Prominin-1), a marker for both neural stem cells and brain cancer stem cells, was not only enriched after irradiation but also capable of initiating xenografts from as few as 500 cells. The authors conclude that CD133-positive tumour cells represent the cellular population responsible for glioma radioresistance and potentially tumour recurrence after radiation therapy [29].
CSCs can also promote tumour heterogeneity by generating different types of CSCs through reversible transformations between stem and non-stem cells [30,31]. This heterogeneity further increases treatment resistance and contributes to cell survival and subsequent repopulation. Preclinical work by Lagadec et al. reports that upregulation of the embryonic transcription factors (such as Sox2, Oct4, Klf4, and Nanog) induced by radiation promotes nontumorigenic cancer cells to acquire CSC-like features [32].
Tumour repopulation itself after radiation therapy can be attributed to several processes. One key factor is the stimulation of surviving cancer cells through a phenomenon known as radiation-induced cytokine release. The irradiated tumour microenvironment can trigger the secretion of growth factors, such as TGF-β and VEGF, which promote the survival and proliferation of cancer cells [33]. Moreover, the radiation-induced DNA damage repair can lead to genetic mutations and clonal expansion of radioresistant cancer cells. This is further compounded by the fact that CSCs have pro-survival pathways that are upregulated and protect these cells from cell death, resulting in CSCs being resistant to radiation damage [34].
2.2. The Effect of Radiotherapy on CSCs
Fractionated radiation therapy, while necessary to mitigate the damage to healthy tissues, is selective in its cell kill. It eliminates, by and large, the differentiated tumour cells and enables the preferential survival of the most radiation therapy resistant and more tumorigenic CSCs, thus generating a so-called radiation-induced CSC phenotype [21,35,36]. This in turn enhances the malignancy of residual tumours. In breast cancer, the proportion of CSCs increases significantly after ionizing radiation, leading to enhanced proliferation, which accelerates tumour repopulation [32,37]. The cell line experiments by Lagadec et al. showed that these radiation-induced CSC phenotypes often exhibit enhanced sphere-forming and tumorigenic abilities. This means that the number of tumorigenic cells increases after treatment, potentially leading to a more rapid tumour recurrence and increased aggressiveness that can lead to metastatic disease [32].
Additionally, the selective killing that enables the radiation induced CSC phenotype is particularly relevant in cases where the overall treatment time is prolonged, which has been associated with a decrease in tumour control rates in certain types of cancers. A seminal study by Withers et al. identified rapid tumour regrowth during extensions of radiotherapy treatment from ~5–8 weeks in almost 500 patients with oropharyngeal cancer. They concluded that for rapidly proliferating cancers such as head and neck, radiotherapy should be completed within the shortest feasible time due to accelerated repopulation of CSCs that occurs while the tumour is still regressing. In view of this, the recommendation is that a delay in treatment initiation is preferable to delays during radiotherapy [18].
2.3. Strategies to Inhibit Tumour Regrowth after Radiotherapy and Future Perspectives
Fast tumour repopulation owing to CSCs gives justification for the implementation of shorter treatment courses and hypofractionation regimens, such as stereotactic ablative radiotherapy (SABR) or hyperfractionated regimens in head and neck cancers, to avoid quick repopulation between two subsequent doses. Where plausible, dose escalations also help to counteract the processes supporting tumour repopulation [38]. Additionally, combining radiation therapy with targeted therapies, such as EGFR inhibitors or immune checkpoint inhibitors, can enhance the radiosensitivity of cancer cells and suppress repopulation.
Other options include the use of protons and heavy ions that generate more complex and direct DNA radiation-induced damage, which may be more independent of oxygen status, overcoming the resistance of hypoxic cells. Chiblak et al. showed that primary human glioma stem cells with resistance to photon therapy could be made sensitive to treatment with carbon ions due to their compromised ability to repair DNA double-strand breaks caused by carbon ion irradiation [39].
The interplay between CSCs and tumour repopulation post-radiotherapy is a complex and multifaceted phenomenon. The large heterogeneity in CSC radiosensitivity which leads to both inter- and intra-patient variability makes a one-size-fits-all treatment approach challenging. Understanding the biology and resistance mechanisms of these cells is critical for advancing cancer treatment strategies. Future research should focus on elucidating the molecular pathways involved and identifying innovative approaches to target clonogenic stem cells, ultimately improving the success rates of radiotherapy as well as combined therapies, thus reducing the risk of tumour recurrence.
Efforts to mitigate tumour repopulation after radiotherapy have led to investigations into strategies that specifically target clonogenic stem cells. These strategies encompass the development of CSC-specific therapies, such as CSC-targeting drugs and combination treatments. While promising, challenges remain in effectively eradicating or sensitizing clonogenic stem cells to radiotherapy.
3. Repopulation during Chemotherapy
3.1. Cancer Stem Cells and Their Role in Repopulation during Chemotherapy
While the role of cancer stem cells in the process of tumour repopulation during radiotherapy is well researched and supported by experimental evidence, accelerated repopulation during the course of chemotherapy is often overlooked, despite similar biological triggers—such as loss of cancer cells—being involved in both types of cancer therapy [40].
In silico studies looking into the mechanisms behind repopulation during drug-based therapies concluded that chemotherapy could trigger tumour regrowth through cell recruitment into the mitotic cycle [41]. This is a potent biological mechanism considering the fact the quiescent cells are not affected by chemotherapy (being non-proliferative), thus the pool of resting cells in the G0 phase is large and viable (Figure 1). Although the onset of tumour repopulation during chemotherapy is a poorly researched factor, the limited experimental data suggests that this process might occur at various degrees and intervals after drug administration, owing to different recruitment rates for each particular tumour type [40]. In silico data showed that early onset of repopulation (during the first week of chemotherapy) has a more powerful effect on the outcome than late onset (during the second half of therapy), an expected result as long as the rate of recruitment remains constant [41]. Nevertheless, treatment outcome is strongly influenced by the recruitment rate, which interplays with the onset of repopulation, thus dictating together the fate of cancer cells [41].
Another mechanism identified as a contributor towards repopulation is abortive division [14], but with a less powerful impact than cell recruitment as it affects differentiated cells with finite proliferating ability (hence the term ‘abortive division’) which only have a short-term, transitory bearing on tumour population [42].
A rather paradoxical phenomenon concerning tumour repopulation and treatment resistance is facilitated by cellular senescence. Senescence, an essential tumour suppressor mechanism, is a cellular response to various stress-inducing stimuli (such as radio/chemotherapy) whereby cells lose their proliferative ability [43]. At the same time, senescent cells can spontaneously evade their condition and regain their proliferative ability re-entering the cell cycle, which is clear proof of cellular plasticity [44]. Thus, instead of tumour suppression, treatment-induced senescence can, depending on microenvironmental factors, lead to an opposite effect, i.e., repopulation and resistance to therapy.
Drug resistance is a very common factor hindering the success of chemotherapy, and it was shown to be linked to tumour repopulation during treatment [45]. Resistance to chemotherapy is a cumulative process that ultimately overpowers tumour regression leading to poor treatment outcome. Therefore, management of drug resistance is one of the key requirements to avoid cancer cell survival that could lead to repopulation during cycles of chemotherapy. Lately, the tumour microenvironment was shown to play an important role in therapy resistance, due to its potential to dictate the maintenance of CSCs through the release of cytokines, growth factors, and other cellular components to promote stemness [46]. While the targeting of CSCs is a critical factor for overcoming repopulation and resistance to treatment, the complex tumour microenvironment together with its pathways to promote stemness require well-targeted, combined approaches to inhibit tumour proliferation.
Recent studies on human colon cancer stem cells highlighted the drug-tolerant characteristics of these CSCs (called persister cells), i.e., their ability to escape cell death induced by a wide range of chemotherapeutic agents [47]. The study enabled the investigation of in vivo CSC dynamics through the tracking of LGR5+ cells with stem-like properties. A quiescent subset of cells expressing the p27 protein has been identified, an expression that is a feature of persister cells. Chemotherapy was shown to disrupt the dormancy of these cells, triggering them into the cell cycle, and thus into the regrowth process. Similar chemotherapy-stimulated transition into the proliferative state was evidenced in previously quiescent glioblastoma cancer stem cells in mouse models [48]. The glioblastoma study confirmed the unique feature of cancer stem cells and the CSC-specific relevance of functional markers in the context of tumour diversity.
