

Dysregulation of Gene Expression of Key Signaling Mediators in PBMCs from People with Type 2 Diabetes Mellitus
Abstract
:1. Introduction
2. Results
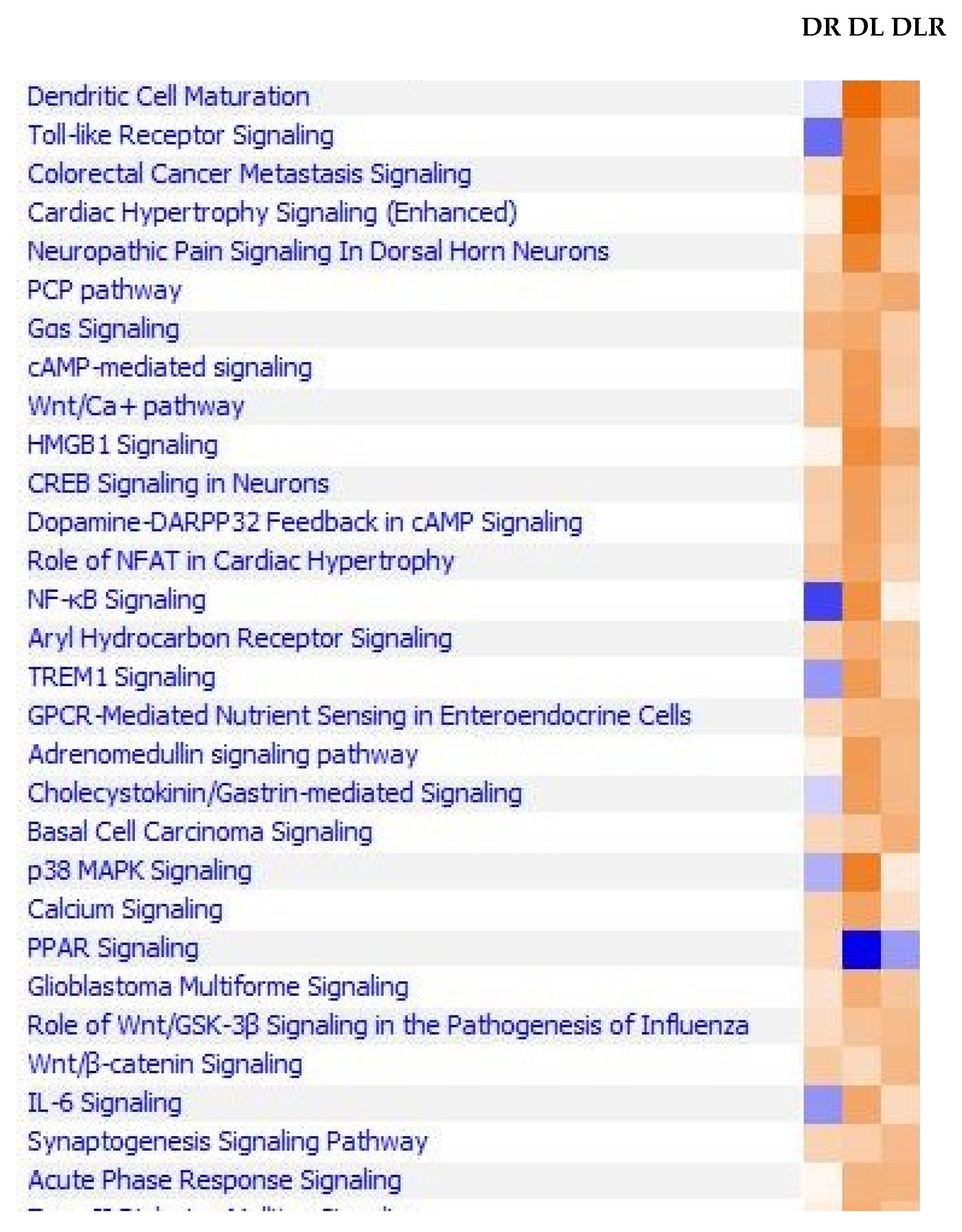
2.1. Next Generation Sequencing (NGS) Profiling Experiment
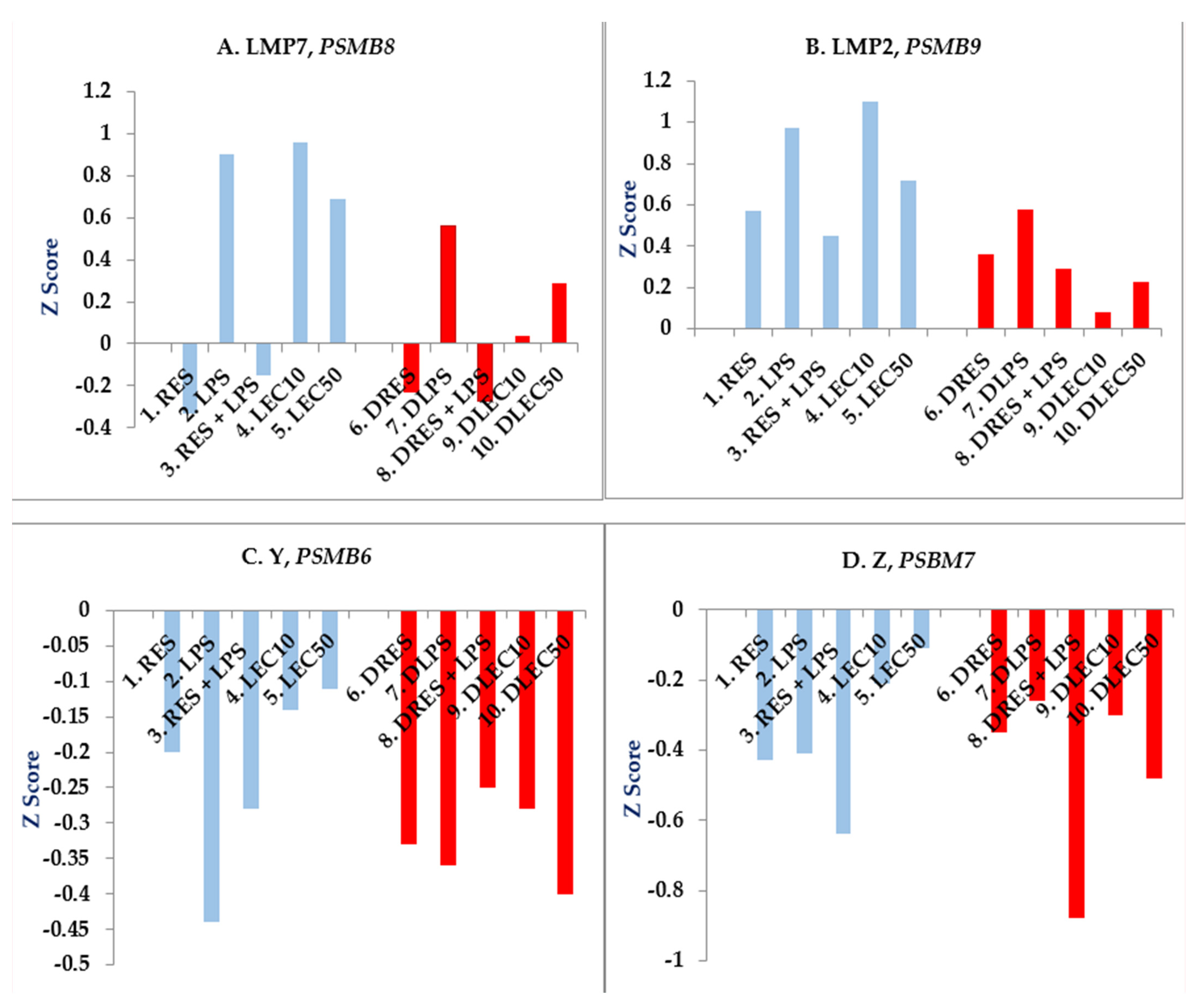
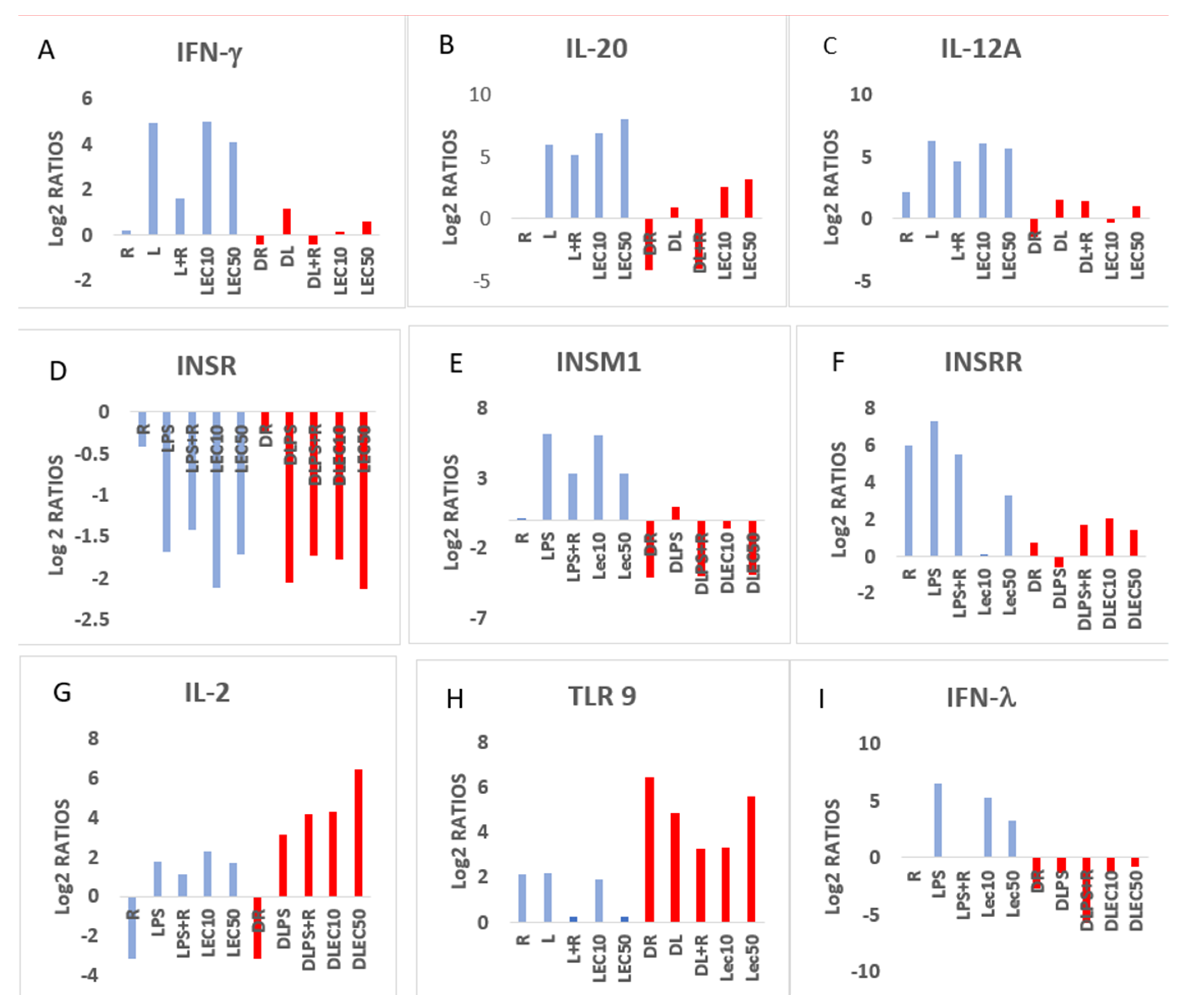
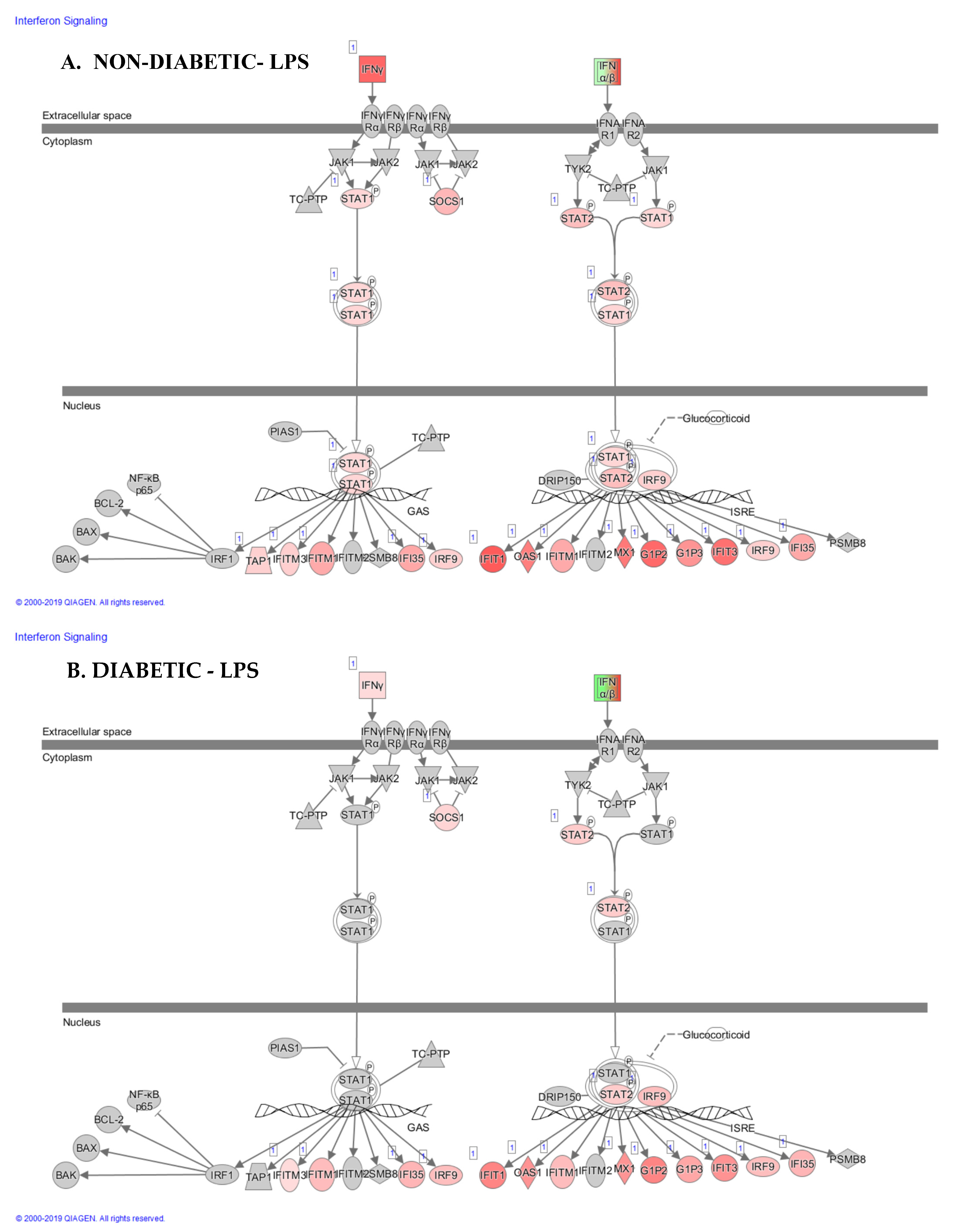
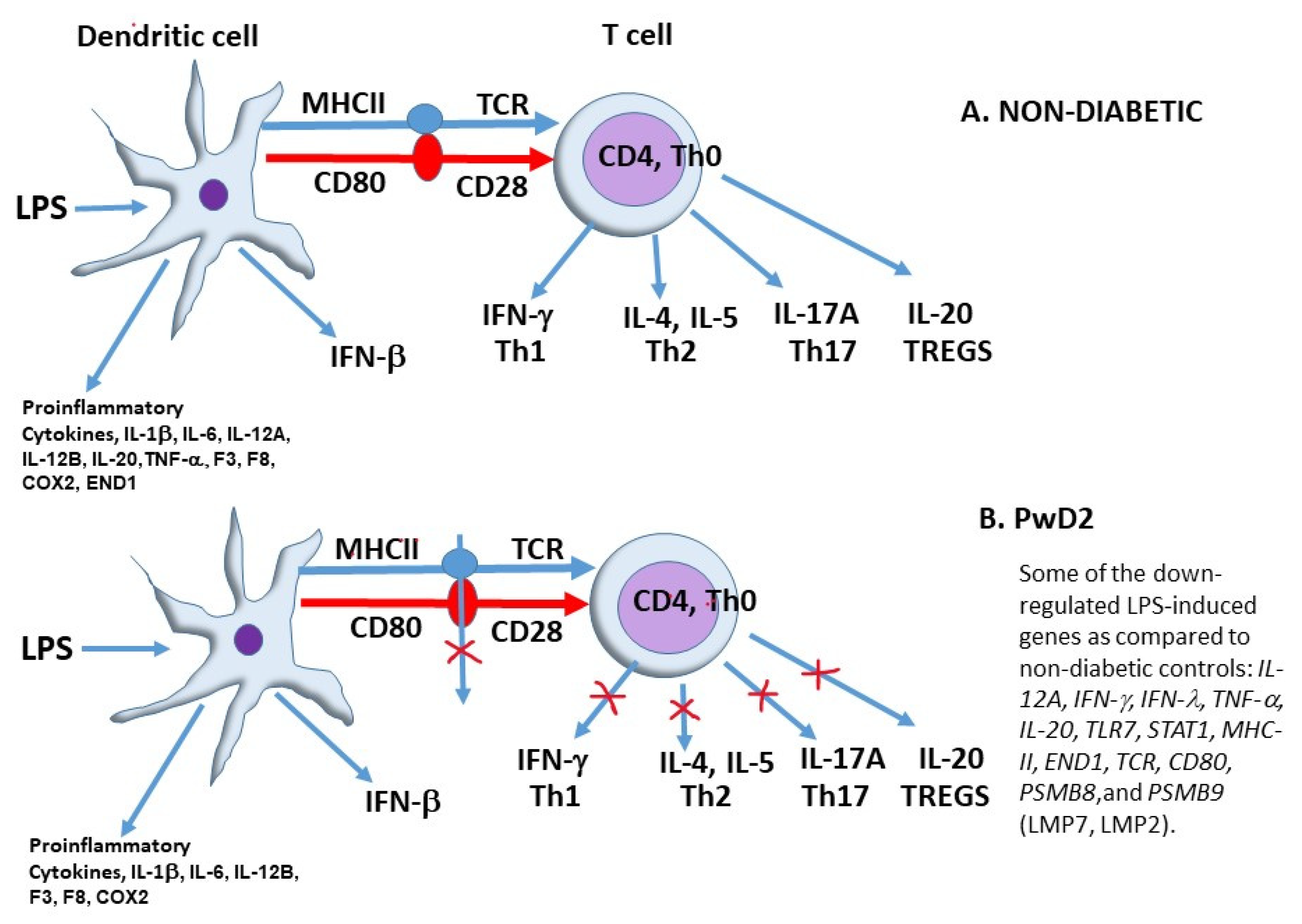
2.2. Expression of Functional Genes after Treatment of PBMCs from Non-Diabetic Controls and PwD2 with LPS, RES, and LEC
3. Discussion
4. Materials and Methods
4.1. Experiments Prior to RNAseq Analysis
4.2. Reagents
4.3. Detection of Cell Viability and Isolation of Total Cellular RNA
4.4. Experiments Prior to RNAseq Analysis
4.5. Total Cellular RNA Isolation and qPCR
4.6. Sample Preparation for RNAseq Analysis
4.7. Data Analysis, Network and Pathway Analysis
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Singh, A.K.; Gupta, R.; Ghosh, A.; Misra, A. Diabetes in COVID-19: Prevalence, pathophysiology, prognosis and practical considerations. Diabetes Metab. Syndr. Clin. Res. Rev. 2020, 14, 303–310. [Google Scholar]
- Jesenak, M.; Brndiarova, M.; Urbancikova, I.; Rennerova, Z.; Vojtkova, J.; Bobcakova, A.; Ostro, R.; Banovcin, P. Immune parameters, and COVID-19 infection-associations with clinical severity and diseases prognosis. Front. Cell. Infect. Microbiology 2020, 10, 364. [Google Scholar] [CrossRef]
- Krug, E.G. Trends in diabetes: Sounding the alarm. Lancet 2016, 387, 1485–1486. [Google Scholar] [CrossRef]
- Wing, S.S. The UPS in diabetes and obesity. BMC Biochem. 2008, 9, S6. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, A.A.; Siddiqui, S.A.; Ahmad, S.; Siddiqui, S.; Ahsan, I.; Sahu, K. Diabetes: Mechanism, pathophysiology, and management-A review. Int. J. Drug Dev. Res. 2013, 5, 1–23. [Google Scholar]
- Leonidou, L.; Mouzaki, A.; Michalaki, M.; DeLastic, A.L.; Kyriazopoulou, V.; Bassaris, H.P.; Gogos, C.A. Cytokine production and hospital mortality in patients with sepsis-induced stress hyperglycemia. J. Infect. 2007, 55, 340–346. [Google Scholar] [CrossRef]
- Falciglia, M.; Freyberg, R.W.; Almenoff, P.L.; D’alessio, D.A.; Render, M.L. Hyperglycemia-related mortality in critically ill patients varies with admission diagnosis. Crit. Care Med. 2009, 37, 3001. [Google Scholar] [CrossRef]
- Kahn, S.E.; Hull, R.L.; Utzschneider, K.M. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 2006, 444, 840–846. [Google Scholar] [CrossRef]
- Koh, G.C.K.W.; Peacock, S.J.; Van der Poll, T.; Wiersinga, W.J. The impact of diabetes on the pathogenesis of sepsis. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 379–388. [Google Scholar] [CrossRef]
- Saisho, Y. Metformin and Inflammation: Its potential beyond glucose-lowering effect. Endocr. Metab. Immune Disord. Drug Targets. 2015, 15, 196–205. [Google Scholar] [CrossRef]
- McCreight, L.J.; Bailey, C.J.; Pearson, E.R. Metformin and the gastrointestinal tract. Diabetologia 2016, 59, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Saad, M.J.; Santos, A.; Prada, P.O. Linking gut microbiota and inflammation to obesity and insulin resistance. Physiology 2016, 31, 283–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandraki, K.; Piperi, C.; Kalofoutis, C.; Singh, J.; Alaveras, A.; Kalofoutis, A. Inflammatory process in type 2 diabetes: The role of cytokines. Ann. N. Y. Acad. Sci. 2006, 1084, 89–117. [Google Scholar] [CrossRef] [PubMed]
