
Granulocyte-Macrophage-Colony-Stimulating-Factor Combined with Prostaglandin E1 Create Dendritic Cells of Leukemic Origin from AML Patients’ Whole Blood and Whole Bone Marrow That Mediate Antileukemic Processes after Mixed Lymphocyte Culture

 , ,
, ,
Abstract
:1. Introduction
1.1. Acute Myeloid Leukemia
1.2. The Physiologic and Leukemic Bone Marrow Niche
1.3. Standard Treatment and Advances in AML Therapy
1.4. DC/DCleu-Based Therapy
1.5. Aim of the Study
- Composition of immune cells in uncultured (as well as after mixed lymphocyte culture (MLC) with Kit-M-treated vs. untreated) WB and WBM
- DC (subsets) from Kit-M-treated vs. untreated leukemic WB and WBM
- Platelet counts in WB and WBM DC culture supernatants under the influence of Kit-M vs. GM-CSF
- (Leukemia-specific/antileukemic) immunoreactive cells (activating/inhibitory cells of the adaptive and innate immune system: NK-, CIK-, iNKT- and T-cell subtypes) were specified using degranulation (DEG), intracellular cytokine (INCYT) as well as cytotoxicity (CTX) assays in uncultured WB and WBM and after MLC with Kit-M-treated vs. untreated WB and WBM
- Differences of results obtained in WB vs. WBM
- Correlation analyses of ex vivo generated data with patients’ allocation to clinical risk groups
2. Results
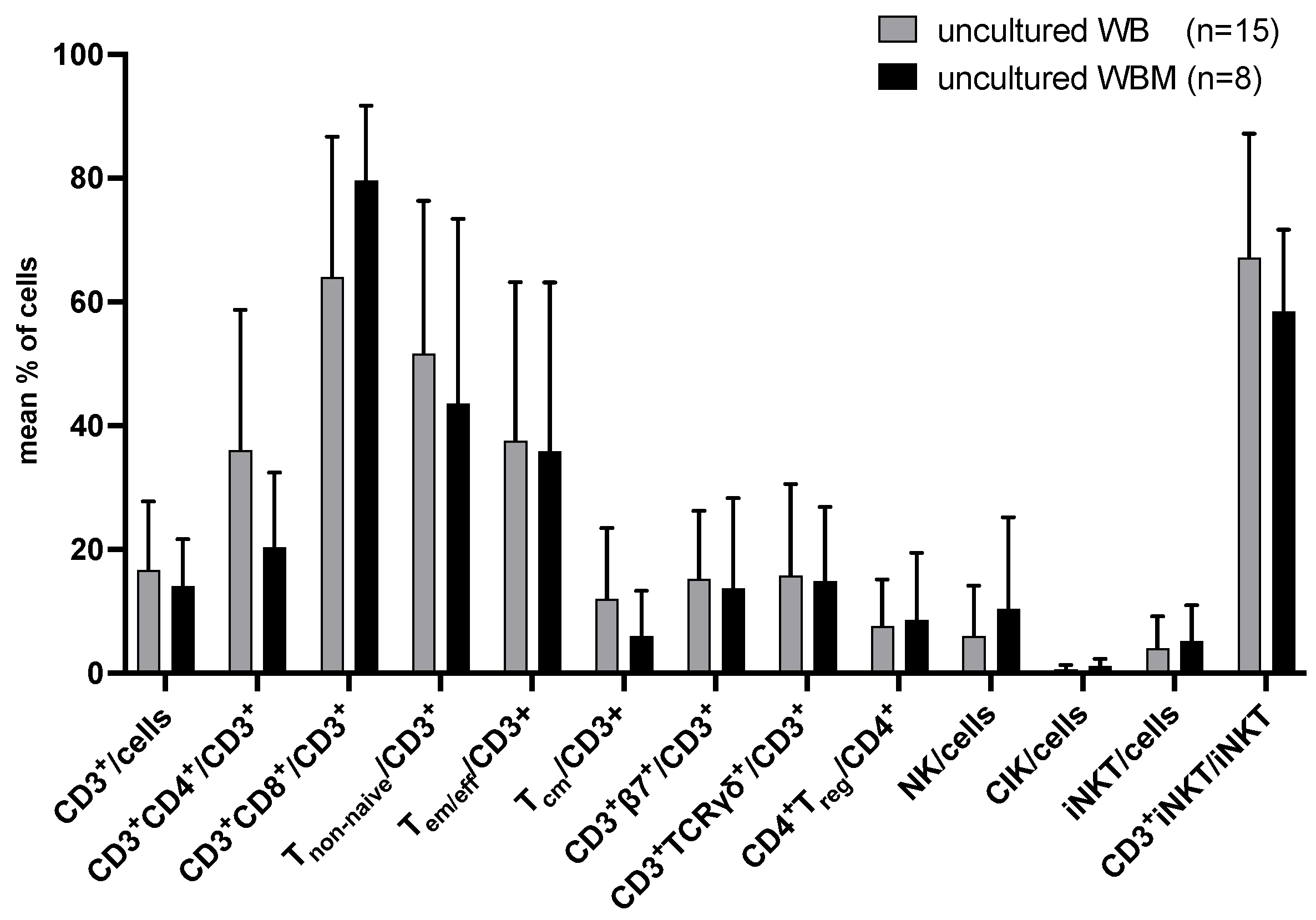
2.1. Uncultured WB and WBM
2.1.1. Composition of T and Innate Immune Cells in Uncultured Leukemic WB and WBM
2.1.2. Composition of (Leukemia-Specific) Degranulating or Intracellularly IFNγ- (and TNFα-) Producing Immunoreactive Cells in Uncultured WB and WBM
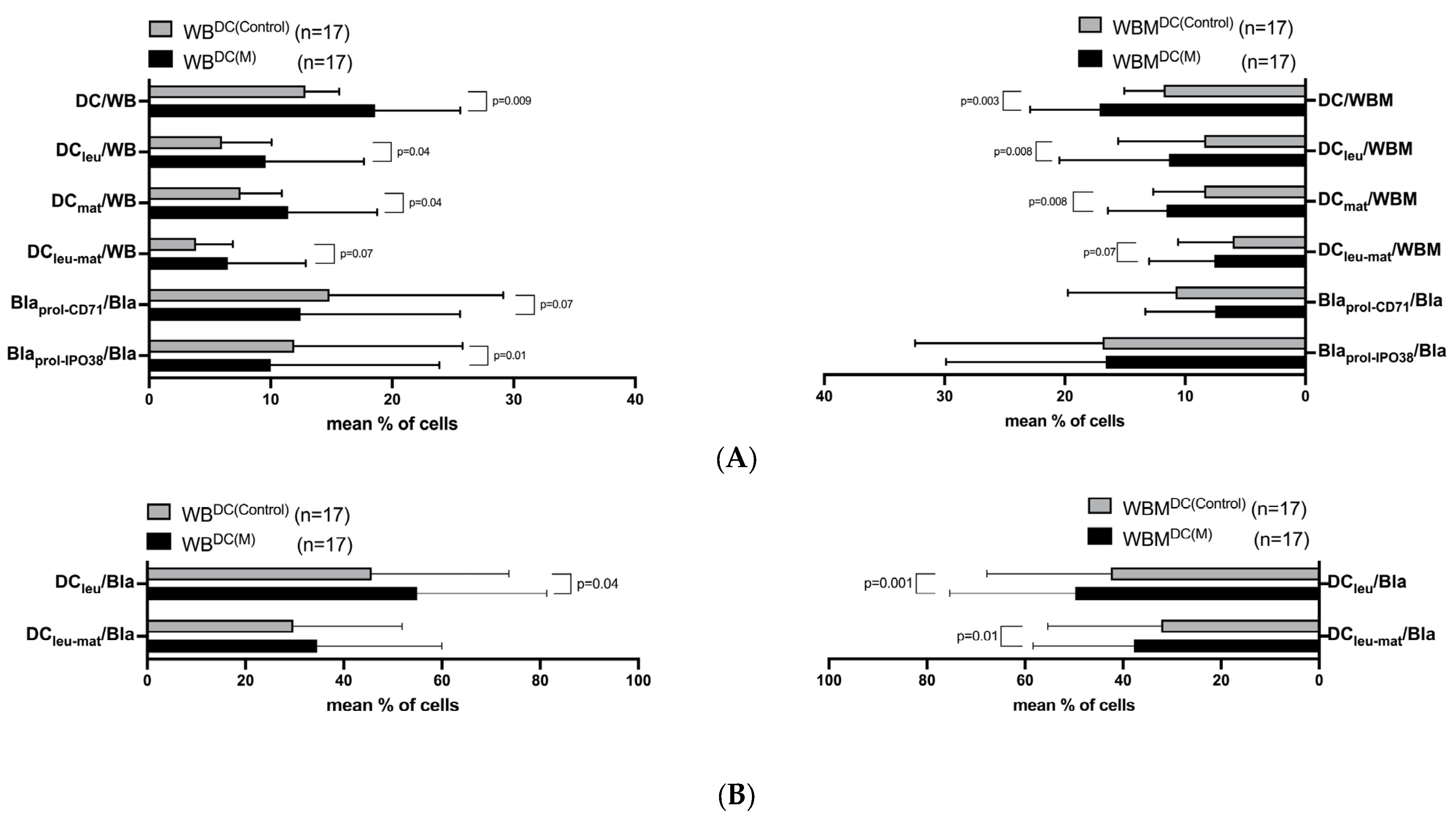
2.2. Effects of Kit-M on the Generation of Mature DC/DCleu from Leukemic WB and WBM
2.3. Effects of Kit-M-Treated WB and WBM on Immunoreactive Cells before and after T Cell-Enriched Mixed Lymphocyte Culture (MLC)
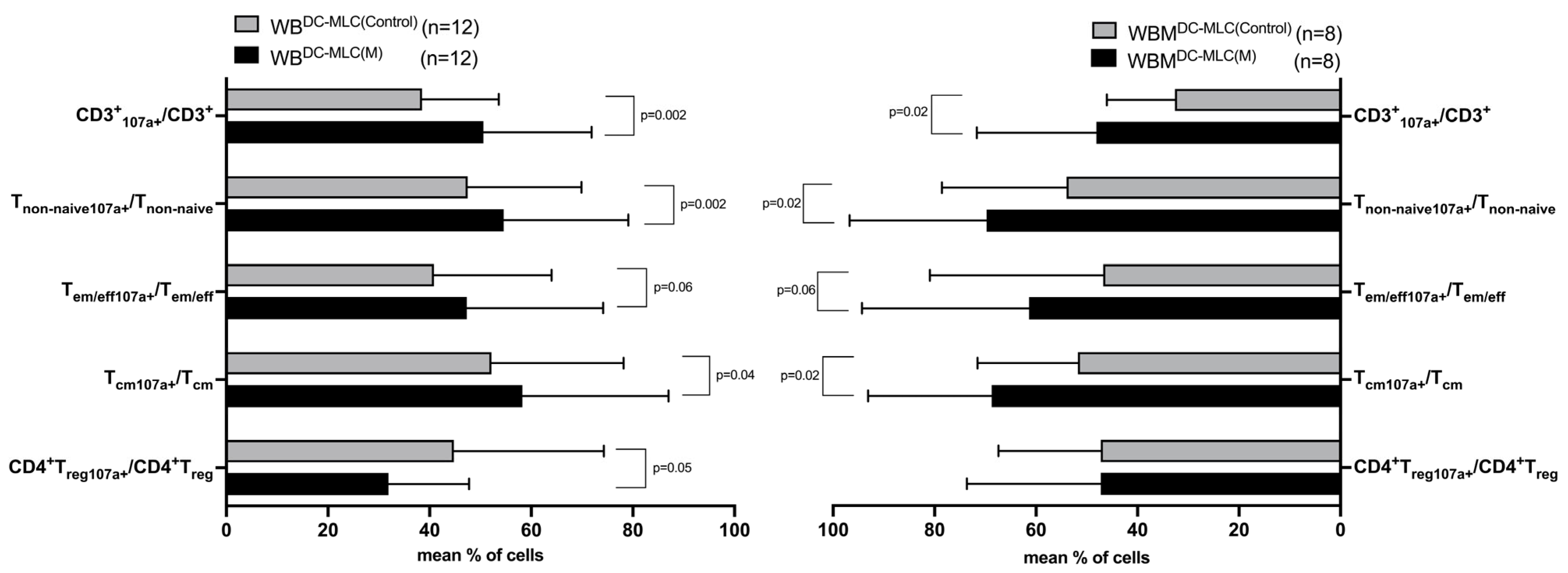
2.4. Effects of Kit-M-Treated WB and WBM on the Degranulation Activity of Immunoreactive Cells after MLC
2.5. Effects of Kit-M-Treated WB and WBM on the Intracellular IFNγ (and TNFα) Production of Immunoreactive Cells after MLC
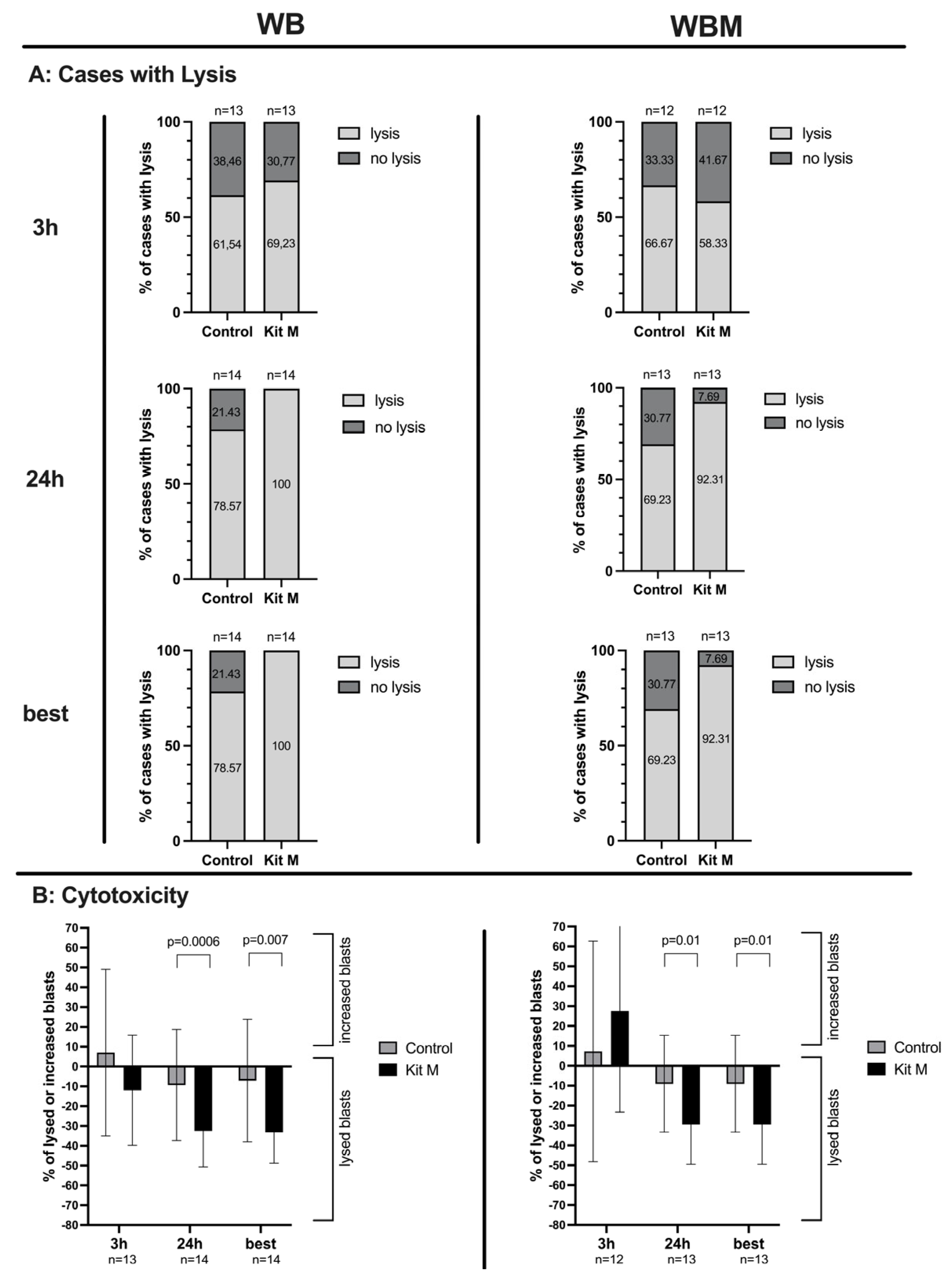
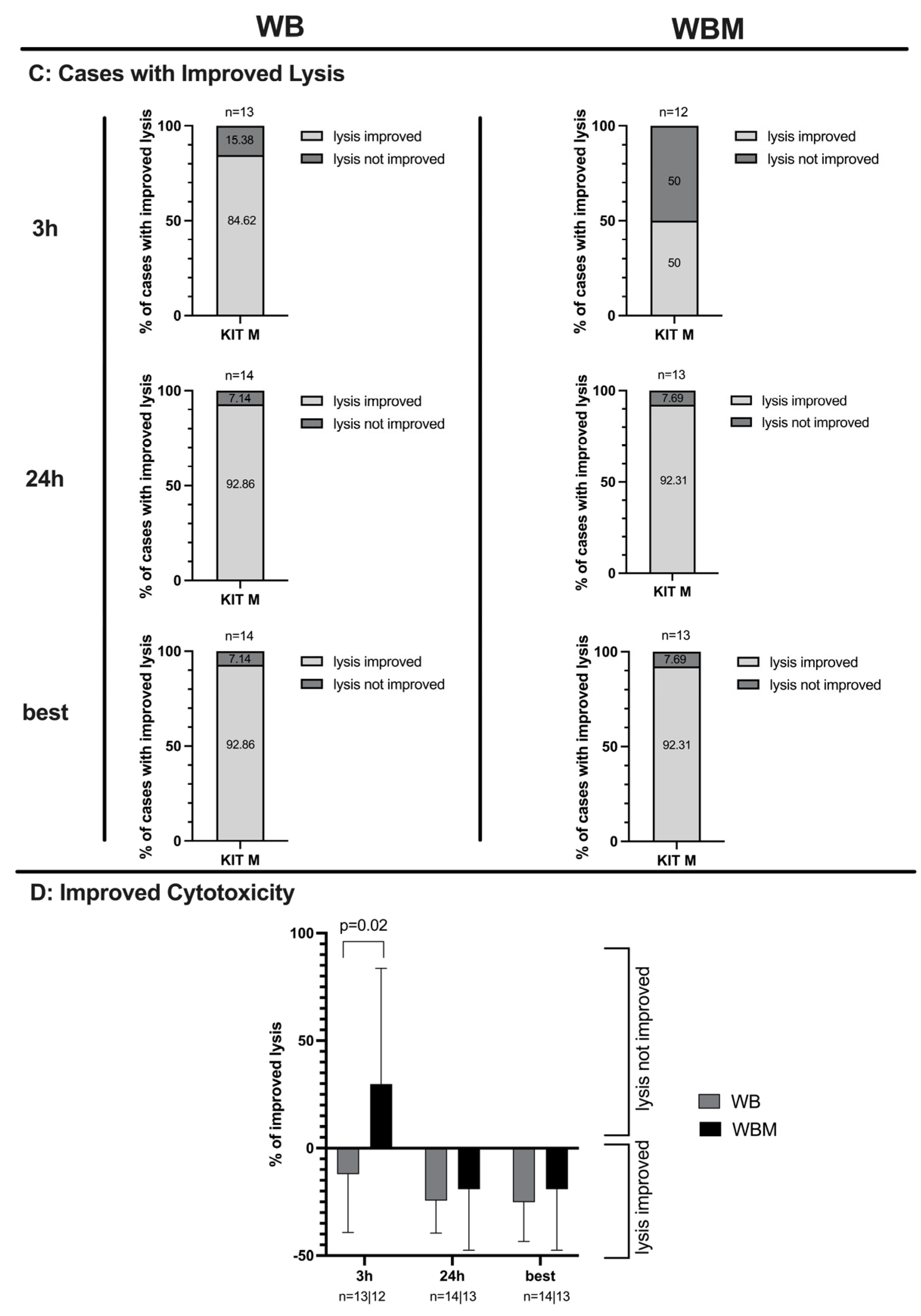
2.6. Effects of Kit-M-Treated WB and WBM on the Anti-Leukemic, Blastolytic Functionality after MLC
2.7. Correlation of Results with Patients’ Allocation to First Diagnosis and to Relapse after SCT
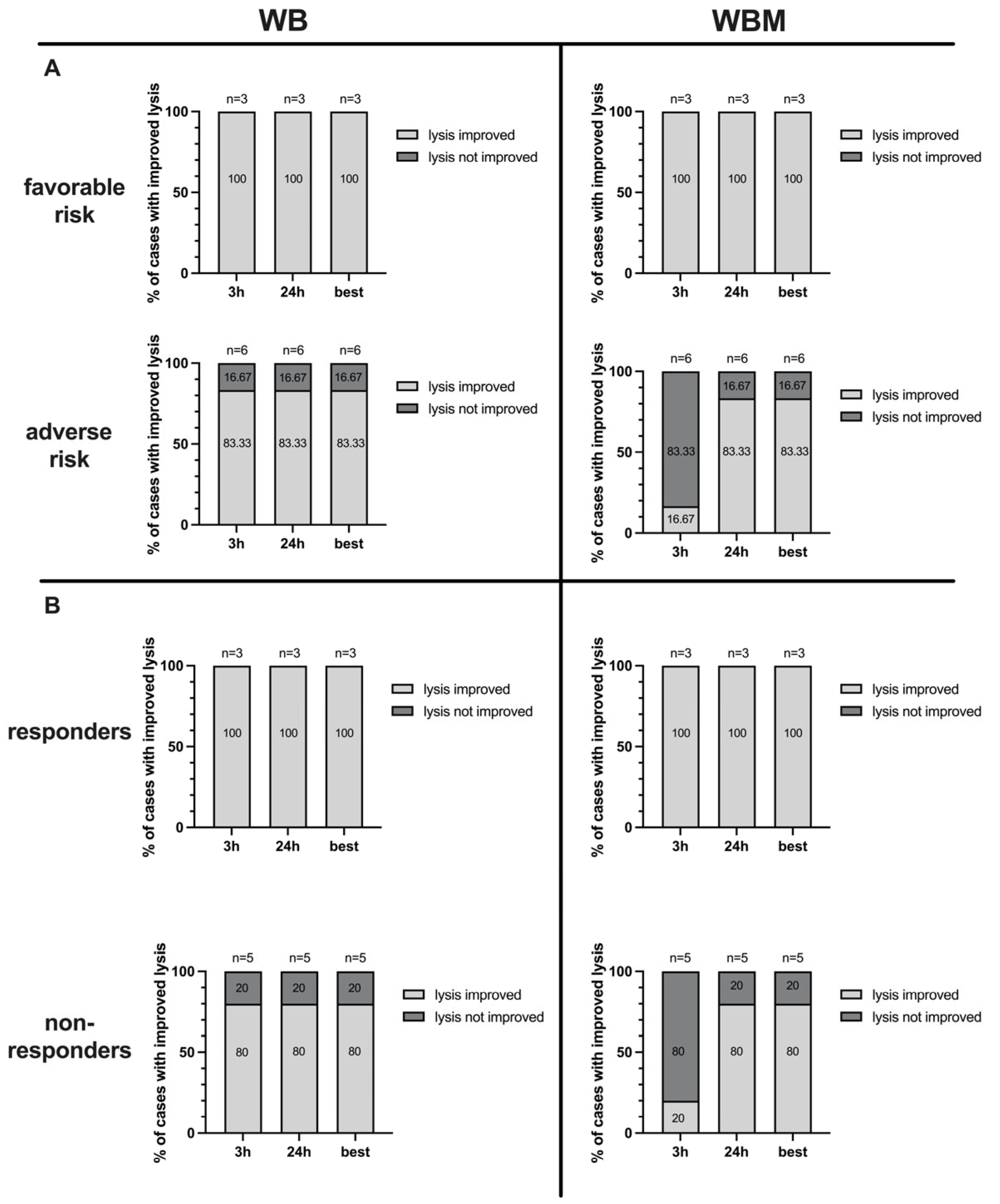
2.8. Correlation of Antileukemic Cytotoxicity with Patients’ Allocation to Risk Groups
2.9. Correlation of Antileukemic Cytotoxicity with Patients’ Allocation to Response to Induction Chemotherapy
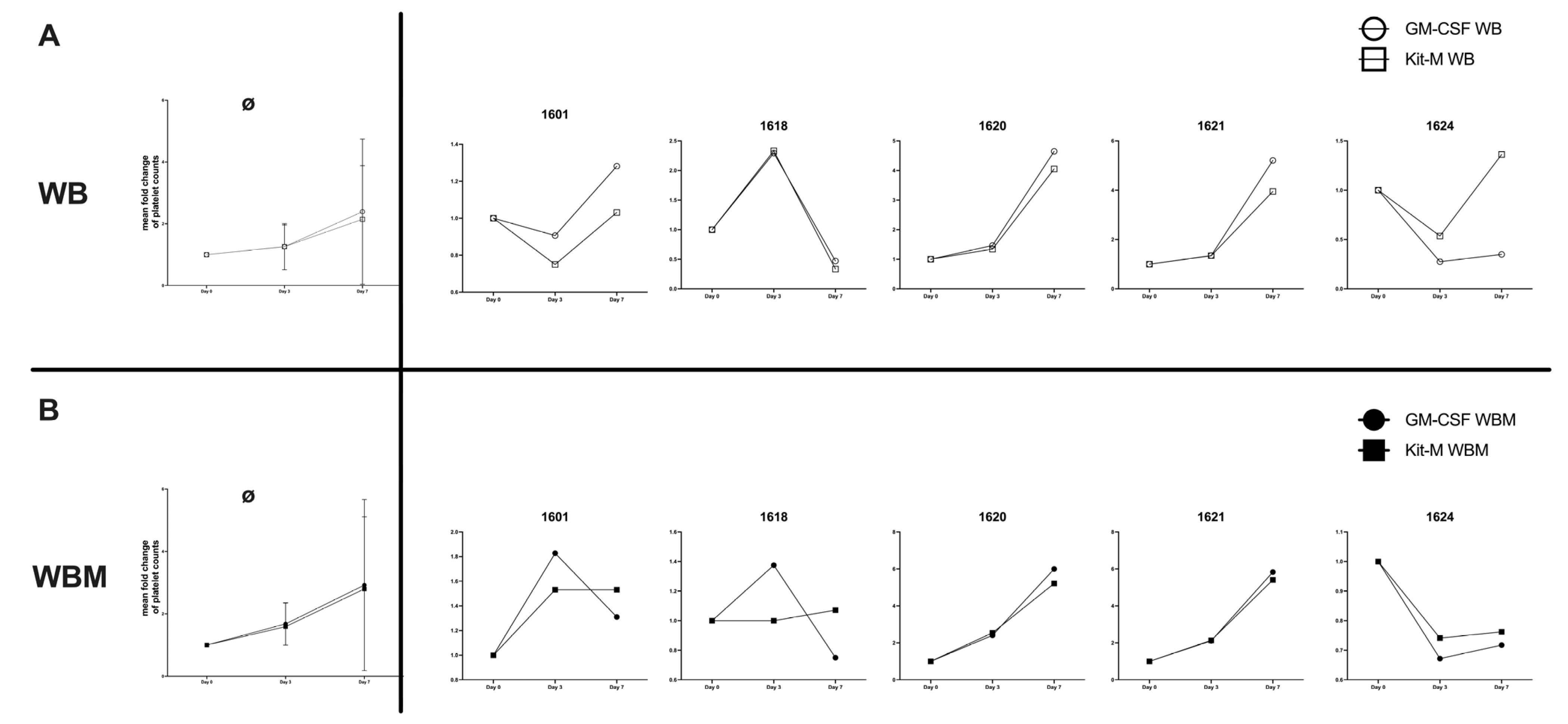
2.10. Effects of Kit-M-Treated WB and WBM on Platelet Counts
3. Discussion
3.1. WBM-the Immunosuppressive Leukemic Microenvironment
3.2. DC-Based Immunotherapy
3.3. Composition of Uncultured WB and WBM
3.4. Generation of (Mature) DC/DCleu in Kit-M-Treated WB and WBM
3.5. Provision of Immunoreactive Cells after MLC in Kit-M-Treated WB and WBM
3.6. Enhanced Degranulation Activity of T Cells after MLC in Kit-M-Treated WB and WBM
3.7. Enhanced IFNγ Production of T Cells after MLC in Kit-M-Treated WB and WBM
3.8. Improved Blastolytic Activity after MLC in Kit-M-treated WB and WBM
3.9. Correlation of Results with Patients’ Allocation to First Diagnosis and to Relapse after SCT
3.10. Influence of Kit-M or GM-CSF on Platelet Counts in WB and WBM
4. Materials and Methods
4.1. Sample Collection
4.2. Patients’ Characteristics