3.2. Clinical Evidence Concerning the Impact of CSC Repopulation on Treatment Outcome
When employing non-conventional chemotherapy regimens to overcome the challenges posed by accelerated repopulation, the results of randomised clinical trials are perhaps the most convincing indication towards tumour regrowth during therapy. Several trials designed for various tumour sites have been conducted to evaluate the effect of non-traditional treatment schedules on tumour response [49,50,51]. The response of unresectable non-small-cell lung cancers (NSCLC) to accelerated treatment regimens was heavily investigated owing to the known potential of accelerated schedules to overcome repopulation during therapy in tumours with short cell doubling time. A phase I trial designed for inoperable NSCLC patients aimed to evaluate the maximum tolerated dose (MTD) of accelerated hypofractionated radiotherapy (3 Gy/fraction, over 5 weeks) concurrently with vinorelbine and carboplatin, in a dose escalation setting (69 Gy was regarded as the MTD) [49]. The short-term response rate was 84.6%, with a 1-year median progression-free survival of 49.4%, showing good local tumour control for this patient group, and thus warranting further investigation.
Maguire et al. reported the results of a randomised phase II trial (SOCCAR trial) that compared sequential with concurrent cisplatin–vinorelbine-based chemotherapy and radical hypofractionated radiotherapy (over 4 weeks) in nonresectable NSCLC patients to assess their effectiveness in minimising accelerated repopulation [50]. The results showed slightly higher 2-year overall survival with the concurrent chemotherapy regimen (50% vs. 46%), with no difference in toxicity between the two arms, concluding that concurrent drug-radiotherapy is a safe and efficient way to counteract the effect of tumour repopulation during treatment.
Since overall treatment time plays a critical role in determining the success of therapy in fast growing tumours, a multicentre, prospective, randomised clinical trial was designed for NSCLC patients, aiming to investigate the effect of accelerated postoperative radiochemotherapy vs. conventionally fractionated therapy in increasing local tumour control by administering seven fractions of two Gy doses per week of either photons or protons [51]. Using this accelerated treatment regimen, it was postulated that the 3-year local tumour control will increase from 70% to 85%. The final results of this trial have not been published so far.
In invasive bladder cancers with fast repopulation of cancer stem cells, Panteliadou et al. conducted a trial employing an accelerated hypofractionation (HypoARC) with 15 fractions of 3.4 Gy to the bladder, together with amifostine to alleviate radiation-induced side effects [52]. The treatment outcome showed an 86.6% complete response rate and 56% 3-year local control, the results being comparable with previous reports. While this schedule seemed safe, particularly due to the administration of amifostine, local tumour control did not present notable differences from more conventional schedules, highlighting the fact that changes in treatment regimens are not always sufficient to overcome tumour repopulation.
3.3. CSC Sensitivity Assay-Guided Chemotherapy
In view of the above, the development of CSC sensitivity assays to guide chemotherapy delivery would allow for treatment personalisation in a variety of cancer types. However, there are several challenges in developing efficient procedures, from tissue sample collection to the length of the test, impacting the clinical implementation [53]. To date, the only US-certified and accredited CSC sensitivity assay is ChemoID, developed to examine the response of CSCs derived from patient samples to conventional chemotherapeutic agents [54,55].
Perhaps one of the most investigated tumours in the context of cancer stem cell assay-guided chemotherapy is glioblastoma, owing to its aggressive nature and resistance to therapy [54,55,56]. Prominin-1, or CD133, is a widely explored CSC biomarker associated with normal neural stem cells, and thus is commonly used in glioblastomas, together with other CSC markers such as CD24, CD44, CXCR4, Oct3/4, and Nanog. ChemoID was employed to evaluate the correlation between drug efficiency (temozolomide) and CSC positivity using tissue samples collected from a cohort of 41 patients treated with standard temozolomide radiotherapy and to assess this association with tumour recurrence [54]. Patients with a positive CSC test (>40% in vitro cell kill) had a median recurrence time of 20 months compared to 3 months for patients with negative CSC tests. The same assay was used in a prospective glioblastoma study to guide primary treatment after analysing the biopsies originating from 14 patients for the most effective drug therapy [55]. Despite unfavourable outcome predictors, patients treated with ChemoID guidance had an 86% overall response rate and a 13.3-month median overall survival compared to the historical median of 9.1 months [55]. These preliminary results were just confirmed by the results of a randomised clinical trial on patients with recurrent glioblastoma, showing the potential of CSC drug response assay to assist with treatment stratification by identifying patients that would benefit from a specific chemotherapy [56].
Another candidate for ChemoID was platinum-resistant ovarian cancer in an attempt to improve treatment outcome in 78 patients affected by the third relapse of their cancer [57]. Beside the choice of drug, the assay assisted in decisions regarding dose escalation/reduction to optimise tumour response and minimise normal tissue toxicity. Patients treated with high cell kill chemotherapy based on the CSC drug response assay showed improved outcomes compared to those with negative CSC tests (15 vs. 6 months overall survival) or when compared to historical cohorts (15 vs. 8.9 months overall survival) [57]. Since this assay demonstrated efficiency in aggressive cancers, it could be considered a tool for preventing recurrence in high-risk patients and for guiding chemotherapy when multiple drugs are clinically available for a particular cancer.
3.4. CSC-Targeting Agents and Mechanisms
As previously stated, non-conventional chemo/radiotherapy regimens have often been employed in clinics to overcome tumour repopulation. However, their limited or tumour-specific success motivated the investigation of other pathways to suppress resistance to treatment caused by CSC repopulation. The Hedgehog pathway is known to play a critical role in several cellular functions, including signal transmission from the membrane to the nucleus for normal tissue development and regeneration. Studies in gastric cancers have identified a subgroup of CSCs as being responsible for aberrant activation in the Hedgehog signalling pathway, leading to chemotherapy resistance [58,59]. Gastric cancers positive for CD44 showed resistance to several traditional drugs, which was reversed in vitro through the inhibition of the Hedgehog signal using smoothened shRNA or vismodegib. Tumour samples from a phase II clinical trial of gastric patients showed a correlation between high CD44 expression and decreased survival, while the addition of vismodegib to chemotherapy to the group presenting high CD44 expression was associated with improved outcome [58].
Another protein identified to interplay with CSCs in a gastric cancer subtype (diffuse gastric cancer) is RhoA, a member of the Rho GTPase family, being involved in varied cellular functions such as proliferation and survival [59]. Based on the increased activity of RhoA found in gastric cancers, Yoon et al. suggested that RhoA is likely to cause resistance to chemotherapy through the promotion of CSCs [59]. To prove their theory, xenografts were grown to be treated with cisplatin and RhoA pathway inhibitors, showing a significant decrease in cells expressing CD44. The study shows not only the complexity of targeting CSCs but their different phenotype that requires tumour-based personalised approaches.
Efficient eradication of cancer stem cells requires their identification and precise targeting [60]. Metformin, a repurposed diabetes drug, was found to be a modulator of cellular growth and metabolism, inducing AMPK activation and inhibition of tumour development [61]. It has been identified as an effective, though selective, CSC-targeting agent in certain cancers, including colorectal and ovarian cancers [61,62]. The results of a nonrandomised phase II clinical trial of metformin combined with platinum-based chemotherapy for patients with advanced epithelial ovarian cancer showed a positive outcome on ALDH+ cancers [62]. The administration of metformin resulted in a 2.4-fold reduction of CSCs in ovarian cancers as well as an alteration of mesenchymal stem cells which in turn eliminated resistance to platinum. Currently, there are 89 active or recruiting clinical trials evaluating the effect of metformin on treatment outcome in a variety of cancers [63].
Blood cancers, such as acute myeloid leukaemia, are also driven by cancer stem cells. This aspect makes them resistant to conventional chemotherapy, which usually leads to recurrence of disease. Preclinical studies have identified possible targets in acute myeloid leukaemia such as cells expressing CD70 and the CD70/CD27 signalling pathway, which were further tested in a phase I/II clinical trial using cusatuzumab—a human αCD70 monoclonal antibody exhibiting enhanced toxicity on leukemic stem cells with promising results [64].
Another approach to interfering with tumour repopulation is to target senescence-induced cancer stem cells. As explained above, senescence is a double-edged sword, being a potent tumour suppressor under certain conditions while promoting cancer stem cells via cellular plasticity in other circumstances [44]. Through the latter process, senescent cells acquire a senescence-associated secretory phenotype (SASP) with pro-tumorigenic effects. Thus, targeting SASP could interfere with the initiation of senescence-induced CSCs. In view of this, a number of preclinical studies have investigated SASP-targeting pathways in combination with chemotherapeutic agents to assess the effect on CSC suppression and inhibition of chemoresistance, pointing towards a feasible clinical application [65,66].