- Spranger, J.; Kroke, A.; Mohlig, M.; Hoffmann, K.; Bergmann, M.M.; Ristow, M.; Boeing, H.; Pfeiffer, A.F. Inflammatory cytokines and the risk to develop type 2 diabetes: Results of the prospective population-based European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam Study. Diabetes 2003, 52, 812–817. [Google Scholar] [CrossRef]
- Correa, F.O.; Gonçalves, D.; Figueredo, C.M.; Bastos, A.S.; Gustafsson, A.; Orrico, S.R. Effect of periodontal treatment on metabolic control, systemic inflammation, and cytokines in patients with type 2 diabetes. J. Clin. Periodontol. 2010, 37, 53–58. [Google Scholar] [CrossRef]
- Pickup, J.C.; Chusney, G.D.; Thomas, S.M.; Burt, D. Plasma interleukin-6, tumour necrosis factor α and blood cytokine production in type 2 diabetes. Life Sci. 2000, 67, 291–300. [Google Scholar] [CrossRef]
- Satoh, N.; Shimatsu, A.; Himeno, A.; Sasaki, Y.; Yamakage, H.; Yamada, K.; Suganami, T.; Ogawa, Y. Unbalanced M1/M2 phenotype of peripheral blood monocytes in obese diabetic patients: Effect of pioglitazone. Diabetes Care. 2010, 33, e7. [Google Scholar] [CrossRef]
- Kelly, B.; O’neill, L.A. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef]
- Kraaij, M.D.; Vereyken, E.J.; Leenen, P.J.; van den Bosch, T.P.; Rezaee, F.; Betjes, M.G.; Baan, C.C.; Rowshani, A.T. Human monocytes produce interferon-gamma upon stimulation with LPS. Cytokine 2014, 67, 7–12. [Google Scholar] [CrossRef]
- Fultz, M.J.; Barber, S.A.; Dieffenbach, C.W.; Vogel, S.N. Induction of IFN-γ in macrophages by lipopolysaccharide. Int. Immunol. 1993, 5, 1383–1392. [Google Scholar] [CrossRef]
- Jin, Y.; Lundkvist, G.; Dons, L.; Kristensson, K.; Rottenberg, M.E. Interferon-γ mediates neuronal killing of intracellular bacteria. Scand. J. Immunol. 2004, 60, 437–448. [Google Scholar] [CrossRef]
- Ouadrhiri, Y.; Scorneaux, B.; Sibille, Y.; Tulkens, P.M. Mechanism of the intracellular killing and modulation of antibiotic susceptibility of Listeria monocytogenes in THP-1 macrophages activated by gamma interferon. Antimicrob. Agents Chemother. 1999, 43, 1242–1251. [Google Scholar] [CrossRef] [Green Version]
- Herbst, S.; Schaible, U.E.; Schneider, B.E. Interferon gamma activated macrophages kill mycobacteria by nitric oxide induced apoptosis. PLoS ONE 2011, 6, e19105. [Google Scholar] [CrossRef]
- Luzina, I.G.; Keegan, A.D.; Heller, N.M.; Rook, G.A.; Shea-Donohue, T.; Atamas, S.P. Regulation of inflammation by interleukin-4: A review of “alternatives”. J. Leukoc. Biol. 2012, 92, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Kammerl, I.E.; Vosyka, O.; Baumann, T.; Yu, Y.; Wu, Y.; Irmler, M.; Overkleeft, H.S.; Beckers, J.; Eickelberg, O.; et al. Immunoproteasome dysfunction augments alternative polarization of alveolar macrophages. Cell Death Differ. 2016, 23, 1026. [Google Scholar] [CrossRef] [PubMed]