4.3. Flow Cytometry
4.4. Sample Preparation
4.5. Dendritic Cell Culture
4.6. T Cell-Enriched Mixed Lymphocyte Culture (MLC)
4.7. Degranulation Assay (DEG)
4.8. Intracellular Cytokine Assay (INTCYT)
4.9. Cytotoxicity Fluorolysis Assay (CTX)
4.10. Platelet Counts
4.11. Statistical Methods
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Estey, E.; Dohner, H. Acute myeloid leukaemia. Lancet 2006, 368, 1894–1907. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.L.; Hege, K.; Yang, J.; Kalpage, H.A.; Su, Y.; Edwards, H.; Huttemann, M.; Taub, J.W.; Ge, Y. Targeting multiple signaling pathways: The new approach to acute myeloid leukemia therapy. Signal Transduct. Target. Ther. 2020, 5, 288. [Google Scholar] [CrossRef] [PubMed]
- Newell, L.F.; Cook, R.J. Advances in acute myeloid leukemia. BMJ 2021, 375, n2026. [Google Scholar] [CrossRef] [PubMed]
- Pelcovits, A.; Niroula, R. Acute Myeloid Leukemia: A Review. Rhode Isl. Med. J. 2020, 103, 38–40. [Google Scholar]
- Lucas, D. Structural organization of the bone marrow and its role in hematopoiesis. Curr. Opin. Hematol. 2021, 28, 36–42. [Google Scholar] [CrossRef]
- Comazzetto, S.; Shen, B.; Morrison, S.J. Niches that regulate stem cells and hematopoiesis in adult bone marrow. Dev. Cell 2021, 56, 1848–1860. [Google Scholar] [CrossRef] [PubMed]
- Horas, K.; Menale, C.; Maurizi, A. Editorial: The bone/bone marrow microenvironment: A hub for immune regulation of the tumor cells fate. Front. Immunol. 2022, 13, 1019489. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Kawamoto, E.; Gaowa, A.; Park, E.J.; Shimaoka, M. Remodeling of Bone Marrow Niches and Roles of Exosomes in Leukemia. Int. J. Mol. Sci. 2021, 22, 1881. [Google Scholar] [CrossRef]
- Ayala, F.; Dewar, R.; Kieran, M.; Kalluri, R. Contribution of bone microenvironment to leukemogenesis and leukemia progression. Leukemia 2009, 23, 2233–2241. [Google Scholar] [CrossRef]
- Tettamanti, S.; Pievani, A.; Biondi, A.; Dotti, G.; Serafini, M. Catch me if you can: How AML and its niche escape immunotherapy. Leukemia 2022, 36, 13–22. [Google Scholar] [CrossRef]
- Zhao, E.; Xu, H.; Wang, L.; Kryczek, I.; Wu, K.; Hu, Y.; Wang, G.; Zou, W. Bone marrow and the control of immunity. Cell Mol. Immunol. 2012, 9, 11–19. [Google Scholar] [CrossRef]
- Mohanty, A.; Polisetti, N.; Vemuganti, G.K. Immunomodulatory properties of bone marrow mesenchymal stem cells. J. Biosci. 2020, 45, 98. [Google Scholar] [CrossRef]
- Shafat, M.S.; Oellerich, T.; Mohr, S.; Robinson, S.D.; Edwards, D.R.; Marlein, C.R.; Piddock, R.E.; Fenech, M.; Zaitseva, L.; Abdul-Aziz, A.; et al. Leukemic blasts program bone marrow adipocytes to generate a protumoral microenvironment. Blood 2017, 129, 1320–1332. [Google Scholar] [CrossRef]
- Yang, X.; Wang, J. Precision therapy for acute myeloid leukemia. J. Hematol. Oncol. 2018, 11, 3. [Google Scholar] [CrossRef]
- de Lima, M.; Roboz, G.J.; Platzbecker, U.; Craddock, C.; Ossenkoppele, G. AML and the art of remission maintenance. Blood Rev. 2021, 49, 100829. [Google Scholar] [CrossRef]
- Nayak, R.K.; Chen, Y.B. Maintenance therapy for AML after allogeneic HCT. Front. Oncol. 2022, 12, 895771. [Google Scholar] [CrossRef]
- Isidori, A.; Cerchione, C.; Daver, N.; DiNardo, C.; Garcia-Manero, G.; Konopleva, M.; Jabbour, E.; Ravandi, F.; Kadia, T.; Burguera, A.F.; et al. Immunotherapy in Acute Myeloid Leukemia: Where We Stand. Front. Oncol. 2021, 11, 656218. [Google Scholar] [CrossRef]