4. Future Avenues in CSC Management
4.1. Imaging
The capacity to observe populations of cancer stem cells (CSC) plays a crucial role in assessing the immediate efficacy of therapeutic measures and making necessary adjustments to enhance treatment results. Utilising quantitative imaging methods for identification and ongoing monitoring holds the promise of real-time tracking of these populations and gauging treatment responses, ultimately facilitating treatment optimisation [67]. Presently, various imaging techniques are employed, including radionuclide imaging methods such as positron emission tomography (PET) and single-photon emission computed tomography (SPECT), magnetic resonance imaging (MRI), intravital imaging, bioluminescence imaging, as well as different forms of fluorescence imaging, including fluorescence-mediated tomography and near-infrared fluorescence reflectance imaging. Researchers are actively exploring advancements in these imaging technologies to unravel the intricate biological aspects of CSC behaviour, with promising implications for clinical applications [68]. Noteworthy reviews have focussed on the application of functional MRI and developments in nuclear medicine imaging techniques, PET, and SPECT, for the identification and tracking of endogenous CSCs [60,69].
PET quantitatively identifies high-energy γ-rays originating from a subject injected with positron-emitting isotopes or isotope-tagged molecular probes. It boasts remarkable sensitivity, offers non-invasive capabilities, enables real-time in vivo monitoring, and functions independently of the emission source’s depth. Nonetheless, at present, there is no PET technology capable of achieving single-cell resolution for the detection of CSC.
Multiple research teams have reported on various innovative imaging agents and tracers designed for CD133, aimed at facilitating the non-invasive detection of CSCs in different types of malignancies. A study by Hu et al. [70] harnessed CM-2, a stabilised variant of the CM peptide that specifically targets CD133, and created a stable tracer named [64Cu]Cu-CM-2 for PET imaging. They assessed its potential for imaging CD133-expressing tissues within a mouse bearing Huh-7 tumours, revealing its accumulation and retention in the tumour regions. In an earlier investigation, Jin et al. [71] explored the possibility of radioimmuno-targeting CSCs using PET or SPECT. They delved into the feasibility of in vivo radionuclide imaging of CSCs by employing Iodine-125-labelled ANC9C5, an anti-human CD133 antibody, in colon carcinoma xenografts. While they did not achieve an ideal biodistribution profile, autoradiography demonstrated overlapping distribution of 125I-labelled ANC9C5 with CD133 immunohistochemistry expression in many regions. Efforts to enhance accuracy, the biocompatibility of tracers, as well as advancements in techniques, imaging resolution, and contrast are ongoing. For instance, a recent development includes a novel and highly specific 68Ga peptide-based PET imaging agent designed for CD133 imaging in colorectal cancer by Liu et al. [72].
MRI is a non-invasive imaging method renowned for its robustness and high spatial resolution. One notable advantage of MRI is its independence from radioactive isotopes, making it particularly suitable for longitudinal studies. To date, various contrast agents, including superparamagnetic or paramagnetic nanoparticles, ultrasmall superparamagnetic nanoparticles, fluorine, gadolinium, and specific reporter genes, have been employed for CSC imaging.
Among these, ultrasmall superparamagnetic iron oxide (SPIO) or superparamagnetic iron oxide labels are commonly utilised for cell tracking via magnetic resonance. Notably, SPIO has yielded promising outcomes, successfully detecting and monitoring transplanted glioblastoma CSCs in vitro [73]. In a recent investigation by Sun et al. [74], researchers explored the use of SPIO-loaded glioblastoma stem cells to enhance radiation therapy and act as a contrast agent for imaging purposes. Their findings showcased heightened DNA damage in glioblastoma cells when combined with radiation therapy. Further development and testing may lead to their incorporation as theranostic agents, serving dual roles as an MRI-based contrast agent and a radiosensitiser, with potential applications in both treatment and imaging.
4.2. Theranostics
Theranostics is a rapidly evolving field, with several integrated approaches and techniques already being implemented in clinical practice. There is a growing body of literature highlighting the use of alpha particles, including astatine-211, actinium-225 (225Ac), lead-212 (212Pb), and thorium-227, all of which induce challenging-to-repair, clustered DNA double-strand breaks. In clinical settings, 177Lu remains the prevailing radioisotope of choice. Beyond safety data on routinely used alpha-emitting radioisotopes, such as radium-223 (223Ra), clinical trials involving 212Pb-DOTAMTATE have exhibited promising safety profiles. The ongoing emphasis on developing new radiopharmaceuticals and the recent advancements in receptor-targeted strategies present an exciting outlook for the theranostics of CSCs.
Recently, new therapeutic strategies such as CSC-targeted therapies and combination treatments have been employed to investigate the effective treatment of CSCs (Figure 2). One of the most promising methods involves the use of monoclonal antibodies (mAbs). Several noteworthy preclinical investigations in the field of theranostics encompass the use of 125I/225At-labelled anti-CXCR4 monoclonal antibody for acute myeloid leukaemia [75], 177Lu-labelled anti-EGFR mAb for breast cancer [76], 111In-NLS-CSL360 for leukaemia [77], 188Re-6D2 for melanoma [78], and 111In-DTPA-CD166tp-G18C for colorectal cancer [79]. While these studies are currently in the preclinical phase, the ongoing emphasis on developing novel radiopharmaceuticals and the recent advancements in receptor-targeted strategies present an exciting outlook for the application of theranostics in CSCs.
Numerous potential markers exist for identifying CSCs across various types of cancers [80]. These CSCs express a range of cell-surface markers, including CD133, CD44, and CD24, as well as ABC transporters such as ABCG2 and ABCB5. These biomarkers are essential for the isolation and analysis of the biological and physiological characteristics specific to CSC populations. Nevertheless, it is important to note that these biomarkers are not exclusive to CSCs; they also mark populations enriched with CSCs and may be present in normal tissue stem cells, rendering them unsuitable for targeted strategies [81].
Efforts have been undertaken to utilise CD133 as a target in cancer theranostics. In a study by Sun et al. [82], an RNA aptamer was linked to cationic liposomes and loaded with siRNA and paclitaxel. The delivery of siRNA and paclitaxel to glioma stem cells induced apoptosis, while increasing the differentiation of CD133+ glioma stem cells into non-stem cells improved the overall survival rate in mice bearing glioma tumours.
The CXCR4 is recognised for its elevated expression in CSCs across more than 20 human tumour types, and it is associated with tumorigenicity, angiogenesis, invasion, and resistance to chemotherapy [83]. High levels of CXCR4 expression are significantly linked to distant metastasis, poor overall survival, and reduced disease-free survival in various cancer types, including breast, prostate, pancreatic, and lung cancers, as well as lymphoma and leukaemia, making it an independent prognostic factor in these diseases [84].
The clinical application of 68Ga-pentixafor for CXCR4-directed PET imaging and the investigation of radionuclide therapies involving 177Lu- and 90Y-Pentixather have shown promising potential in theranostics. Additionally, a newly developed CXCR4 antagonist, PRX177561, has demonstrated a significant reduction in glioblastoma tumour growth and the potential to enhance the effects of anticancer chemotherapy and radiotherapy. In addition, PRX177561 has extended disease-free survival and overall survival in mice carrying orthotopic brain xenografts [85].
Further research is essential to validate and explore the therapeutic potential of these markers and factors in various cancer types.
4.3. Nanotheranostics
Nanotheranostics represents an innovative fusion of therapeutic and diagnostic imaging within a single agent, seamlessly connected and integrated by nanoparticles [86]. In recent years, advancements in nanotechnology-based approaches have paved the way for targeted therapy breakthroughs, offering a fresh approach to tackling chemo- and radioresistance. Nanoparticles can be tailored to specifically address the fundamental characteristics and mechanisms of CSCs, thereby paving the way for more efficacious treatment strategies. Nevertheless, numerous challenges persist in the widespread adoption of these formulations. With the expanding horizons of nanotechnology, the potential of nanotheranostics, a convergence of diagnostics and therapy, holds great promise [87]. For instance, consider the utilisation of RNAi-delivering nanoplatforms for targeting stem cells in CSC-focused anticancer therapy [88].