- Lecker, S.H.; Goldberg, A.L.; Mitch, W.E. Protein degradation by the ubiquitin–proteasome pathway in normal and disease states. J. Am. Soc. Nephrol. 2006, 17, 1807–1819. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, N.; Perera, P.Y.; Shen, J.; Zhang, G.; Lenschat, A.; Splitter, G.; Morrison, D.C.; Vogel, S.N. The proteasome as a lipopolysaccharide-binding protein in macrophages: Differential effects of proteasome inhibition on lipopolysaccharide-induced signaling events. J. Immunol. 2003, 171, 1515–1525. [Google Scholar] [CrossRef]
- Shen, J.; Gao, J.J.; Zhang, G.; Tan, X.; Morrison, D.C.; Papasian, C.J.; Vogel, S.N.; Qureshi, N. Proteasome inhibitor, lactacystin blocks CpG DNA-and peptidoglycan induced inflammatory genes, cytokines and mitogen-activated protein kinases in macrophages. Shock 2006, 25, 594599.5. [Google Scholar] [CrossRef]
- Shen, J.; Reis, J.; Morrison, D.C.; Papasian, C.; Raghavakaimal, S.; Kolbert, C.; Qureshi, A.A.; Vogel, S.N.; Qureshi, N. Key inflammatory signaling pathways are regulated by the proteasome. Shock 2006, 25, 472–484. [Google Scholar] [CrossRef]
- Qureshi, N.; Morrison, D.C.; Reis, J. Proteasome protease mediated regulation of cytokine induction and inflammation. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2012, 1823, 2087–2093. [Google Scholar] [CrossRef]
- Silswal, N.; Reis, J.; Qureshi, A.A.; Papasian, C.; Qureshi, N. Of Mice and Men: Proteasome’s role in LPS-induced inflammation and tolerance. Shock 2017, 47, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Rockwell, C.E.; Monaco, J.J.; Qureshi, N. A critical role for the inducible proteasomal subunits LMP7 and MECL1 in cytokine production by activated murine splenocytes. Pharmacology 2012, 89, 117–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reis, J.; Tan, X.; Yang, R.; Rockwell, C.E.; Papasian, C.J.; Vogel, S.N.; Morrison, D.C.; Qureshi, A.A.; Qureshi, N. A combination of proteasome inhibitors and antibiotics prevents lethality in a septic shock model. Innate Immun. 2008, 14, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, N.; Khan, D.A.; Zuberi, A.; Vernon, K.; Kaja, S.; Drees, B.M.; Qureshi, A.A.; Van Way, C.W.; Morrison, D.C.; Silswal, N. Levels of proteasome subunit expression provide information about host’s immune system status. Intern. Med. Rev. 2017, 3, 22. [Google Scholar] [CrossRef]
- Reis, J.; Hassan, F.; Guan, X.Q.; Shen, J.; Monaco, J.J.; Papasian, C.J.; Qureshi, A.A.; Van Way, C.W.; Vogel, S.N.; Morrison, D.C.; et al. The immunoproteasomes regulate LPS-induced TRIF/TRAM signaling pathway in murine macrophages. Cell Biochem. Biophys. 2011, 60, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Reis, J.; Guan, X.Q.; Kisselev, A.F.; Papasian, C.J.; Qureshi, A.A.; Morrison, D.C.; Van Way, C.W.; Vogel, S.N.; Qureshi, N. LPS-induced formation of immunoproteasomes: TNF-α and nitric oxide production are regulated by altered composition of proteasome-active sites. Cell Biochem. Biophys. 2011, 60, 77–88. [Google Scholar] [CrossRef]
- Morozov, A.V.; Karpov, V.L. Proteasomes and several aspects of their heterogeneity relevant to cancer. Front. Oncol. 2019, 9, 761. [Google Scholar] [CrossRef]
- Qureshi, A.A.; Guan, X.Q.; Reis, J.C.; Papasian, C.J.; Jabre, S.; Morrison, D.C.; Qureshi, N. Inhibition of nitric oxide and inflammatory cytokines in LPS-stimulated murine macrophages by resveratrol, a potent proteasome inhibitor. Lipids Health Dis. 2012, 11, 76. [Google Scholar] [CrossRef]