- Albinger, N.; Pfeifer, R.; Nitsche, M.; Mertlitz, S.; Campe, J.; Stein, K.; Kreyenberg, H.; Schubert, R.; Quadflieg, M.; Schneider, D.; et al. Primary CD33-targeting CAR-NK cells for the treatment of acute myeloid leukemia. Blood Cancer J. 2022, 12, 61. [Google Scholar] [CrossRef]
- Vago, L.; Gojo, I. Immune escape and immunotherapy of acute myeloid leukemia. J. Clin. Investig. 2020, 130, 1552–1564. [Google Scholar] [CrossRef] [PubMed]
- Kohler, N.; Ruess, D.A.; Kesselring, R.; Zeiser, R. The Role of Immune Checkpoint Molecules for Relapse After Allogeneic Hematopoietic Cell Transplantation. Front. Immunol. 2021, 12, 634435. [Google Scholar] [CrossRef]
- Stone, R.M.; Larson, R.A.; Dohner, H. Midostaurin in FLT3-Mutated Acute Myeloid Leukemia. N. Engl. J. Med. 2017, 377, 1903. [Google Scholar] [CrossRef]
- Liu, H. Emerging agents and regimens for AML. J. Hematol. Oncol. 2021, 14, 49. [Google Scholar] [CrossRef]
- Dohner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef]
- Haslauer, T.; Greil, R.; Zaborsky, N.; Geisberger, R. CAR T-Cell Therapy in Hematological Malignancies. Int. J. Mol. Sci. 2021, 22, 8996. [Google Scholar] [CrossRef]
- Gardner, A.; de Mingo Pulido, Á.; Ruffell, B. Dendritic Cells and Their Role in Immunotherapy. Front. Immunol. 2020, 11, 924. [Google Scholar] [CrossRef]
- Yang, L.; Li, S.; Chen, L.; Zhang, Y. Emerging roles of plasmacytoid dendritic cell crosstalk in tumor immunity. Cancer Biol. Med. 2023, 20, 728–747. [Google Scholar] [CrossRef] [PubMed]
- Patente, T.A.; Pinho, M.P.; Oliveira, A.A.; Evangelista, G.C.M.; Bergami-Santos, P.C.; Barbuto, J.A.M. Human Dendritic Cells: Their Heterogeneity and Clinical Application Potential in Cancer Immunotherapy. Front. Immunol. 2018, 9, 3176. [Google Scholar] [CrossRef] [PubMed]
- Collin, M.; Bigley, V. Human dendritic cell subsets: An update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Chrisikos, T.T.; Zhou, Y.; Slone, N.; Babcock, R.; Watowich, S.S.; Li, H.S. Molecular regulation of dendritic cell development and function in homeostasis, inflammation, and cancer. Mol. Immunol. 2019, 110, 24–39. [Google Scholar] [CrossRef]
- Yu, J.; Sun, H.; Cao, W.; Song, Y.; Jiang, Z. Research progress on dendritic cell vaccines in cancer immunotherapy. Exp. Hematol. Oncol. 2022, 11, 3. [Google Scholar] [CrossRef]
- Kremser, A.; Kufner, S.; Konhaeuser, E.; Kroell, T.; Hausmann, A.; Tischer, J.; Kolb, H.J.; Zitzelsberger, H.; Schmetzer, H. Combined immunophenotyping and fluorescence in situ hybridization with chromosome-specific DNA probes allows quantification and differentiation of ex vivo generated dendritic cells, leukemia-derived dendritic cells and clonal leukemic cells in patients with acute myeloid leukemia. Leuk. Lymphoma 2013, 54, 1297–1308. [Google Scholar] [CrossRef]
- Schmetzer, H.M.; Kremser, A.; Loibl, J.; Kroell, T.; Kolb, H.J. Quantification of ex vivo generated dendritic cells (DC) and leukemia-derived DC contributes to estimate the quality of DC, to detect optimal DC-generating methods or to optimize DC-mediated T-cell-activation-procedures ex vivo or in vivo. Leukemia 2007, 21, 1338–1341. [Google Scholar] [CrossRef] [PubMed]
- Plett, C.; Klauer, L.K.; Amberger, D.C.; Ugur, S.; Rabe, A.; Fischer, Z.; Deen, D.; Hirn-Lopez, A.; Gunsilius, C.; Werner, J.O.; et al. Immunomodulatory kits generating leukaemia derived dendritic cells do not induce blast proliferation ex vivo: IPO-38 as a novel marker to quantify proliferating blasts in acute myeloid leukaemia. Clin. Immunol. 2022, 242, 109083. [Google Scholar] [CrossRef] [PubMed]