Multifunctional hybrid nanoparticles represent a burgeoning class of drug delivery carriers capable of facilitating targeted therapy at specific sites, mitigating side effects, and delivering combined therapies concurrently to combat drug resistance. Smiley et al. [89] recently explored initial CD133 labelling of nanoparticles for glioblastoma. The study demonstrated a noticeable trend in enhanced CSC elimination in vitro when compared to CD133-deficient nanoparticles. The incorporation of the PET radiotracer, 89Zr, into these multifunctional CD133-targetable nanoparticles introduces exciting possibilities for theranostic applications, allowing real-time in vivo tracking of their fate.
4.4. Machine Learning
In recent years, the scientific landscape for image analysis has been swiftly revolutionised by the rapid advancements in machine learning (ML) and artificial intelligence (AI). The progress in these fields has been greatly expedited by the emergence of high-performance computers [90]. Research endeavours have started to incorporate ML techniques in the characterisation of histopathological image analysis, particularly in the context of in vivo CSC imaging [91].
Most clinical AI studies are conducted on existing retrospective datasets, focusing on improving algorithm performance for both internal and external datasets. In a recent study by Yang et al. [92], a deep learning radiomic model based on MRI data was developed and validated from data collected retrospectively for 257 patients. This model exhibited an area under the curve (AUC) of 0.726 and 0.790 in two external validation cohorts. The study investigated Cytokeratin 19 (CK19), a stem cell marker associated with the progenitor subtype, which has been linked to heightened recurrence and therapy resistance in hepatocellular carcinoma.
Other investigations have explored AI-driven modelling to predict individual patient responses based on imaging data. Wei et al. [93] developed a gastric CSC-related score using a series of ML algorithms, enabling the prediction of immunotherapy response with an AUC of 0.736. Most recently, Zhou et al. [94] conducted research into novel immunofluorescence and AI-based comprehensive analyses to quantify and spatially analyse CD8+ T cells and CD133+ CSCs, aiming to predict the survival outcomes of patients with pancreatic adenocarcinoma.
5. Final Thoughts
Today, the research and clinical communities are still trying to elucidate the complex mechanisms behind one of the main causes of treatment failure in radiation oncology—tumour repopulation during treatment. Thirty-five years have passed since the landmark publication authored by R. Withers et al. on The hazard of accelerated tumour clonogen repopulation during radiotherapy (Acta Oncologica 27:131) [18], a paper that discusses accelerated tumour regrowth in head and neck carcinomas, and presents quantitative parameters for the clonogen doubling rate and the onset of repopulation during radiotherapy.
Currently, there is broad evidence towards the existence of cancer stem cells that are responsible for tumour repopulation, dissemination, and distant metastases in most solid cancers. For a successful management of most cancers, CSCs need identification through specific imaging techniques that can assist novel technologies to target and eradicate them.
Research is currently in the preclinical phase, and determining the suitability of these findings for conventional clinical application will necessitate further extensive preclinical studies and clinical trials. Recent advancements in preclinical oncology, particularly in immunology, are expected to identify novel molecular targets that may be utilised in future theranostic approaches such as those highlighted in the review. Further investigations are required in all directions of cancer stem cell research to shed more light on their complex properties and to identify the optimal therapies for their control.
Author Contributions
Conceptualization, L.G.M.; methodology, L.G.M., M.D. and E.B.; writing—original draft preparation, L.G.M., M.D. and E.B.; writing—review and editing, L.G.M., M.D. and E.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is available from the authors upon request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef]
- Walcher, L.; Kistenmacher, A.K.; Suo, H.; Kitte, R.; Dluczek, S.; Strauß, A.; Blaudszun, A.R.; Yevsa, T.; Fricke, S.; Kossatz-Boehlert, U. Cancer Stem Cells-Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front. Immunol. 2020, 11, 1280. [Google Scholar] [CrossRef]
- Yu, Z.; Pestell, T.G.; Lisanti, M.P.; Pestell, R.G. Cancer stem cells. Int. J. Biochem. Cell Biol. 2012, 44, 2144–2151. [Google Scholar] [CrossRef]
- Baccelli, I.; Trumpp, A. The evolving concept of cancer and metastasis stem cells. J. Cell Biol. 2012, 198, 281–293. [Google Scholar] [CrossRef]
- Reid, P.; Marcu, L.G.; Olver, I.; Moghaddasi, L.; Staudacher, A.H.; Bezak, E. Diversity of cancer stem cells in head and neck carcinomas: The role of HPV in cancer stem cell heterogeneity, plasticity and treatment response. Radiother. Oncol. 2019, 135, 1–12. [Google Scholar] [CrossRef]
- Gong, L.P.; Chen, J.N.; Dong, M.; Xiao, Z.D.; Feng, Z.Y.; Pan, Y.H.; Zhang, Y.; Du, Y.; Zhang, J.Y.; Bi, Y.H.; et al. Epstein-Barr virus-derived circular RNA LMP2A induces stemness in EBV-associated gastric cancer. EMBO Rep. 2020, 21, e49689. [Google Scholar] [CrossRef]
- Yasui, M.; Kunita, A.; Numakura, S.; Uozaki, H.; Ushiku, T.; Fukayama, M. Cancer stem cells in Epstein-Barr virus-associated gastric carcinoma. Cancer Sci. 2020, 111, 2598–2607. [Google Scholar] [CrossRef]
- Al Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Borovski, T.; De Sousa EMelo, F.; Vermeulen, L.; Medema, J.P. Cancer stem cell niche: The place to be. Cancer Res. 2011, 71, 634–639. [Google Scholar] [CrossRef]
- Ju, F.; Atyah, M.M.; Horstmann, N.; Gul, S.; Vago, R.; Bruns, C.J.; Zhao, Y.; Dong, Q.Z.; Ren, N. Characteristics of the cancer stem cell niche and therapeutic strategies. Stem Cell Res. Ther. 2022, 13, 233. [Google Scholar] [CrossRef]
- Paul, R.; Dorsey, J.F.; Fan, Y. Cell plasticity, senescence, and quiescence in cancer stem cells: Biological and therapeutic implications. Pharmacol. Ther. 2022, 231, 107985. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Withers, H.R. Treatment-Induced Accelerated Human Tumour Growth. Semin. Radiat. Oncol. 1993, 3, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Dörr, W. Three A’s of repopulation during fractionated irradiation of squamous epithelia: Asymmetry loss, acceleration of stem-cell divisions and abortive divisions. Int. J. Radiat. Biol. 1997, 72, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Marcu, L.; van Doorn, T.; Olver, I. Modelling of post irradiation accelerated repopulation in squamous cell carcinomas. Phys. Med. Biol. 2004, 49, 3676–3779. [Google Scholar] [CrossRef]