- Qureshi, A.A.; Tan, X.; Reis, J.C.; Badr, M.Z.; Papasian, C.J.; Morrison, D.C.; Qureshi, N. Suppression of nitric oxide induction and pro-inflammatory cytokines by novel proteasome inhibitors in various experimental models. Lipids Health Dis. 2011, 10, 177. [Google Scholar] [CrossRef]
- Silswal, N.; Reddy, N.S.; Qureshi, A.A.; Qureshi, N. Resveratrol downregulates biomarkers of sepsis Via inhibition of proteasome's proteases. Shock 2018, 50, 579–588. [Google Scholar] [CrossRef]
- Qureshi, A.A.; Zuvanich, E.G.; Khan, D.A.; Mushtaq, S.; Silswal, N.; Qureshi, N. Proteasome inhibitors modulate anticancer and anti-proliferative properties via NF-kB signaling, and ubiquitin-proteasome pathways in cancer cell lines of different organs. Lipids Health Dis. 2018, 17, 62. [Google Scholar] [CrossRef] [PubMed]
- Cianciulli, A.; Calvello, R.; Cavallo, P.; Dragone, T.; Carofiglio, V.; Panaro, M.A. Modulation of NF-kB activation by resveratrol in LPS treated human intestinal cells results in downregulation of PGE2 production and COX-2 expression. Toxicol. Vitr. 2012, 26, 1122–1128. [Google Scholar] [CrossRef] [PubMed]
- Birrell, M.A.; McCluskie, K.; Wong, S.; Donnelly, L.E.; Barnes, P.J.; Belvisi, M.G. Resveratrol, an extract of red wine, inhibits lipopolysaccharide induced airway neutrophilia and inflammatory mediators through an NF-κB-independent mechanism. FASEB J. 2005, 19, 840–841. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.E.; Damle, S.S.; Wancewicz, E.V.; Mukhopadhyay, S.; Hart, C.E.; Mazur, C.; Swayze, E.E.; Kamme, F.A. modular analysis of microglia gene expression, insights into the aged phenotype. BMC Genom. 2019, 20, 164. [Google Scholar] [CrossRef]
- Holthoff, J.H.; Wang, Z.; Seely, K.A.; Gokden, N.; Mayeux, P.R. Resveratrol improves renal microcirculation, protects the tubular epithelium, and prolongs survival in a mouse model of sepsis-induced acute kidney injury. Kidney Int. 2012, 81, 370–378. [Google Scholar] [CrossRef]
- Zong, Y.; Sun, L.; Liu, B.; Deng, Y.S.; Zhan, D.; Chen, Y.L.; He, Y.; Liu, J.; Zhang, Z.J.; Sun, J.; et al. Resveratrol inhibits LPS-induced MAPKs activation via activation of the phosphatidylinositol 3-kinase pathway in murine RAW 264.7 macrophage cells. PLoS ONE 2012, 7, e44107. [Google Scholar] [CrossRef]
- Jiang, L.; Zhang, L.; Kang, K.; Fei, D.; Gong, R.; Cao, Y.; Pan, S.; Zhao, M.; Zhao, M. Resveratrol ameliorates LPS-induced acute lung injury via NLRP3 inflammasome modulation. Biomed. Pharmacother. 2016, 84, 130–138. [Google Scholar] [CrossRef]
- Wadsworth, T.L.; Koop, D.R. Effects of the wine polyphenolics quercetin and resveratrol on pro-inflammatory cytokine expression in RAW 264.7 macrophages. Biochem. Pharmacol. 1999, 57, 941–949. [Google Scholar] [CrossRef] [PubMed]
- Liener, I. The Lectins: Properties, Functions, and Applications in Biology and Medicine; Liener, I., Ed.; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Benjamin, C.F.; Figueiredo, R.C.; Henriques, M.G.; Barja-Fidalgo, C. Inflammatory and anti-inflammatory effects of soybean agglutinin. Braz. J. Med. Biol. 1997, 30, 873–881. [Google Scholar] [CrossRef]
- Gong, T.; Wang, X.; Yang, Y.; Yan, Y.; Yu, C.; Zhou, R.; Jiang, W. Plant lectins activate the NLRP3 inflammasome to promote inflammatory disorders. J. Immunol. 2017, 198, 2082–2092. [Google Scholar] [CrossRef]