- Klauer, L.K.; Schutti, O.; Ugur, S.; Doraneh-Gard, F.; Amberger, D.C.; Rogers, N.; Kramer, D.; Rank, A.; Schmid, C.; Eiz-Vesper, B.; et al. Interferon Gamma Secretion of Adaptive and Innate Immune Cells as a Parameter to Describe Leukaemia-Derived Dendritic-Cell-Mediated Immune Responses in Acute Myeloid Leukaemia in vitro. Transfus. Med. Hemother 2021, 49, 44–61. [Google Scholar] [CrossRef] [PubMed]
- Schwepcke, C.; Klauer, L.K.; Deen, D.; Amberger, D.C.; Fischer, Z.; Doraneh-Gard, F.; Gunsilius, C.; Hirn-Lopez, A.; Kroell, T.; Tischer, J.; et al. Generation of Leukaemia-Derived Dendritic Cells (DCleu) to Improve Anti-Leukaemic Activity in AML: Selection of the Most Efficient Response Modifier Combinations. Int. J. Mol. Sci. 2022, 23, 8333. [Google Scholar] [CrossRef]
- Amberger, D.C.; Doraneh-Gard, F.; Gunsilius, C.; Weinmann, M.; Mobius, S.; Kugler, C.; Rogers, N.; Bock, C.; Kodel, U.; Werner, J.O.; et al. PGE1-Containing Protocols Generate Mature (Leukemia-Derived) Dendritic Cells Directly from Leukemic Whole Blood. Int. J. Mol. Sci. 2019, 20, 4590. [Google Scholar] [CrossRef]
- Schutti, O.; Schmetzer, H. Intracellular Cytokine (IFNγ, TNFα) Assays in Combination with a Degranulation Assay Contribute Significantly to Detect and Quantify Potentially Leukemia-Specific Cells of Several Lines in Leukemia Patients’ Blood or Culture Settings; Department of Medicine III, University Hospital of Munich: Munich, Germany, 2023; in preparation. [Google Scholar]
- Boyman, O.; Sprent, J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat. Rev. Immunol. 2012, 12, 180–190. [Google Scholar] [CrossRef]
- Duarte, D.; Hawkins, E.D.; Lo Celso, C. The interplay of leukemia cells and the bone marrow microenvironment. Blood 2018, 131, 1507–1511. [Google Scholar] [CrossRef]
- Kim, Y.W.; Koo, B.K.; Jeong, H.W.; Yoon, M.J.; Song, R.; Shin, J.; Jeong, D.C.; Kim, S.H.; Kong, Y.Y. Defective Notch activation in microenvironment leads to myeloproliferative disease. Blood 2008, 112, 4628–4638. [Google Scholar] [CrossRef]
- Kode, A.; Manavalan, J.S.; Mosialou, I.; Bhagat, G.; Rathinam, C.V.; Luo, N.; Khiabanian, H.; Lee, A.; Murty, V.V.; Friedman, R.; et al. Leukaemogenesis induced by an activating beta-catenin mutation in osteoblasts. Nature 2014, 506, 240–244. [Google Scholar] [CrossRef]
- Pozzobon, T.; Goldoni, G.; Viola, A.; Molon, B. CXCR4 signaling in health and disease. Immunol. Lett. 2016, 177, 6–15. [Google Scholar] [CrossRef]
- Zhou, W.; Guo, S.; Liu, M.; Burow, M.E.; Wang, G. Targeting CXCL12/CXCR4 Axis in Tumor Immunotherapy. Curr. Med. Chem. 2019, 26, 3026–3041. [Google Scholar] [CrossRef]
- Stelmach, P.; Trumpp, A. Leukemic stem cells and therapy resistance in acute myeloid leukemia. Haematologica 2023, 108, 353–366. [Google Scholar] [CrossRef]
- Van Acker, H.H.; Versteven, M.; Lichtenegger, F.S.; Roex, G.; Campillo-Davo, D.; Lion, E.; Subklewe, M.; Van Tendeloo, V.F.; Berneman, Z.N.; Anguille, S. Dendritic Cell-Based Immunotherapy of Acute Myeloid Leukemia. J. Clin. Med. 2019, 8, 579. [Google Scholar] [CrossRef]
- Rackl, E.; Li, L.; Klauer, L.K.; Ugur, S.; Pepeldjiyska, E.; Seidel, C.L.; Gunsilius, C.; Weinmann, M.; Doraneh-Gard, F.; Reiter, N.; et al. Dendritic Cell-Triggered Immune Activation Goes along with Provision of (Leukemia-Specific) Integrin Beta 7-Expressing Immune Cells and Improved Antileukemic Processes. Int. J. Mol. Sci. 2022, 24, 463. [Google Scholar] [CrossRef] [PubMed]