- Harriss-Phillips, W.M.; Bezak, E.; Yeoh, E.K. Monte Carlo radiotherapy simulations of accelerated repopulation and reoxygenation for hypoxic head and neck cancer. Br. J. Radiol. 2011, 84, 903–918. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Levy, D. Role of symmetric and asymmetric division of stem cells in developing drug resistance. Proc. Natl. Acad. Sci. USA 2010, 107, 16766–16771. [Google Scholar] [CrossRef]
- Withers, H.R.; Taylor, J.M.; Maciejewski, B. The hazard of accelerated tumor clonogen repopulation during radiotherapy. Acta Oncol. 1988, 27, 131–146. [Google Scholar] [CrossRef]
- Lagadec, C.; Vlashi, E.; Alhiyari, Y.; Phillips, T.M.; Bochkur Dratver, M.; Pajonk, F. Radiation-induced Notch signaling in breast cancer stem cells. Int. J. Radiat. Oncol. Biol. Phys. 2013, 87, 609–618. [Google Scholar] [CrossRef]
- Bae, J.H.; Park, S.H.; Yang, J.H.; Yang, K.; Yi, J.M. Stem cell-like gene expression signature identified in ionizing radiation-treated cancer cells. Gene 2015, 572, 285–291. [Google Scholar] [CrossRef]
- Martins-Neves, S.R.; Cleton-Jansen, A.M.; Gomes, C.M.F. Therapy-induced enrichment of cancer stem-like cells in solid human tumors: Where do we stand? Pharmacol. Res. 2018, 137, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Krause, M.; Dubrovska, A.; Linge, A.; Baumann, M. Cancer stem cells: Radioresistance, prediction of radiotherapy outcome and specific targets for combined treatments. Adv. Drug Deliv. Rev. 2017, 109, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Marzagalli, M.; Fontana, F.; Raimondi, M.; Limonta, P. Cancer Stem Cells-Key Players in Tumour Relapse. Cancers 2021, 13, 376. [Google Scholar] [CrossRef] [PubMed]
- Le, N.H.; Franken, P.; Fodde, R. Tumour-stroma interactions in colorectal cancer: Converging on beta-catenin activation and cancer stemness. Br. J. Cancer 2008, 98, 1886–1893. [Google Scholar] [CrossRef] [PubMed]
- Arnold, C.R.; Mangesius, J.; Skvortsova, I.I.; Ganswindt, U. The Role of Cancer Stem Cells in Radiation Resistance. Front. Oncol. 2020, 10, 164. [Google Scholar] [CrossRef] [PubMed]
- Olivares-Urbano, M.A.; Grinan-Lison, C.; Marchal, J.A.; Nunez, M.I. CSC Radioresistance: A Therapeutic Challenge to Improve Radiotherapy Effectiveness in Cancer. Cells 2020, 9, 1651. [Google Scholar] [CrossRef] [PubMed]
- Lagadec, C.; Vlashi, E.; Della Donna, L.; Meng, Y.; Dekmezian, C.; Kim, K.; Pajonk, F. Survival and self-renewing capacity of breast cancer initiating cells during fractionated radiation treatment. Breast Cancer Res. 2010, 12, R13. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, M.; Luo, J.; Zhou, H. Radiotherapy targeting cancer stem cells “awakens” them to induce tumour relapse and metastasis in oral cancer. Int. J. Oral Sci. 2020, 12, 19. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Brueckmann, I.; Scheel, C.; Kaestli, A.J.; Wiggins, P.A.; Rodrigues, L.O.; Brooks, M.; Reinhardt, F.; Su, Y.; Polyak, K.; et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc. Natl. Acad. Sci. USA 2011, 108, 7950–7955. [Google Scholar] [CrossRef]
- Gao, X.; Sishc, B.J.; Nelson, C.B.; Hahnfeldt, P.; Bailey, S.M.; Hlatky, L. Radiation-Induced Reprogramming of Pre-Senescent Mammary Epithelial Cells Enriches Putative CD44(+)/CD24(-/low) Stem Cell Phenotype. Front. Oncol. 2016, 6, 138. [Google Scholar] [CrossRef] [PubMed]
- Lagadec, C.; Vlashi, E.; Della Donna, L.; Dekmezian, C.; Pajonk, F. Radiation-induced reprogramming of breast cancer cells. Stem Cells 2012, 30, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogenet 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [PubMed]
- Peitzsch, C.; Kurth, I.; Kunz-Schughart, L.; Baumann, M.; Dubrovska, A. Discovery of the cancer stem cell related determinants of radioresistance. Radiother. Oncol. 2013, 108, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Casal, R.; Bhattacharya, C.; Ganesh, N.; Bailey, L.; Basse, P.; Gibson, M.; Epperly, M.; Levina, V. Non-small cell lung cancer cells survived ionizing radiation treatment display cancer stem cell and epithelial-mesenchymal transition phenotypes. Mol. Cancer 2013, 12, 94. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Jeong, E.K.; Ju, M.K.; Jeon, H.M.; Kim, M.Y.; Kim, C.H.; Park, H.G.; Han, S.I.; Kang, H.S. Induction of metastasis, cancer stem cell phenotype, and oncogenic metabolism in cancer cells by ionizing radiation. Mol. Cancer 2017, 16, 10. [Google Scholar] [CrossRef] [PubMed]
- Phillips, T.M.; McBride, W.H.; Pajonk, F. The response of CD24(-/low)/CD44+ breast cancer-initiating cells to radiation. J. Natl. Cancer Inst. 2006, 98, 1777–1785. [Google Scholar] [CrossRef]
- Wang, J.Z.; Li, X.A. Impact of tumor repopulation on radiotherapy planning. Int. J. Radiat. Oncol. Biol. Phys. 2005, 61, 220–227. [Google Scholar] [CrossRef]
- Chiblak, S.; Tang, Z.; Campos, B.; Gal, Z.; Unterberg, A.; Debus, J.; Herold-Mende, C.; Abdollahi, A. Radiosensitivity of Patient-Derived Glioma Stem Cell 3-Dimensional Cultures to Photon, Proton, and Carbon Irradiation. Int. J. Radiat. Oncol. Biol. Phys. 2016, 95, 112–119. [Google Scholar] [CrossRef]
- Davis, A.J.; Tannock, J.F. Repopulation of tumour cells between cycles of chemotherapy: A neglected factor. Lancet Oncol. 2000, 1, 86–93. [Google Scholar] [CrossRef]
- Marcu, L.; Bezak, E. Modelling of tumour repopulation after chemotherapy. Australas. Phys. Eng. Sci. Med. 2010, 33, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Marcu, L.G. Tumour repopulation and the role of abortive division in squamous cell carcinomas during chemotherapy. Cell Prolif. 2014, 47, 318–325. [Google Scholar] [CrossRef]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [PubMed]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Dabritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Marcu, L.; Bezak, E.; Olver, I.; van Doorn, T. Tumour resistance to cisplatin: A modelling approach. Phys. Med. Biol. 2005, 50, 93–102. [Google Scholar] [CrossRef]
- Nallasamy, P.; Nimmakayala, R.K.; Parte, S.; Are, A.C.; Batra, S.K.; Ponnusamy, M.P. Tumour microenvironment enriches the stemness features: The architectural event of therapy resistance and metastasis. Mol. Cancer 2022, 21, 225. [Google Scholar] [CrossRef] [PubMed]
- Ohta, Y.; Fujii, M.; Takahashi, S.; Takano, A.; Nanki, K.; Matano, M.; Hanyu, H.; Saito, M.; Shimokawa, M.; Nishikori, S.; et al. Cell-matrix interface regulates dormancy in human colon cancer stem cells. Nature 2022, 608, 784–794. [Google Scholar] [CrossRef]