- Wang, K.; Liu, C.; Hou, Y.; Zhou, H.; Wang, X.; Mai, K.; He, G. Differential apoptotic and mitogenic effects of lectins in zebrafish. Front. Endocrinol. 2019, 10, 356. [Google Scholar] [CrossRef]
- Mishra, A.; Behura, A.; Mawatwal, S.; Kumar, A.; Naik, L.; Mohanty, S.S.; Manna, D.; Dokania, P.; Mishra, A.; Patra, S.K.; et al. Structure-function, and application of plant lectins in disease biology and immunity. Food Chem. Toxicol. 2019, 134, 110827. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, N.; Desousa, J.; Siddiqui, A.Z.; Morrison, D.C.; Qureshi, A.A. Reprograming of Gene Expression of Key Inflammatory Signaling Pathways in Human Peripheral Blood Mononuclear Cells by Soybean Lectin and Resveratrol. Int. J. Mol. Sci. 2022, 23, 12946. [Google Scholar] [CrossRef] [PubMed]
- Hulme, M.A.; Wasserfall, C.H.; Atkinson, M.A.; Brusko, T.M. Central role for interleukin-2 in type 1 diabetes. Diabetes 2012, 61, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Al Dubayee, M.; Alshahrani, A.; Aljada, D.; Zahra, M.; Alotaibi, A.; Ababtain, I.; Alnaim, M.; Alahmari, A.; Aljarallah, A.; Elahi, M.A.; et al. Gene expression profiling of apoptotic proteins in circulating peripheral blood mononuclear cells in type II diabetes mellitus and modulation by metformin. Diabetes Metab. Syndr. Obes. Targets Ther. 2021, 14, 1129. [Google Scholar] [CrossRef] [PubMed]
- Titov, A.A.; Baker, H.V.; Brusko, T.M.; Sobel, E.S.; Morel, L. Metformin inhibits the type 1 IFN response in human CD4+ T cells. J. Immunol. 2019, 203, 338–348. [Google Scholar] [CrossRef] [PubMed]
- de Kreutzenberg, S.V.; Ceolotto, G.; Cattelan, A.; Pagnin, E.; Mazzucato, M.; Garagnani, P.; Borelli, V.; Bacalini, M.G.; Franceschi, C.; Fadini, G.P.; et al. Metformin improves putative longevity effectors in peripheral mononuclear cells from subjects with prediabetes. A randomized controlled trial. Nutr. Metab. Cardiovasc. Dis. 2015, 25, 686–693. [Google Scholar] [CrossRef]
- Rabinovitch, A.; Suarez-Pinzon, W.L.; Sorensen, O.; Bleackley, R.C.; Power, R.F. IFN-gamma gene expression in pancreatic islet-infiltrating mononuclear cells correlates with autoimmune diabetes in nonobese diabetic mice. J. Immunol. 1995, 154, 874–882. [Google Scholar] [CrossRef]
- Pharmacologic Approaches to Glycemic Treatment. Am. Diabetes Assoc. Diabetes Care 2021, 44 (Suppl. 1), S111–S124. [CrossRef]
- Kartika, R.; Purnamasari, D.; Pradipta, S.; Larasati, R.A.; Wibowo, H. Impact of low interferon-γ and il-10 levels on TNF-α and il-6 production by PHA-induced pbmcs in type 2 diabetes mellitus. J. Inflamm. Res. 2020, 13, 187. [Google Scholar] [CrossRef]
- Tsiavou, A.; Hatziagelaki, E.; Chaidaroglou, A.; Koniavitou, K.; Degiannis, D.; Raptis, S.A. Correlation between intracellular interferon-γ (IFN-γ) production by CD4+ and CD8+ lymphocytes and IFN-γ gene polymorphism in patients with type 2 diabetes mellitus and latent autoimmune diabetes of adults (LADA). Cytokine 2005, 31, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Willmer, T.; Cooper, A.; Peres, J.; Omar, R.; Prince, S. The T-Box transcription factor 3 in development and cancer. Biosci. Trends 2017, 11, 254–266. [Google Scholar] [CrossRef] [Green Version]
