
Understanding the Roles of the Hedgehog Signaling Pathway during T-Cell Lymphopoiesis and in T-Cell Acute Lymphoblastic Leukemia (T-ALL)
 ,
,  ,
,
Abstract
:1. Introduction
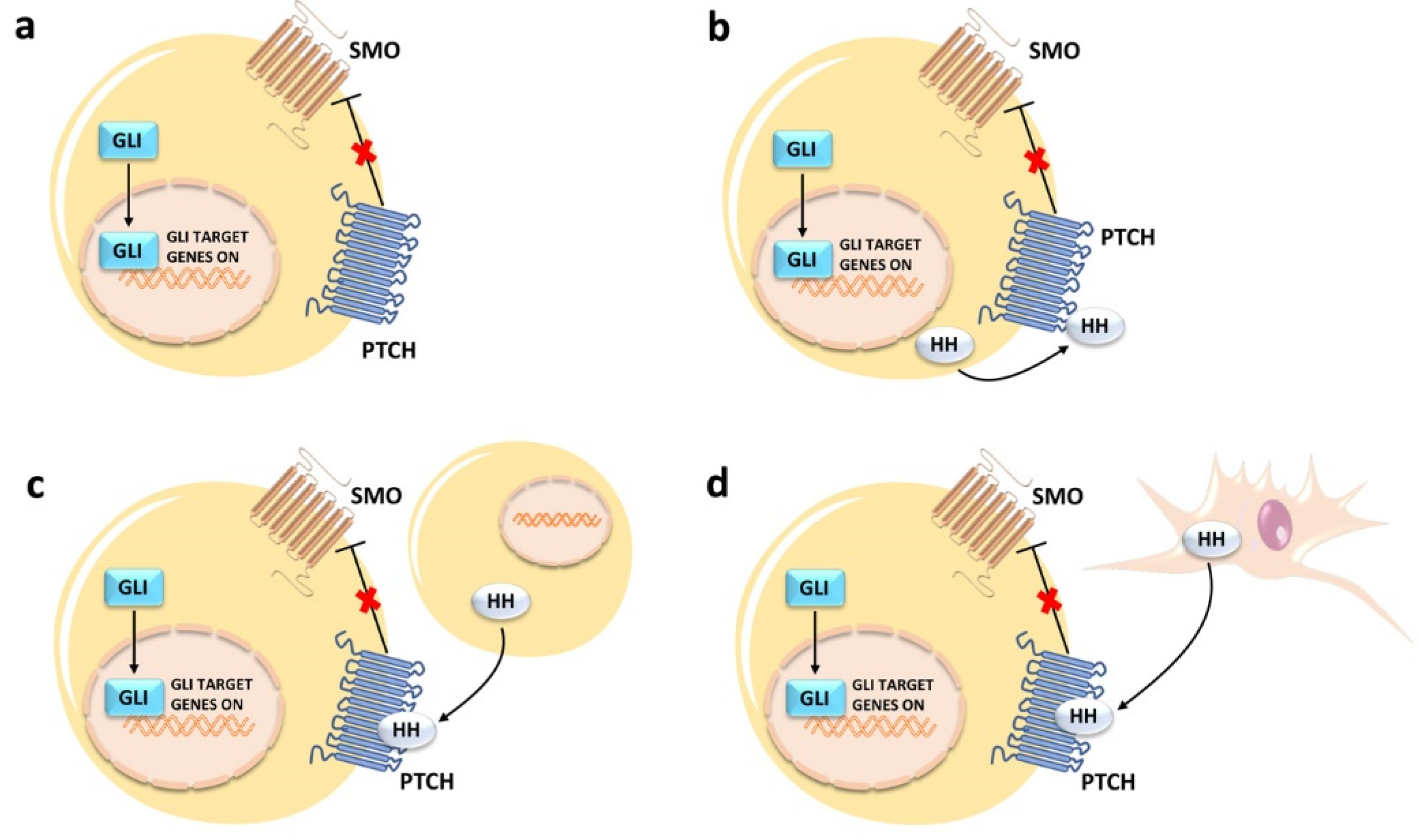
2. An Overview of Canonical HH Signaling Pathway
3. Deregulated Canonical and Noncanonical HH Signaling in Cancer Cells
3.1. Canonical Activation
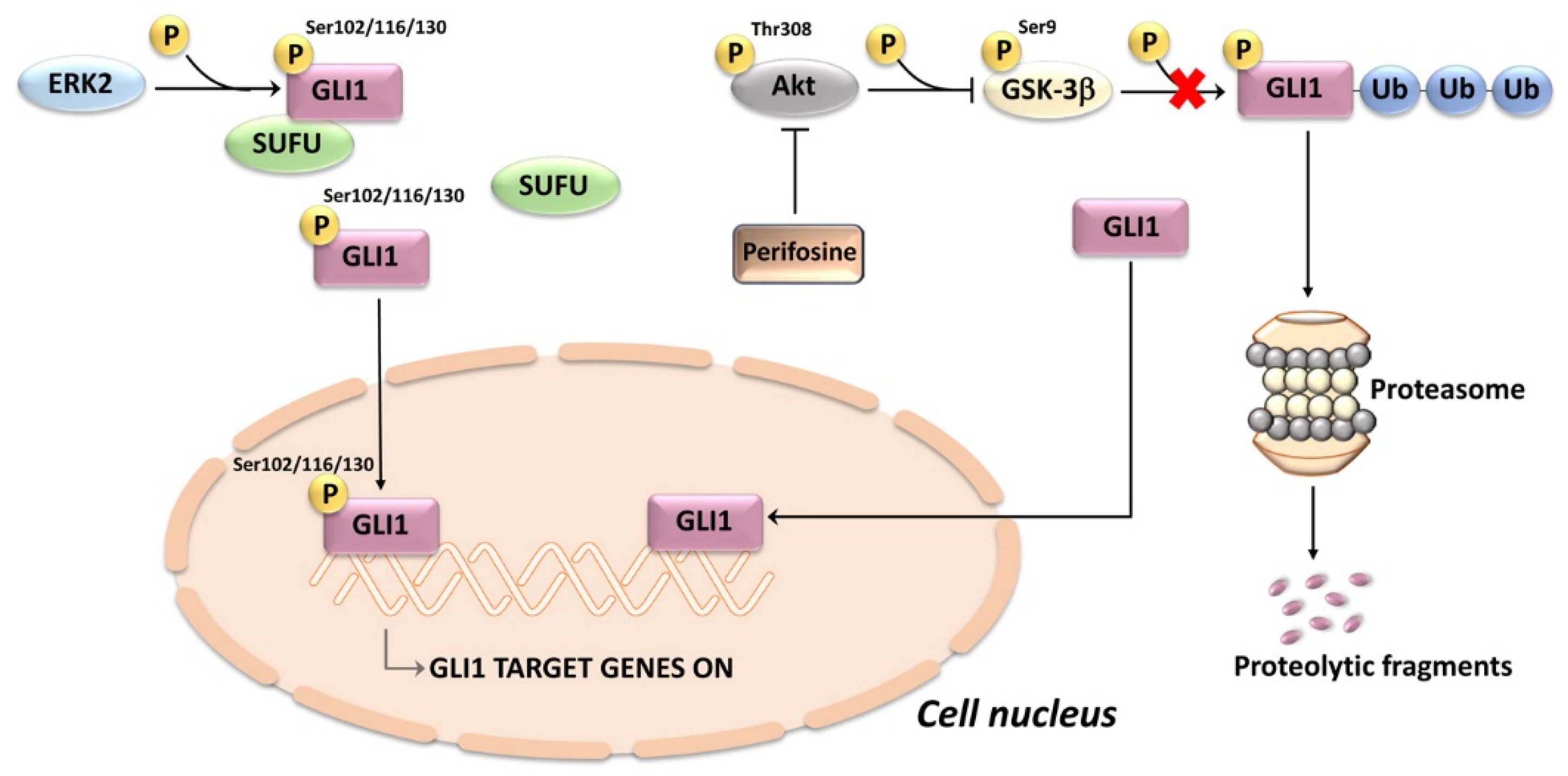
3.2. Noncanonical Activation
4. Therapeutic Targeting of HH in Cancer Cells
4.1. SMO Inhibitors
4.2. Targeting GLI Transcription Factors
4.3. Targeting HH Signaling via Nanoparticles
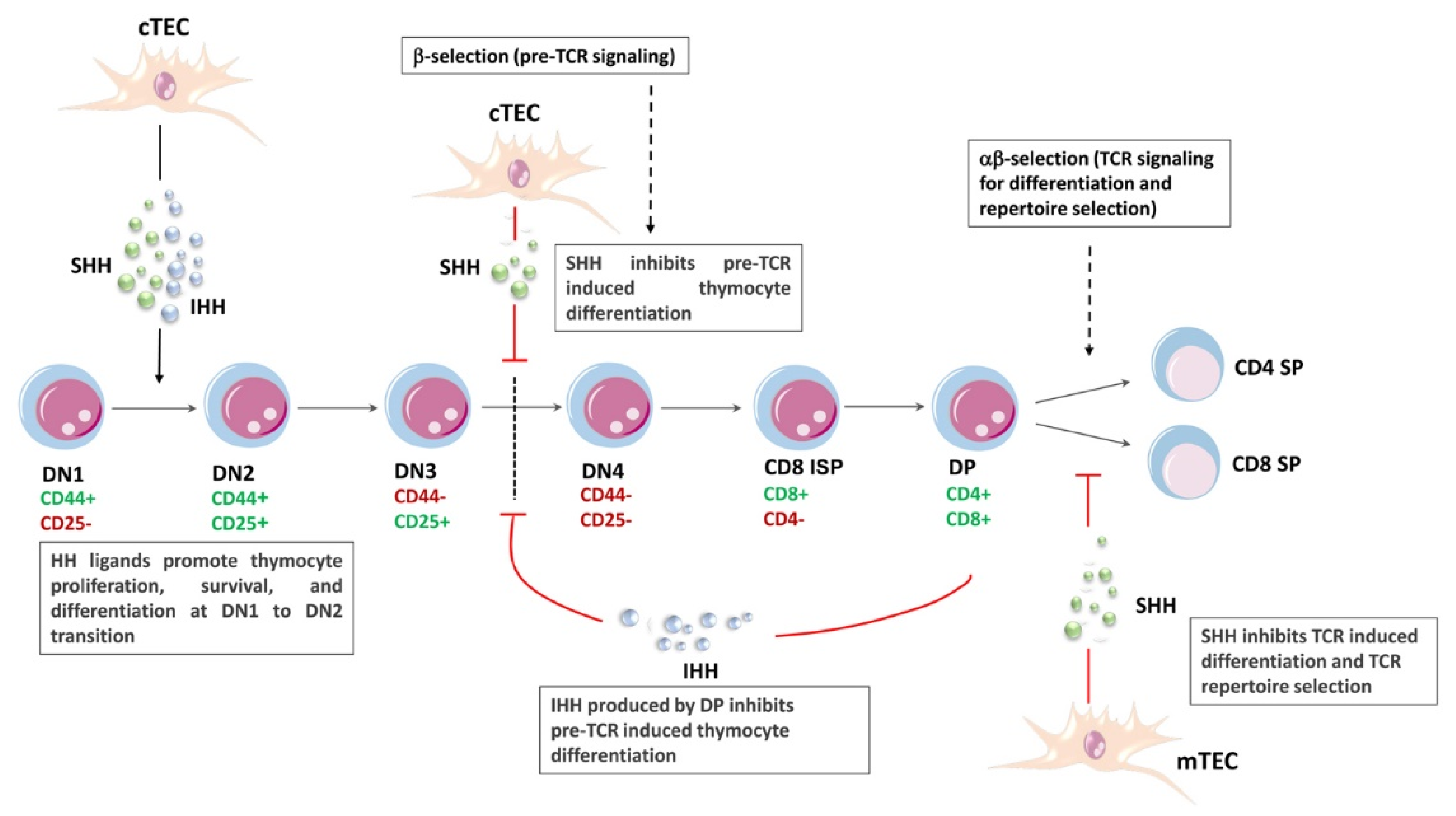
5. HH Signaling and T-Cell Lymphopoiesis
6. T-ALL
7. HH Signaling in T-ALL
7.1. Mechanisms of HH Signaling Activation in T-ALL
7.1.1. Activating Mutations
7.1.2. Autocrine Mechanism of HH Activation
7.1.3. Noncanonical Mechanisms of HH Activation and Crosstalk with Other Networks
7.1.4. Regulation through Non-Coding RNAs
7.2. Oncogenic Potential of HH Mutations in T-ALL
7.3. Therapeutic Targeting of HH Signaling in T-ALL