- Xie, X.P.; Laks, D.R.; Sun, D.; Ganbold, M.; Wang, Z.; Pedraza, A.M.; Bale, T.; Tabar, V.; Brennan, C.; Zhou, X.; et al. Quiescent human glioblastoma cancer stem cells drive tumor initiation, expansion, and recurrence following chemotherapy. Dev. Cell 2022, 10, 32–46.e8. [Google Scholar] [CrossRef]
- Lin, Q.; Liu, Y.E.; Ren, X.C.; Wang, N.; Chen, X.J.; Wang, D.Y.; Zong, J.; Peng, Y.; Guo, Z.J.; Hu, J. Dose escalation of accelerated hypofractionated three-dimensional conformal radiotherapy (at 3 Gy/fraction) with concurrent vinorelbine and carboplatin chemotherapy in unresectable stage III non-small-cell lung cancer: A phase I trial. Radiat. Oncol. 2013, 8, 201. [Google Scholar] [CrossRef]
- Maguire, J.; Khan, I.; McMenemin, R.; O'Rourke, N.; McNee, S.; Kelly, V.; Peedell, C.; Snee, M. SOCCAR: A randomised phase II trial comparing sequential versus concurrent chemotherapy and radical hypofractionated radiotherapy in patients with inoperable stage III Non-Small Cell Lung Cancer and good performance status. Eur. J. Cancer. 2014, 50, 2939–2949. [Google Scholar] [CrossRef]
- Bütof, R.; Simon, M.; Löck, S.; Troost, E.G.C.; Appold, S.; Krause, M.; Baumann, M. PORTAF—Postoperative radiotherapy of non-small cell lung cancer: Accelerated versus conventional fractionation—Study protocol for a randomized controlled trial. Trials 2017, 18, 608. [Google Scholar] [CrossRef]
- Panteliadou, M.; Giatromanolaki, A.; Touloupidis, S.; Destouni, E.; Tsoutsou, P.G.; Pantelis, P.; Abatzoglou, I.; Sismanidou, K.; Koukourakis, M.I. Treatment of invasive bladder cancer with conformal hypofractionated accelerated radiotherapy and amifostine (HypoARC). Urol. Oncol. 2012, 30, 813–820. [Google Scholar] [CrossRef] [PubMed]
- D’Arcangelo, M.; Todaro, M.; Salvini, J.; Benfante, A.; Colorito, M.L.; D'Incecco, A.; Landi, L.; Apuzzo, T.; Rossi, E.; Sani, S.; et al. Cancer Stem Cells Sensitivity Assay (STELLA) in Patients with Advanced Lung and Colorectal Cancer: A Feasibility Study. PLoS ONE 2015, 10, e0125037. [Google Scholar] [CrossRef] [PubMed]
- Howard, C.M.; Valluri, J.; Alberico, A.; Julien, T.; Mazagri, R.; Marsh, R.; Alastair, H.; Cortese, A.; Griswold, M.; Wang, W.; et al. Analysis of Chemopredictive Assay for Targeting Cancer Stem Cells in Glioblastoma Patients. Transl. Oncol. 2017, 10, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, T.; Howard, C.M.; Yu, A.; Xu, L.; Aziz, K.; Jho, D.; Leonardo, J.; Hameed, M.A.; Karlovits, S.M.; Wegner, R.E.; et al. Cancer Stem Cell Chemotherapeutics Assay for Prospective Treatment of Recurrent Glioblastoma and Progressive Anaplastic Glioma: A Single-Institution Case Series. Transl. Oncol. 2020, 13, 100755. [Google Scholar] [CrossRef]
- Ranjan, T.; Sengupta, S.; Glantz, M.J.; Green, R.M.; Yu, A.; Aregawi, D.; Chaudhary, R.; Chen, R.; Zuccarello, M.; Lu-Emerson, C.; et al. Cancer stem cell assay-guided chemotherapy improves survival of patients with recurrent glioblastoma in a randomized trial. Cell Rep. Med. 2023, 4, 101025. [Google Scholar] [CrossRef] [PubMed]
- Howard, C.M.; Bush S 2nd Zgheib, N.B.; Lirette, S.T.; Cortese, A.; Mollo, A.; Valluri, J.; Claudio, P.P. Cancer Stem Cell Assay for the Treatment of Platinum-Resistant Recurrent Ovarian Cancer. HSOA J. Stem Cells Res. Dev. Ther. 2021, 7, 076. [Google Scholar] [PubMed]
- Yoon, C.; Park, D.J.; Schmidt, B.; Thomas, N.J.; Lee, H.J.; Kim, T.S.; Janjigian, Y.Y.; Cohen, D.J.; Yoon, S.S. CD44 expression denotes a subpopulation of gastric cancer cells in which Hedgehog signaling promotes chemotherapy resistance. Clin. Cancer Res. 2014, 20, 3974–3988. [Google Scholar] [CrossRef]
- Yoon, C.; Cho, S.J.; Aksoy, B.A.; Park, D.J.; Schultz, N.; Ryeom, S.W.; Yoon, S.S. Chemotherapy Resistance in Diffuse-Type Gastric Adenocarcinoma Is Mediated by RhoA Activation in Cancer Stem-Like Cells. Clin. Cancer Res. 2016, 22, 971–983. [Google Scholar] [CrossRef]
- Marcu, L.G.; Moghaddasi, L.; Bezak, E. Cannot Target What Cannot Be Seen: Molecular Imaging of Cancer Stem Cells. Int. J. Mol. Sci. 2023, 24, 1524. [Google Scholar] [CrossRef]
- Seo, Y.; Kim, J.; Park, S.J.; Park, J.J.; Cheon, J.H.; Kim, W.H.; Kim, T.I. Metformin Suppresses Cancer Stem Cells through AMPK Activation and Inhibition of Protein Prenylation of the Mevalonate Pathway in Colorectal Cancer. Cancers 2020, 12, 2554. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.R.; Chan, D.K.; Shank, J.J.; Griffith, K.A.; Fan, H.; Szulawski, R.; Yang, K.; Reynolds, R.K.; Johnston, C.; McLean, K.; et al. Phase II clinical trial of metformin as a cancer stem cell-targeting agent in ovarian cancer. JCI Insight 2020, 5, e133247. [Google Scholar] [CrossRef] [PubMed]
- Available online: www.clinicaltrials.gov (accessed on 12 October 2023).
- Riether, C.; Pabst, T.; Höpner, S.; Bacher, U.; Hinterbrandner, M.; Banz, Y.; Müller, R.; Manz, M.G.; Gharib, W.H.; Francisco, D.; et al. Targeting CD70 with cusatuzumab eliminates acute myeloid leukemia stem cells in patients treated with hypomethylating agents. Nat. Med. 2020, 26, 1459–1467. [Google Scholar] [CrossRef] [PubMed]
- Nacarelli, T.; Fukumoto, T.; Zundell, J.A.; Fatkhutdinov, N.; Jean, S.; Cadungog, M.G.; Borowsky, M.E.; Zhang, R. NAMPT Inhibition Suppresses Cancer Stem-like Cells Associated with Therapy-Induced Senescence in Ovarian Cancer. Cancer Res. 2020, 80, 890–900. [Google Scholar] [CrossRef] [PubMed]
- Takasugi, M.; Yoshida, Y.; Hara, E.; Ohtani, N. The role of cellular senescence and SASP in tumour microenvironment. FEBS J. 2023, 290, 1348–1361. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.B.; Chaffer, C.L.; Weinberg, R.A. Cancer stem cells: Mirage or reality? Nat. Med. 2009, 15, 1010–1012. [Google Scholar] [CrossRef]
- Mahajan, A.; Goh, V.; Basu, S.; Vaish, R.; Weeks, A.J.; Thakur, M.H.; Cook, G.J. Bench to bedside molecular functional imaging in translational cancer medicine: To image or to imagine? Clin. Radiol. 2015, 70, 1060–1082. [Google Scholar] [CrossRef]
- Marcu, L.G.; Moghaddasi, L.; Bezak, E. Imaging of Tumour Characteristics and Molecular Pathways With PET: Developments Over the Last Decade Toward Personalized Cancer Therapy. Int. J. Radiat. Oncol. Biol. Phys. 2018, 102, 1165–1182. [Google Scholar] [CrossRef]
- Hu, K.; Ma, X.; Xie, L.; Zhang, Y.; Hanyu, M.; Obata, H.; Zhang, L.; Nagatsu, K.; Suzuki, H.; Shi, R.; et al. Development of a Stable Peptide-Based PET Tracer for Detecting CD133-Expressing Cancer Cells. ACS Omega 2021, 7, 334–341. [Google Scholar] [CrossRef]