- Munafo, M.R.; Thiselton, D.L.; Clark, T.G.; Flint, J. Association of the NRG1 gene and schizophrenia: A meta-analysis. Mol. Psychiatry 2006, 11, 539–546. [Google Scholar] [CrossRef]
- Poggi, M.; Bastelica, D.; Gual, P.; Iglesias, M.A.; Gremeaux, T.; Knauf, C.; Peiretti, F.; Verdier, M.; Juhan-Vague, I.; Tanti, J.F.; et al. C3H/HeJ mice carrying a toll-like receptor 4 mutation are protected against the development of insulin resistance in white adipose tissue in response to a high-fat diet. Diabetologia 2007, 50, 1267–1276. [Google Scholar] [CrossRef]
- Sugita, H.; Kaneki, M.; Tokunaga, E.; Sugita, M.; Koike, C.; Yasuhara, S.; Tompkins, R.G.; Martyn, J.J. Inducible nitric oxide synthase plays a role in LPS-induced hyperglycemia and insulin resistance. Am. J. Physiol.-Endocrinol. Metab. 2002, 282, E386–E394. [Google Scholar] [CrossRef]
- Lassenius, M.I.; Pietiläinen, K.H.; Kaartinen, K.; Pussinen, P.J.; Syrjänen, J.; Forsblom, C.; Pörsti, I.; Rissanen, A.; Kaprio, J.; Mustonen, J.; et al. Bacterial endotoxin activity in human serum is associated with dyslipidemia, insulin resistance, obesity, and chronic inflammation. Diabetes Care. 2011, 34, 1809–1815. [Google Scholar] [CrossRef] [PubMed]
- Jayashree, B.; Bibin, Y.S.; Prabhu, D.; Shanthirani, C.S.; Gokulakrishnan, K.; Lakshmi, B.S.; Mohan, V.; Balasubramanyam, M. Increased circulatory levels of lipopolysaccharide (LPS) and zonulin signify novel biomarkers of proinflammation in patients with type 2 diabetes. Mol. Cell. Biochem. 2014, 388, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Gierl, M.S.; Karoulias, N.; Wende, H.; Strehle, M.; Birchmeier, C. The zinc-finger factor Insm1 (IA-1) is essential for the development of pancreatic β cells and intestinal endocrine cells. Genes Dev. 2006, 20, 2465–2478. [Google Scholar] [CrossRef]
- Hersoug, L.G.; Møller, P.; Loft, S. Gut microbiota-derived lipopolysaccharide uptake and trafficking to adipose tissue: Implications for inflammation and obesity. Obes. Rev. 2016, 17, 297–312. [Google Scholar] [CrossRef]
- Carvalho, B.M.; Guadagnini, D.; Tsukumo, D.M.; Schenka, A.A.; Latuf-Filho, P.; Vassallo, J.; Dias, J.C.; Kubota, L.T.; Carvalheira, J.B.; Saad, M.J. Modulation of gut microbiota by antibiotics improves insulin signaling in high-fat fed mice. Diabetologia 2012, 55, 2823–2834. [Google Scholar] [CrossRef]
- Qureshi, A.A.; Khan, D.A.; Mahjabeen, W.; Silswal, N.; Qureshi, N. A novel mixture of d-tocotrienol, vitamin D3, and resveratrol significantly decreases diabetes biomarkers including inflammatory, in people with type 2 diabetes. J. Diabetes Clin. Stud. 2021, 5, 2258. [Google Scholar]
- Pusztai, A.; Bardocz, S.; Ewen, S.W. Uses of plant lectins in bioscience and biomedicine. Front. Biosci. 2008, 13, 1130–1140. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, N.; Takayama, K.; Mascagni, P.; Honovich, J.; Wong, R.O.; Cotter, R.J. Complete structural determination of lipopolysaccharide obtained from deep rough mutant of Escherichia coli. Purification by high performance liquid chromatography and direct analysis by plasma desorption mass spectrometry. J. Biol. Chem. 1988, 263, 1971–1976. [Google Scholar] [CrossRef]