- Platt, A.M.; Randolph, G.J. Dendritic cell migration through the lymphatic vasculature to lymph nodes. Adv. Immunol. 2013, 120, 51–68. [Google Scholar] [CrossRef] [PubMed]
- Pepeldjiyska, E.; Li, L.; Gao, J.; Seidel, C.L.; Blasi, C.; Ozkaya, E.; Schmohl, J.; Kraemer, D.; Schmid, C.; Rank, A.; et al. Leukemia derived dendritic cell (DCleu) mediated immune response goes along with reduced (leukemia-specific) regulatory T-cells. Immunobiology 2022, 227, 152237. [Google Scholar] [CrossRef]
- Grabrucker, C.; Liepert, A.; Dreyig, J.; Kremser, A.; Kroell, T.; Freudenreich, M.; Schmid, C.; Schweiger, C.; Tischer, J.; Kolb, H.J.; et al. The quality and quantity of leukemia-derived dendritic cells from patients with acute myeloid leukemia and myelodysplastic syndrome are a predictive factor for the lytic potential of dendritic cells-primed leukemia-specific T cells. J. Immunother. 2010, 33, 523–537. [Google Scholar] [CrossRef]
- Logan, C.; Koura, D.; Taplitz, R. Updates in infection risk and management in acute leukemia. Hematology Am. Soc. Hematol. Educ. Program. 2020, 2020, 135–139. [Google Scholar] [CrossRef]
- Hobo, W.; Hutten, T.J.A.; Schaap, N.P.M.; Dolstra, H. Immune checkpoint molecules in acute myeloid leukaemia: Managing the double-edged sword. Br. J. Haematol. 2018, 181, 38–53. [Google Scholar] [CrossRef] [PubMed]
- Rowshanravan, B.; Halliday, N.; Sansom, D.M. CTLA-4: A moving target in immunotherapy. Blood 2018, 131, 58–67. [Google Scholar] [CrossRef]
- Boeck, C.L.; Amberger, D.C.; Doraneh-Gard, F.; Sutanto, W.; Guenther, T.; Schmohl, J.; Schuster, F.; Salih, H.; Babor, F.; Borkhardt, A.; et al. Significance of Frequencies, Compositions, and/or Antileukemic Activity of (DC-stimulated) Invariant NKT, NK and CIK Cells on the Outcome of Patients With AML, ALL and CLL. J. Immunother. 2017, 40, 224–248. [Google Scholar] [CrossRef]
- Yu, Z.; Liu, W.; He, Y.; Sun, M.; Yu, J.; Jiao, X.; Han, Q.; Tang, H.; Zhang, B.; Xian, Y.; et al. HLA-A2.1-restricted ECM1-derived epitope LA through DC cross-activation priming CD8(+) T and NK cells: A novel therapeutic tumour vaccine. J. Hematol. Oncol. 2021, 14, 71. [Google Scholar] [CrossRef] [PubMed]
- Ugur, S.; Schmetzer, H.M. ‘Kit’-Mediated Blastmodulation to Leukemia-Derived DC Significantly Improves Antileukemic Activities in Whole Blood Independent of AML-Patients’ Subtypes; Department of Medicine III, University Hospital of Munich: Munich, Germany, 2023; in preparation. [Google Scholar]
- Baudrexler, T.; Boeselt, T.; Li, L.; Bohlscheid, S.; Boas, U.; Schmid, C.; Rank, A.; Schmohl, J.; Koczulla, R.; Schmetzer, H.M. Volatile Phases Derived from Serum, DC, or MLC Culture Supernatants to Deduce a VOC-Based Diagnostic Profiling Strategy for Leukemic Diseases. Biomolecules 2023, 13, 989. [Google Scholar] [CrossRef]
- Shenghui, Z.; Yixiang, H.; Jianbo, W.; Kang, Y.; Laixi, B.; Yan, Z.; Xi, X. Elevated frequencies of CD4(+) CD25(+) CD127lo regulatory T cells is associated to poor prognosis in patients with acute myeloid leukemia. Int. J. Cancer 2011, 129, 1373–1381. [Google Scholar] [CrossRef]
- Hassin, D.; Garber, O.G.; Meiraz, A.; Schiffenbauer, Y.S.; Berke, G. Cytotoxic T lymphocyte perforin and Fas ligand working in concert even when Fas ligand lytic action is still not detectable. Immunology 2011, 133, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Doucette, K.; Karp, J.; Lai, C. Advances in therapeutic options for newly diagnosed, high-risk AML patients. Ther. Adv. Hematol. 2021, 12, 20406207211001138. [Google Scholar] [CrossRef] [PubMed]