- Jin, Z.H.; Sogawa, C.; Furukawa, T.; Saito, Y.; Aung, W.; Fujibayashi, Y.; Saga, T. Basic studies on radioimmunotargeting of CD133-positive HCT116 cancer stem cells. Mol. Imaging 2012, 11, 445–450. [Google Scholar] [CrossRef]
- Liu, Y.; Yao, X.; Wang, C.; Wang, M.; Wang, Y.; Ye, M.; Liu, Y. Peptide-based 68Ga-PET radiotracer for imaging CD133 expression in colorectal cancer. Nucl. Med. Commun. 2021, 42, 1144–1150. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wei, F.; Liu, A.; Wang, L.; Wang, J.C.; Ren, L.; Liu, W.; Tu, Q.; Li, L.; Wang, J. Cancer stem cell labeling using poly(L-lysine)-modified iron oxide nanoparticles. Biomaterials 2012, 33, 3719–3732. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Joh, D.Y.; Al-Zaki, A.; Stangl, M.; Murty, S.; Davis, J.J.; Baumann, B.C.; Alonso-Basanta, M.; Kaol, G.D.; Tsourkas, A.; et al. Theranostic Application of Mixed Gold and Superparamagnetic Iron Oxide Nanoparticle Micelles in Glioblastoma Multiforme. J. Biomed. Nanotechnol. 2016, 12, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Oriuchi, N.; Aoki, M.; Ukon, N.; Washiyama, K.; Tan, C.; Shimoyama, S.; Nishijima, K.I.; Takahashi, K.; Ito, H.; Ikezoe, T.; et al. Possibility of cancer-stem-cell-targeted radioimmunotherapy for acute myelogenous leukemia using 211At-CXCR4 monoclonal antibody. Sci. Rep. 2020, 10, 6810. [Google Scholar] [CrossRef] [PubMed]
- Al-Ejeh, F.; Shi, W.; Miranda, M.; Simpson, P.T.; Vargas, A.C.; Song, S.; Wiegmans, A.P.; Swarbrick, A.; Welm, A.L.; Brown, M.P.; et al. Treatment of triple-negative breast cancer using anti-EGFR-directed radioimmunotherapy combined with radiosensitizing chemotherapy and PARP inhibitor. J. Nucl. Med. 2013, 54, 913–921. [Google Scholar] [CrossRef]
- Leyton, J.V.; Gao, C.; Williams, B.; Keating, A.; Minden, M.; Reilly, R.M. A radiolabeled antibody targeting CD123(+) leukemia stem cells—Initial radioimmunotherapy studies in NOD/SCID mice engrafted with primary human AML. Leuk. Res. Rep. 2015, 4, 55–59. [Google Scholar] [CrossRef]
- Jandl, T.; Revskaya, E.; Jiang, Z.; Harris, M.; Dorokhova, O.; Tsukrov, D.; Casadevall, A.; Dadachova, E. Melanoma stem cells in experimental melanoma are killed by radioimmunotherapy. Nucl. Med. Biol. 2013, 40, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Weng, D.; Jin, X.; Qin, S.; Lan, X.; Chen, C.; Sun, X.; She, X.; Dong, C.; An, R. Radioimmunotherapy for CD133(+) colonic cancer stem cells inhibits tumor development in nude mice. Oncotarget 2017, 8, 44004–44014. [Google Scholar]
- Ghaderi, F.; Jokar, N.; Gholamrezanezhad, A.; Assadi, M.; Ahmadzadehfar, H. Toward radiotheranostics in cancer stem cells: A promising initial step for tumour eradication. Clin. Translat. Imag. 2021, 9, 561–578. [Google Scholar] [CrossRef]
- Nakanishi, Y.; Seno, H.; Fukuoka, A.; Ueo, T.; Yamaga, Y.; Maruno, T.; Nakanishi, N.; Kanda, K.; Komekado, H.; Kawada, M.; et al. Dclk1 distinguishes between tumor and normal stem cells in the intestine. Nat. Genet. 2013, 45, 98–103. [Google Scholar] [CrossRef]
- Sun, X.; Chen, Y.; Zhao, H.; Qiao, G.; Liu, M.; Zhang, C.; Cui, D.; Ma, L. Dual-modified cationic liposomes loaded with paclitaxel and survivin siRNA for targeted imaging and therapy of cancer stem cells in brain glioma. Drug Deliv. 2018, 25, 1718–1727. [Google Scholar] [CrossRef] [PubMed]
- Hadebe, B.; Sathekge, M.M.; Aldous, C.; Vorster, M. Current Status of 68Ga-Pentixafor in Solid Tumours. Diagnostics 2022, 12, 2135. [Google Scholar] [CrossRef] [PubMed]
- Schottelius, M.; Osl, T.; Poschenrieder, A.; Hoffmann, F.; Beykan, S.; Hänscheid, H.; Schirbel, A.; Buck, A.K.; Kropf, S.; Schwaiger, M.; et al. [177Lu]pentixather: Comprehensive Preclinical Characterization of a First CXCR4-directed Endoradiotherapeutic Agent. Theranostics 2017, 7, 2350–2362. [Google Scholar] [CrossRef] [PubMed]
- Gravina, G.L.; Mancini, A.; Colapietro, A.; Vitale, F.; Vetuschi, A.; Pompili, S.; Rossi, G.; Marampon, F.; Richardson, P.J.; Patient, L.; et al. The novel CXCR4 antagonist, PRX177561, reduces tumor cell proliferation and accelerates cancer stem cell differentiation in glioblastoma preclinical models. Tumour Biol. 2017, 39, 1010428317695528. [Google Scholar] [CrossRef]
- Ladju, R.B.; Ulhaq, Z.S.; Soraya, G.V. Nanotheranostics: A powerful next-generation solution to tackle hepatocellular carcinoma. World J. Gastroenterol. 2022, 28, 176–187. [Google Scholar] [CrossRef]
- Kola, P.; Nagesh, P.K.B.; Roy, P.K.; Deepak, K.; Reis, R.L.; Kundu, S.C.; Mandal, M. Innovative nanotheranostics: Smart nanoparticles based approach to overcome breast cancer stem cells mediated chemo- and radioresistances. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2023, 15, e1876. [Google Scholar] [CrossRef]
- Tang, Y.; Chen, Y.; Zhang, Z.; Tang, B.; Zhou, Z.; Chen, H. Nanoparticle-Based RNAi Therapeutics Targeting Cancer Stem Cells: Update and Prospective. Pharmaceutics 2021, 13, 2116. [Google Scholar] [CrossRef]
- Smiley, S.B.; Yun, Y.; Ayyagari, P.; Shannon, H.E.; Pollok, K.E.; Vannier, M.W.; Das, S.K.; Veronesi, M.C. Development of CD133 Targeting Multi-Drug Polymer Micellar Nanoparticles for Glioblastoma—In Vitro Evaluation in Glioblastoma Stem Cells. Pharm. Res. 2021, 38, 1067–1079. [Google Scholar] [CrossRef]
- Koh, D.M.; Papanikolaou, N.; Bick, U.; Illing, R.; Kahn, C.E., Jr.; Kalpathi-Cramer, J.; Matos, C.; Martí-Bonmatí, L.; Miles, A.; Mun, S.K.; et al. Artificial intelligence and machine learning in cancer imaging. Commun. Med. 2022, 2, 133. [Google Scholar] [CrossRef]
- Zhang, Z.; Ishihata, H.; Maruyama, R.; Kasai, T.; Kameda, H.; Sugiyama, T. Deep Learning of Phase-Contrast Images of Cancer Stem Cells Using a Selected Dataset of High Accuracy Value Using Conditional Generative Adversarial Networks. Int. J. Mol. Sci. 2023, 24, 5323. [Google Scholar] [CrossRef]
- Yang, F.; Wan, Y.; Xu, L.; Wu, Y.; Shen, X.; Wang, J.; Lu, D.; Shao, C.; Zheng, S.; Niu, T.; et al. MRI-Radiomics Prediction for Cytokeratin 19-Positive Hepatocellular Carcinoma: A Multicenter Study. Front. Oncol. 2021, 11, 672126. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Chen, M.; Deng, W.; Bie, L.; Ma, Y.; Zhang, C.; Liu, K.; Shen, W.; Wang, S.; Yang, C.; et al. Characterization of gastric cancer stem-like molecular features, immune and pharmacogenomic landscapes. Brief. Bioinform. 2022, 23, bbab386. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Man, Q.; Li, X.; Xie, Y.; Hou, X.; Wang, H.; Yan, J.; Wei, X.; Bai, W.; Liu, Z.; et al. Artificial intelligence-based comprehensive analysis of immune-stemness-tumor budding profile to predict survival of patients with pancreatic adenocarcinoma. Cancer Biol. Med. 2023, 20, 196–217. [Google Scholar] [CrossRef] [PubMed]
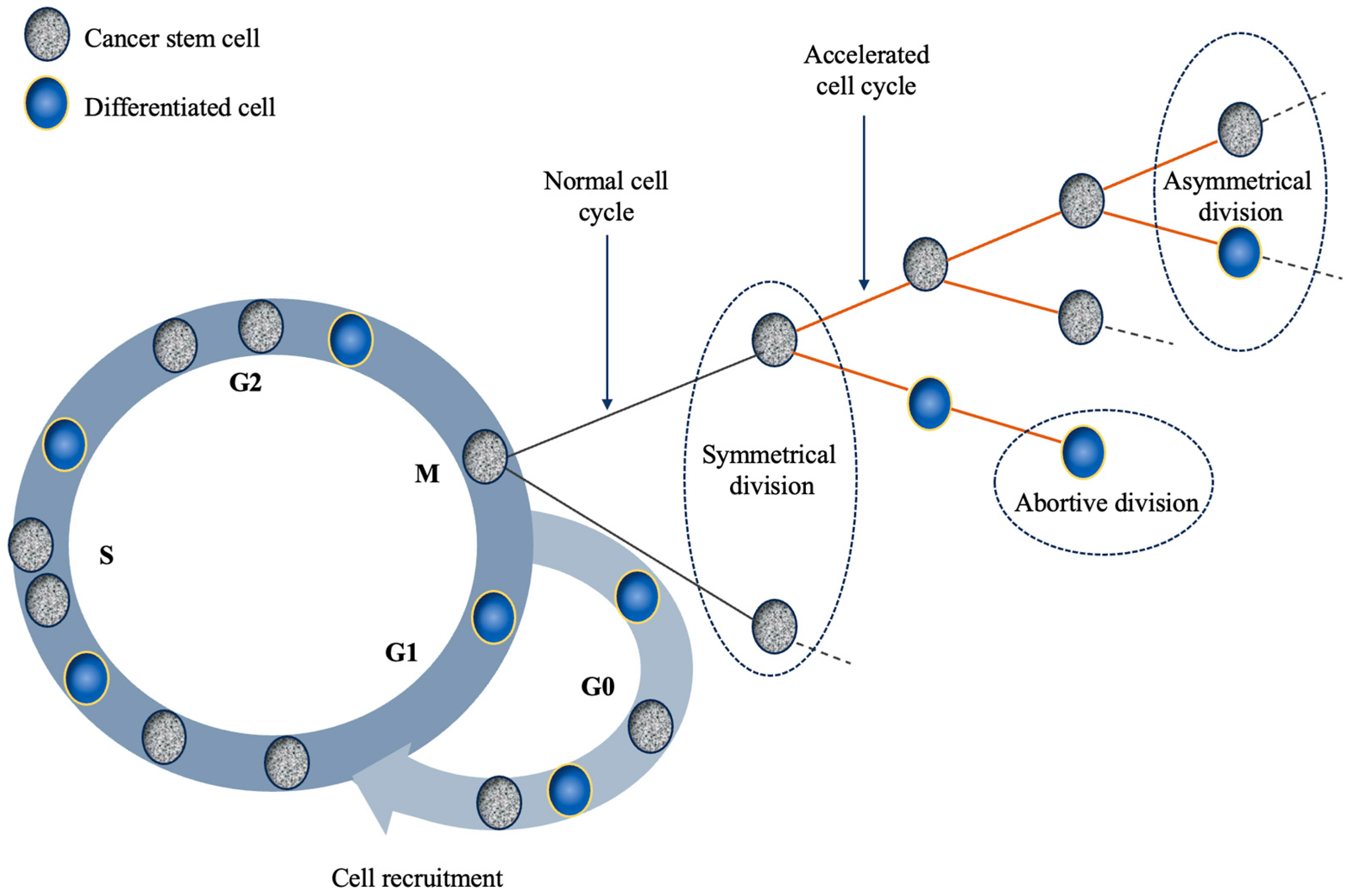
Figure 1.