- Qureshi, N.; Takayama, K.; Ribi, E. Purification, and structural determination of nontoxic lipid A from the rough mutant of Salmonella typhimurium. J. Biol. Chem. 1982, 257, 11808–11815. [Google Scholar] [CrossRef]
- Qureshi, N.; Takayama, K.; Heller, D.; Fenselau, C. Position of ester groups in the lipid A backbone of lipopolysaccharides obtained from Salmonella typhimurium. J. Biol. Chem. 1983, 258, 12947–12951. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Treatments | Total DEGS | Upregulated | Downregulated |
| 1 | Control non-diabetics | |||
| 2 | Resveratrol (RES) | 26 | 4 | 22 |
| 3 | Lipopolysaccharide (LPS) | 680 | 555 | 125 |
| 4 | LPS + RES | 424 | 290 | 133 |
| 5 | Lectin10 (LEC10) | 295 | 207 | 28 |
| 6 | LEC10 + LPS | 640 | 534 | 106 |
| 7 | LEC50 | 570 | 492 | 78 |
| 8 | LEC50 + LPS | 698 | 588 | 110 |
| Treatments | Total DEGS | Upregulated | Downregulated | |
| 9 | Control PwD2 * | |||
| 10 | Diabetes-Resveratrol (DRES) | 40 | 14 | 26 |
| 11 | Diabetes-Lipopolysaccharide (DLPS) | 472 | 377 | 95 |
| 12 | DLPS + RES | 426 | 275 | 151 |
| 13 | Diabetes-Lectin10 (DLEC10) | 287 | 249 | 38 |
| 14 | DLEC10 + LPS | 469 | 382 | 87 |
| 15 | Diabetes-Lectin 50 (DLEC50) | 489 | 384 | 105 |
| 16 | DLEC50 + LPS | 494 | 383 | 111 |
| CR | DR | CL | DL | CLR | DLR | CLEC10 | DLEC10 | |
|---|---|---|---|---|---|---|---|---|
| IL6 | −0.095 | 0.75 | 9.67 | 9.729 | 10.139 | 10.38 | 9.724 | 9.691 |
| IL1B | −0.956 | −1.096 | 7.304 | 6.026 | 7.467 | 6.401 | 7.38 | 6.32 |
| PTGS2 | −0.541 | −0.338 | 7.018 | 6.09 | 7.396 | 6.733 | 7.012 | 6.349 |
| TNF | −0.043 | 0.074 | 5.790 | 3.108 | 5.392 | 3.488 | 5.986 | 3.38 |
| CXCL10 | 0.873 | −0.303 | 4.645 | 1.922 | 2.06 | −0.226 | 4.560 | 1.245 |
| IFNG | 0.190 | −0.453 | 4.913 | 1.123 | 1.586 | −0.45 | 4.953 | 0.144 |
| CXCL8 | −0.893 | −0.267 | 3.913 | 2.696 | 3.845 | 2.894 | 3.974 | 2.832 |
| IL12B | 9.513 | 9.276 | 8.337 | 8.162 | 9.761 | 7.834 | ||
| HMOX1 | 0.321 | 0.42 | −3.322 | −3.731 | −2.866 | −3.902 | −3.399 | −3.252 |
| IL12A | 2.13 | −1.496 | 6.216 | 1.556 | 4.646 | 1.435 | 6.08 | −0.314 |
| NLRP3 | 0.361 | 0.315 | 3.019 | 2.53 | 4.083 | 3.449 | 3.124 | 3.126 |
| TLR7 | −0.116 | −0.294 | 3.156 | 2.313 | 1.854 | 1.369 | 3.089 | 1.665 |
| SOD2 | 0.783 | 0.839 | 2.787 | 2.462 | 2.704 | 2.335 | 2.821 | 2.321 |
| CD80 | 0.936 | −0.139 | 3.120 | 1.125 | 1.541 | 0.629 | 3.183 | 1.631 |
| CD40 | 0.383 | 0.422 | 1.284 | 0.61 | 1.495 | 1.232 | 1.188 | 0.274 |
| SLC1A2 | −0.23 | −1.222 | 3.167 | 2.706 | 0.523 | 0.084 | 2.843 | 1.875 |
| IFNB1 | 8.443 | 6.524 | 7.453 | 7.167 | 8.194 | 6.013 | ||
| IRF7 | 0.178 | 0.573 | 2.492 | 1.989 | 1.519 | 1.083 | 2.556 | 1.112 |
| HLA-DOB | 1.360 | 0.887 | 3.122 | 1.723 | 2.148 | 0.853 | 3.206 | 0.606 |
| TGFB2 | 1.636 | −1.054 | 3.349 | 0.607 | 1.609 | −0.374 | 3.377 | 1.654 |
| NFKB1 | −0.063 | −0.142 | 1.412 | 1.211 | 1.458 | 1.198 | 1.449 | 1.145 |
| FAS | −0.392 | −0.555 | 1.405 | 0.9 | 0.739 | 0.26 | 1.473 | 0.441 |
| IRAK2 | 0.039 | 0.158 | 1.358 | 1.127 | 1.015 | 0.677 | 1.453 | 1.165 |
| STAT1 | 0.09 | −0.429 | 1.356 | 0.897 | −0.206 | −0.656 | 1.249 | 0.245 |
| TICAM1 | −0.432 | −0.352 | 1.335 | 0.986 | 0.433 | 0.098 | 1.339 | 0.695 |
| PLA2G4A | −1.374 | −1.702 | 1.317 | 0.145 | 0.181 | −0.109 | 1.088 | −0.101 |