- Gulati, S.; Bennett, C.; Phillips, J.; Van-Poznak, C. GM-CSF as an adjunct to autologous bone marrow transplantation. Stem Cells 1993, 11, 20–25. [Google Scholar] [CrossRef]
- Negrin, R.S.; Greenberg, P.L. The use of colony stimulating factors in clinical bone marrow transplantation. Curr. Opin. Immunol. 1992, 4, 567–570. [Google Scholar] [CrossRef]
- McIntosh, B.; Kaushansky, K. Transcriptional regulation of bone marrow thrombopoietin by platelet proteins. Exp. Hematol. 2008, 36, 799–806. [Google Scholar] [CrossRef]
- Ugolini, A.; Nuti, M. CD137(+) T-Cells: Protagonists of the Immunotherapy Revolution. Cancers 2021, 13, 456. [Google Scholar] [CrossRef] [PubMed]
- Aktas, E.; Kucuksezer, U.C.; Bilgic, S.; Erten, G.; Deniz, G. Relationship between CD107a expression and cytotoxic activity. Cell Immunol. 2009, 254, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Morath, A.; Schamel, W.W. alphabeta and gammadelta T cell receptors: Similar but different. J. Leukoc. Biol. 2020, 107, 1045–1055. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stage | Patient No. | Age at Sampling/Sex | FAB/WHO Classification | ELN (2017)-Risk-Stratification | Cyto-, Molecular Genetics | Blast Phenotype (CD) | IC in WB/WBM (%) * | Response to Induction Chemotherapy | Experiments Conducted with WB | Experiments Conducted with WBM |
|---|---|---|---|---|---|---|---|---|---|---|
| First Diagnosis | 1615 | 78/F | sAML | Intermediate | IDH1, ASXL1, JAK2, SRSF2 | 34, 117, 13 | 53/65 | no | DC, MLC, Deg (UC, C), InCyt (UC, C), CTX | DC, MLC, CTX |
| 1618 | 63/M | pAML/M1 | Favorable | NPM1 | 117, 33, 13, 15 | 10/7 | yes | DC, MLC, Deg (UC, C), InCyt (UC, C), CTX | DC, MLC, CTX | |
| 1620 | 61/F | sAML | Adverse | t(2;3)(p23;q26), del (7)(q21), del(7)(q31), MECOM rearrangement, ASXL1, RUNX1 | 34, 117, 33, 4 | 4/12 | no | DC, MLC, Deg (UC), InCyt (UC), CTX | DC, MLC, CTX | |
| 1621 | 71/M | sAML | Adverse | TP53, ASXL1, SRSF2, SETPBP1, KRAS | 34, 117, 5, 13 | 18/24 | no | DC, MLC, Deg (UC, C), InCyt (UC, C), CTX | DC, MLC, CTX | |
| 1624 | 77/F | sAML | Adverse | monosomy 7, der(7)t(1;7), der (2)t(2;3), del(7)(q31), ampl(3q26), IDH2, STAG2, DNMT3A, MPL, NRAS, SRSF2 | 34, 117, 13 | 30/32 | yes | DC, MLC, Deg (UC), InCyt (UC), CTX | DC, MLC, CTX | |
| 1630 | 29/M | pAML | Favorable | FLT3-TKD, NPM1, PTPN11 | 34, 117, 13, 33, 15, 64, 65 | 20/27 | yes | DC, MLC, Deg (UC, C), InCyt (UC, C), CTX | DC, MLC, Deg (C), InCyt (C), CTX | |
| 1638 | 68/M | sAML | Adverse | del(5)(q31), TP53, RUNX1, RUNX1T1 | 34, 117, 33, 13, 56, 4 | 60/54 | no | DC, MLC, Deg (UC, C), InCyt (UC, C), CTX | DC, MLC, Deg (UC, C), InCyt (UC, C), CTX | |
| 1642 | 63/F | sAML | Adverse | IDH2, complex aberrant | 34, 117, 33, 13 | 1/19 | yes | DC, MLC, Deg (UC, C), InCyt (UC, C), CTX | DC, MLC, Deg (UC, C), InCyt (UC, C), CTX | |
| 1643 | 68/M | pAML | Favorable | NPM1, IDH2 | 117, 33, 64, 13 | 23/39 | no | DC, MLC, Deg (UC, C), InCyt (UC, C), CTX | DC, MLC, Deg (UC, C), InCyt (UC, C), CTX | |
| 1645 | 58/F | pAML | Favorable | IDH2, NPM1 | 117, 33, 56 | 36/47 | yes | DC, MLC, Deg (UC), InCyt (UC) | DC, MLC, Deg (UC), InCyt (UC) | |
| Persisting Disease | 1601 | 75/F | sAML | Adverse | none detected | 34, 117, 33, 13 | 61/49 | DC, MLC, Deg (UC), InCyt (UC), CTX | DC, MLC, CTX | |
| Chimerism (%) | ||||||||||
| Relapse After Stem Cell Transplantation | 1628 | 22/F | tAML/M5 | RUNX1, WT1, FLT3-ITD, KMT2A-PTD | 34, 117, 65, 33, 56, 64 | 3/31 | 77 | DC, Deg (UC), InCyt (UC) | DC | |