Schematic representation of the main mechanisms responsible for tumour repopulation during therapy. The four main mechanisms discussed in the text that are depicted in this illustration are the following: (1) cell recruitment: G0 cells re-enter the cell cycle; (2) symmetrical division: a stem cell divides into two stem cells; (3) abortive division: non-stem cells exhibit finite proliferative ability; (4) accelerated division: stem cells shorten the duration of their mitotic cycle.
Figure 1.
Schematic representation of the main mechanisms responsible for tumour repopulation during therapy. The four main mechanisms discussed in the text that are depicted in this illustration are the following: (1) cell recruitment: G0 cells re-enter the cell cycle; (2) symmetrical division: a stem cell divides into two stem cells; (3) abortive division: non-stem cells exhibit finite proliferative ability; (4) accelerated division: stem cells shorten the duration of their mitotic cycle.
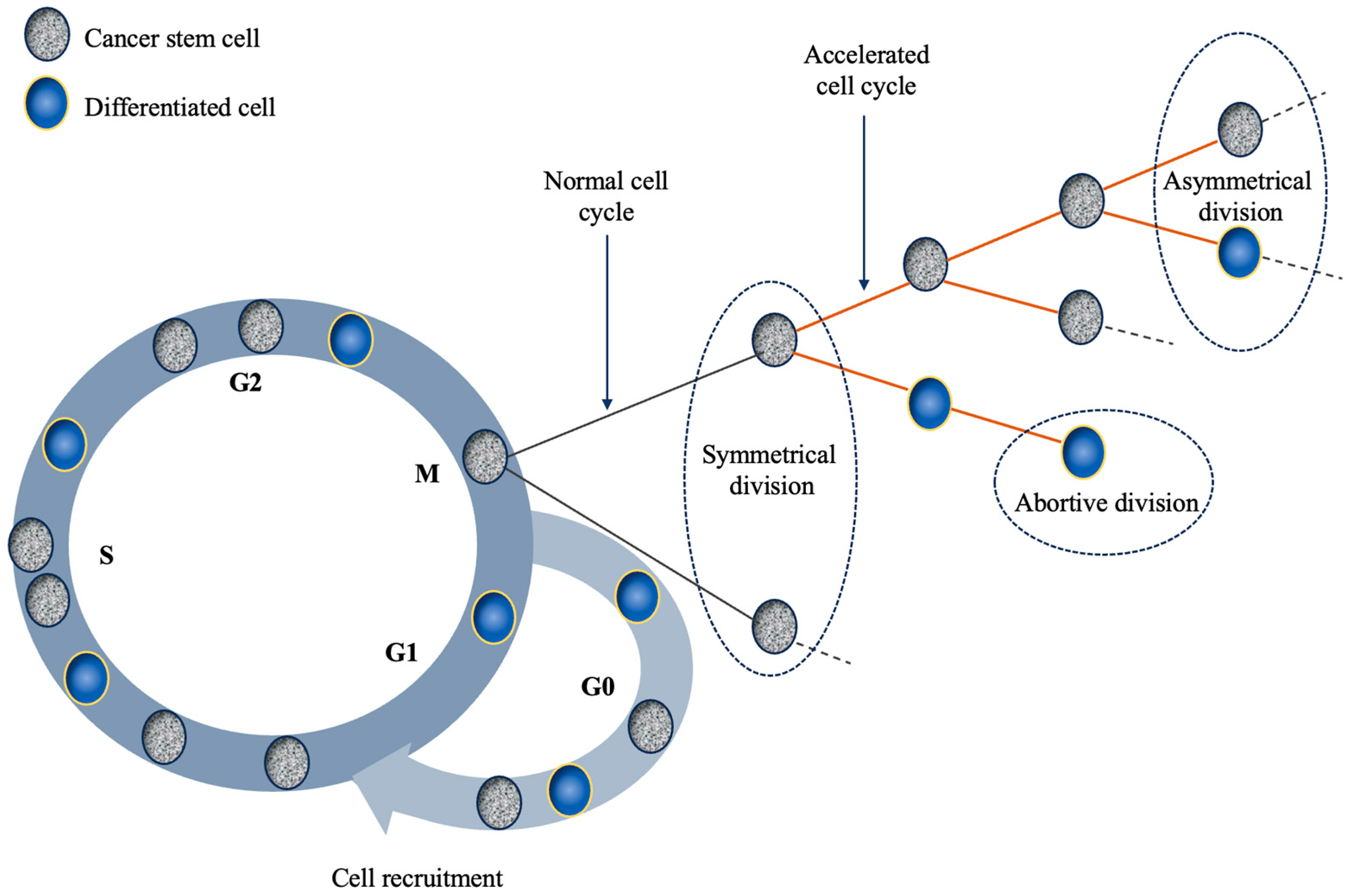
Figure 2.
Diagram illustrating the overlap of future CSC management techniques. Initiatives have commenced to address CSC by employing targeting cell-surface markers or monoclonal antibodies, coupled with progress in the delivery of nanoparticles and radiopharmaceuticals.
Figure 2.
Diagram illustrating the overlap of future CSC management techniques. Initiatives have commenced to address CSC by employing targeting cell-surface markers or monoclonal antibodies, coupled with progress in the delivery of nanoparticles and radiopharmaceuticals.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Marcu, L.G.; Dell’Oro, M.; Bezak, E. Opportunities in Cancer Therapies: Deciphering the Role of Cancer Stem Cells in Tumour Repopulation. Int. J. Mol. Sci. 2023, 24, 17258. https://doi.org/10.3390/ijms242417258
AMA Style
Marcu LG, Dell’Oro M, Bezak E. Opportunities in Cancer Therapies: Deciphering the Role of Cancer Stem Cells in Tumour Repopulation. International Journal of Molecular Sciences. 2023; 24(24):17258. https://doi.org/10.3390/ijms242417258
Chicago/Turabian StyleMarcu, Loredana G., Mikaela Dell’Oro, and Eva Bezak. 2023. "Opportunities in Cancer Therapies: Deciphering the Role of Cancer Stem Cells in Tumour Repopulation" International Journal of Molecular Sciences 24, no. 24: 17258. https://doi.org/10.3390/ijms242417258
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.