| ZBTB12 | 1.68 | 0.759 | 2.26 | 1.236 | 2.055 | 1.81 | 1.262 | 1.175 |
| IL1R1 | −0.904 | −1.196 | 1.184 | 0.76 | 0.095 | −0.138 | 1.17 | 0.242 |
| ACVR1C | 0.034 | −1.107 | 1.495 | 0.388 | 0.464 | −0.167 | 1.303 | 0.457 |
| JAK3 | 0.123 | 0.108 | 1.118 | 0.888 | 0.904 | 0.883 | 1.131 | 0.779 |
| TGFB3 | −0.614 | −0.287 | 1.424 | 0.306 | 0.052 | −0.159 | 0.377 | 0.206 |
| GLS | 0.31 | 0.185 | 1.060 | 0.597 | 0.688 | 0.583 | 0.893 | 0.422 |
| MYD88 | −0.249 | −0.367 | 1.05 | 0.412 | −0.134 | −0.413 | 0.975 | 0.282 |
| FASLG | −0.542 | −1.166 | 1.085 | 0.461 | 0.076 | −0.813 | 1.355 | −0.275 |
| RIPK1 | 0.128 | 0.08 | 1.031 | 0.566 | 0.485 | 0.413 | 1.012 | 0.156 |
| PIK3R3 | −0.266 | −1.235 | 1.331 | 0.261 | −1.642 | −2.063 | 1.167 | −0.67 |
| CTLA4 | −0.119 | −0.436 | 1.171 | 0.604 | 0.451 | −0.204 | 1.181 | 0.651 |
| END1 | 1.4 | −0.547 | 4.901 | 2.506 | 1.302 | 1.176 | 4.967 | 2.248 |
| ATG7 | −0.38 | −0.202 | 1.167 | 1.374 | 0.767 | 1.184 | 1.228 | 1.310 |
| TRIP10 | −0.192 | 0.239 | 2.73 | 3.317 | 2.621 | 3.132 | 2.741 | 2.721 |
| PSMB8 | −0.325 | −0.147 | 0.903 | 0.385 | −0.154 | −0.424 | 0.964 | 0.415 |
| PSMB9 | 0.574 | 0.084 | 1.097 | 0.437 | 0.446 | 0.673 | 1.103 | 0.378 |
| Expt 1 | Sex | Age | Race | Weight | Drug/Metformin |
|---|---|---|---|---|---|
| PwD2 PBMCs | Male | 49 yrs | Caucasian | 90 kg | Yes |
| Non-diabetic control PBMCs | Male | 27 yrs | African Amer. | 112 kg | No |
| Expt 2 | |||||
| PwD2 PBMCs | Female | 52 yrs | Caucasian | 110 kg | Yes |
| Non-diabetic control PBMCs | Female | 49 yrs | Caucasian | 63 kg | No |
| Expt 3 | |||||
| PwD2 PBMCs | Male | 49 yrs | Caucasian | 90 kg | Yes |
| Non-diabetic control PBMCs | Male | 54 yrs | Caucasian | 105 kg | No |
| Expt 4 | |||||
| PwD2 PBMCs | Female | 52 yrs | Caucasian | 110 kg | Yes |
| Non-diabetic control PBMCs | Female | 48 yrs | Caucasian | 106 kg | No |
| Expt 5 | |||||
| PwD2 PBMCs | Male | 65 yrs | Caucasian | 108 kg | Yes |
| Non-diabetic control PBMCs | Male | 60 yrs | Caucasian | 92 kg | No |
| 1. | Control | 9. | Control |
| 2. | RES 80 µM | 10. | RES 80 µM |
| 3. | LPS 10 ng | 11. | LPS 10 ng |
| 4. | LPS + RES | 12. | LPS + RES |
| 5. | LEC10 µg | 13. | LEC10 µg |
| 6. | LEC10 + LPS | 14. | LEC10 + LPS |
| 7. | LEC50 µg | 15. | LEC50 µg |
| 8. | LEC50 + LPS | 16. | LEC50 + LPS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qureshi, N.; Desousa, J.; Siddiqui, A.Z.; Drees, B.M.; Morrison, D.C.; Qureshi, A.A. Dysregulation of Gene Expression of Key Signaling Mediators in PBMCs from People with Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2023, 24, 2732. https://doi.org/10.3390/ijms24032732
Qureshi N, Desousa J, Siddiqui AZ, Drees BM, Morrison DC, Qureshi AA. Dysregulation of Gene Expression of Key Signaling Mediators in PBMCs from People with Type 2 Diabetes Mellitus. International Journal of Molecular Sciences. 2023; 24(3):2732. https://doi.org/10.3390/ijms24032732
Chicago/Turabian StyleQureshi, Nilofer, Julia Desousa, Adeela Z. Siddiqui, Betty M. Drees, David C. Morrison, and Asaf A. Qureshi. 2023. "Dysregulation of Gene Expression of Key Signaling Mediators in PBMCs from People with Type 2 Diabetes Mellitus" International Journal of Molecular Sciences 24, no. 3: 2732. https://doi.org/10.3390/ijms24032732