| 1640 | 73/F | pAML | del(9q), NMP1, IDH2, WT1, DNMT3A | 34, 117, 33, 13, 7 | <1/<1 | 56 | DC, MLC, Deg (UC, C), InCyt (UC, C), CTX | DC, MLC, Deg (UC, C), InCyt (UC, C), CTX | ||
| 1641 | 64/F | tAML | dup(13)(q12q33), DLEU signal, FLT3-ITD, EZH2, WT1 | 34, 117, 33, 13, 64, 65, 15 | 6/16 | 62 | DC, MLC, Deg (UC, C), InCyt (UC, C), CTX | DC, MLC, Deg (UC, C), InCyt (UC, C), CTX | ||
| 1650 | 64/F | AML-MRC | RUNX1, BCOR, KMT2A-PTD, TP53 | 34, 117, 13 | 3/8 | 86 | DC, MLC, Deg (UC, C), InCyt (UC, C), CTX | DC, Deg (UC), InCyt (UC) | ||
| 1654 | 71/M | pAML/M2 | der(16)t(16;17), NPM1 | 117, 56, 13, 34 | 3/3 | 97 | DC, MLC, Deg (UC, C), InCyt (UC, C), CTX | DC, MLC, Deg (UC, C), InCyt (UC, C), CTX | ||
| 1656 | 42/F | pAML | MECOM rearrangement, complex aberrant, PTPN11 | 34, 117, 33, 56, 13 | 7/14 | 86 | DC, MLC, Deg (UC, C), InCyt (UC, C), CTX | DC, MLC, Deg (C), InCyt (C), CTX |
| Cell Type | Name of Subgroups | Abbreviation of Subgroups | Surface Marker | Referred to | Abbreviation | Reference |
|---|---|---|---|---|---|---|
| Subtypes of blasts and DC | ||||||
| Blast cells | Leukemic blasts | Bla | Bla+ (e.g., CD34+, CD177+) | WB or WBM | Bla/WB or/WBM | [32] |
| Proliferating blasts | Blaprol-CD71 | Bla+DC−CD71+ | Bla | Blaprol-CD71/Bla | [33] | |
| Proliferating blasts | Blaprol-Ipo38 | Bla+DC−Ipo38+ | Bla | Blaprol-Ipo38/Bla | [33] | |
| Dendritic cells | Dendritic cells | DC | DC+ (CD80+, CD206+) | WB or WBM | DC/WB or/WBM | [35] |
| Leukemia derived DC | DCleu | DC+Bla+ | WB or WBM | DCleu/WB or/WBM | [35] | |
| Bla | DCleu/Bla | |||||
| Mature DC | DCmat | DC+CD197+ | WB or WBM | DCmat/WB or/WBM | [35] | |
| Mature DCleu | DCleu-mat | DC+CD197+Bla+ | WB or WBM | DCleu-mat/WB or/WBM | [35] | |
| Bla | DCleu-mat/Bla | [35] | ||||
| Subtypes of Immune Reactive Cells | ||||||
| T cells | CD3+ pan T cells | CD3+ | CD3+ | cells | CD3+/cells | [36] |
| CD4+coexpressing T cells | CD3+CD4+ | CD3+CD4+ | CD3+ | CD3+CD4+/CD3+ | [36] | |
| CD8+coexpressing T cells | CD3+CD8+ | CD3+CD8+ | CD3+ | CD3+CD8+/CD3+ | [36] | |
| Proliferating T cells-early | Tprol-early | CD3+CD69+ | CD3+ | Tprol-early/CD3+ | [36] | |
| Non-naive T cells | Tnon-naive | CD3+CD45RO+ | CD3+ | Tnon-naive/CD3+ | [34] | |
| Effector (memory) T cells | Tem/eff | CD3+CD45RO+CD197− | CD3+ | Tem/eff/CD3+ | [34] | |
| Central (memory) T cells | Tcm | CD3+CD45RO+CD197+ | CD3+ | Tcm/CD3+ | [34] | |
| CD137+ coexpressing T cells | CD3+CD137+ | CD3+CD137+ | CD3+ | CD3+CD137+/CD3+ | [63] | |
| Integrinβ7+ coexpressing T cells | CD3+β7+ | CD3+Integrinβ7+ | CD3+ | CD3+β7+/CD3+ | [46] | |
| TCRγδ+ coexpressing T cells | CD3+TCRγδ | CD3+TCRγδ+ | CD3+ | CD3+TCRγδ+/CD3+ | [46] | |
| CD152+ coexpressing T cells | CD3+CD152+ | CD3+CD152+ | CD3+ | CD3+CD152+/CD3+ | [52] | |
| Regulatory T cells | CD4+Treg | CD3+CD4+CD25+CD127low | CD3+CD4+ | CD4+Treg/CD4+ | [48] | |
| Natural killer cells | CD3−CD56+NK cells | NK | CD3−CD56+ | Cells | NK/cells | [53] |
| Cytokine induced killer cells | CD3+CD56+ CIK cells | CIK | CD3+CD56+ | Cells | CIK/cells | [53] |
| Invariant natural killer T cells | 6B11+ iNKT cells | iNKT | 6B11+ | Cells | iNKT/cells | [53] |
| CD3+ coexpressing 6B11+ iNKT cells | CD3+iNKT | CD3+6B11+ | 6B11+ | CD3+iNKT/iNKT | [53] | |
| Subtypes of different degranulating (CD107a+) cells as evaluated by flow cytometry | ||||||
| T cells | CD3+ pan T cells | CD3+107a+ | CD107a+CD3+ | CD3+ | CD3+107a+/CD3+ | [64] |
| Non-naive T cells | Tnon-naive107a+ | CD107a+CD3+CD45RO+ | Tnon-naive | Tnon-naive107a+/Tnon-naive | [64] | |
| Effector (memory) T cells | Tem/eff107a+ | CD107a+CD3+CD45RO+CD197− | Tem/eff | Tem/eff107a+/Tem/eff | [64] | |
| Central (memory) T cells | Tcm107a+ | CD107a+CD3+CD45RO+CD197+ | Tcm | Tcm107a+/Tcm | [64] | |
| Integrinβ7+ coexpressing T cells | CD3+β7+107a+ | CD107a+CD3+Integrinβ7+ | CD3+ | CD3+β7+107a+/CD3+ | [46] | |
| TCRγδ+ coexpressing T cells | CD3+TCRγδ+107a+ | CD107a+CD3+TCRγδ+ | CD3+TCRγδ+ | CD3+TCRγδ+107a+/CD3+ TCRγδ+ | [65] | |
| Regulatory T cells | CD4+Treg107a+ | CD107a+CD4+CD25+CD127low | CD4+Treg | CD4+Treg107a+/CD4+Treg | [48] | |
| NK cells | CD3−CD56+NK cells | NK107a+ | CD107a+CD3−CD56+ | NKcell | NK107a+/NK | [64] |
| CIK cells | CD3+CD56+ CIK cells | CIK107a+ | CD107a+CD3+CD56+ | CIKcell | CIK107a+/CIK | [64] |
| Subtypes of different intracellularly IFNγ or TNFα-producing cells as evaluated by flow cytometry | ||||||
| T cells | CD3+ pan T cells | CD3+IFNγ/TNFα | IFNγ+/TNFα+CD3+ | CD3+ | CD3+IFNγ/TNFα/CD3+ | [34] |
| CD4+-coexpressing T cells | CD3+CD4+IFNγ/TNFα | IFNγ+/TNFα+CD3+CD4+ | CD3+CD4+ | CD3+CD4+IFNγ/TNFα/CD3+CD4+ | [34] | |
| CD8+-coexpressing T cells | CD3+CD8+IFNγ/TNFα | IFNγ+/TNFα+CD3+CD8+ | CD3+CD8+ | CD3+CD4−IFNγ/TNFα/CD3+CD8+ | [34] | |
| Non-naive T cells | Tnon-naive IFNγ/TNFα | IFNγ+/TNFα+CD3+CD45RO+ | Tnon-naive | Tnon-naive IFNγ/TNFα/Tnon-naive | [34] | |
| Effector (memory) T cells | Tem/eff IFNγ/TNFα/Tem/eff | IFNγ+/TNFα+CD3+CD45RO+CD197− | Tem/eff | Tem/eff IFNγ/TNFα/Tem/eff | [34] | |
| Central (memory) T cells | Tcm IFNγ/TNFα | IFNγ+/TNFα+CD3+CD45RO+CD197+ | Tcm | Tcm IFNγ/TNFα/Tcm | [34] | |
| Integrinβ7+ coexpressing T cells | CD3+β7+IFNγ/TNFα | IFNγ+/TNFα+CD3+β7+ | CD3+β7+ | CD3+β7+IFNγ/TNFα/CD3+β7+ | [46] | |
| NK cells | CD3−CD56+NK cells | CD3−CD56+IFNγ/TNFα | IFNγ+/TNFα+CD3−CD56+ | NK | NKIFNγ/TNFα/NK | [34] |
| CIK cells | CD3+CD56+ CIK cells | CIIFNγ/TNFα | IFNγ+/TNFα+CD3+CD56+ | CIK | CIKIFNγ/TNFα/CIK | [34] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Unterfrauner, M.; Rejeski, H.A.; Hartz, A.; Bohlscheid, S.; Baudrexler, T.; Feng, X.; Rackl, E.; Li, L.; Rank, A.; Filippini Velázquez, G.; et al. Granulocyte-Macrophage-Colony-Stimulating-Factor Combined with Prostaglandin E1 Create Dendritic Cells of Leukemic Origin from AML Patients’ Whole Blood and Whole Bone Marrow That Mediate Antileukemic Processes after Mixed Lymphocyte Culture. Int. J. Mol. Sci. 2023, 24, 17436. https://doi.org/10.3390/ijms242417436
Unterfrauner M, Rejeski HA, Hartz A, Bohlscheid S, Baudrexler T, Feng X, Rackl E, Li L, Rank A, Filippini Velázquez G, et al. Granulocyte-Macrophage-Colony-Stimulating-Factor Combined with Prostaglandin E1 Create Dendritic Cells of Leukemic Origin from AML Patients’ Whole Blood and Whole Bone Marrow That Mediate Antileukemic Processes after Mixed Lymphocyte Culture. International Journal of Molecular Sciences. 2023; 24(24):17436. https://doi.org/10.3390/ijms242417436
Chicago/Turabian StyleUnterfrauner, Marianne, Hazal Aslan Rejeski, Anne Hartz, Sophia Bohlscheid, Tobias Baudrexler, Xiaojia Feng, Elias Rackl, Lin Li, Andreas Rank, Giuliano Filippini Velázquez, and et al. 2023. "Granulocyte-Macrophage-Colony-Stimulating-Factor Combined with Prostaglandin E1 Create Dendritic Cells of Leukemic Origin from AML Patients’ Whole Blood and Whole Bone Marrow That Mediate Antileukemic Processes after Mixed Lymphocyte Culture" International Journal of Molecular Sciences 24, no. 24: 17436. https://doi.org/10.3390/ijms242417436