8. Perspectives and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nusslein-Volhard, C.; Wieschaus, E. Mutations affecting segment number and polarity in Drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes. Dev. 2001, 15, 3059–3087. [Google Scholar] [CrossRef]
- Varjosalo, M.; Taipale, J. Hedgehog: Functions and mechanisms. Genes. Dev. 2008, 22, 2454–2472. [Google Scholar] [CrossRef] [PubMed]
- Doheny, D.; Manore, S.G.; Wong, G.L.; Lo, H.W. Hedgehog Signaling and Truncated GLI1 in Cancer. Cells 2020, 9, 2114. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, C.; Martinelli, G.; Papayannidis, C.; Cortes, J.E. Hedgehog Pathway Inhibitors: A New Therapeutic Class for the Treatment of Acute Myeloid Leukemia. Blood Cancer Discov. 2020, 1, 134–145. [Google Scholar] [CrossRef]
- Kantarjian, H.; Short, N.J.; DiNardo, C.; Stein, E.M.; Daver, N.; Perl, A.E.; Wang, E.S.; Wei, A.; Tallman, M. Harnessing the benefits of available targeted therapies in acute myeloid leukaemia. Lancet. Haematol. 2021, 8, e922–e933. [Google Scholar] [CrossRef]
- Pui, C.H.; Robison, L.L.; Look, A.T. Acute lymphoblastic leukaemia. Lancet. 2008, 371, 1030–1043. [Google Scholar] [CrossRef]
- Bongiovanni, D.; Saccomani, V.; Piovan, E. Aberrant Signaling Pathways in T-Cell Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2017, 18, 1904. [Google Scholar] [CrossRef]
- Dagklis, A.; Pauwels, D.; Lahortiga, I.; Geerdens, E.; Bittoun, E.; Cauwelier, B.; Tousseyn, T.; Uyttebroeck, A.; Maertens, J.; Verhoef, G.; et al. Hedgehog pathway mutations in T-cell acute lymphoblastic leukemia. Haematologica. 2015, 100, e102–e105. [Google Scholar] [CrossRef]
- Dagklis, A.; Demeyer, S.; De Bie, J.; Radaelli, E.; Pauwels, D.; Degryse, S.; Gielen, O.; Vicente, C.; Vandepoel, R.; Geerdens, E.; et al. Hedgehog pathway activation in T-cell acute lymphoblastic leukemia predicts response to SMO and GLI1 inhibitors. Blood 2016, 128, 2642–2654. [Google Scholar] [CrossRef] [Green Version]
- Sigafoos, A.N.; Paradise, B.D.; Fernandez-Zapico, M.E. Hedgehog/GLI Signaling Pathway: Transduction, Regulation, and Implications for Disease. Cancers. 2021, 13, 3410. [Google Scholar] [CrossRef]
- Jaiswal, A.; Singh, R. Homeostases of epidermis and hair follicle, and development of basal cell carcinoma. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188795. [Google Scholar] [CrossRef]
- Locker, M.; Agathocleous, M.; Amato, M.A.; Parain, K.; Harris, W.A.; Perron, M. Hedgehog signaling and the retina: Insights into the mechanisms controlling the proliferative properties of neural precursors. Genes. Dev. 2006, 20, 3036–3048. [Google Scholar] [CrossRef]
- Li, X.; Li, Y.; Li, S.; Li, H.; Yang, C.; Lin, J. The role of Shh signalling pathway in central nervous system development and related diseases. Cell Biochem. Funct. 2021, 39, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.Y.; Sun, L.; Veltmaat, J.M. Hedgehog and Gli signaling in embryonic mammary gland development. J. Mammary. Gland. Biol. Neoplasia. 2013, 18, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Fendrich, V.; Esni, F.; Garay, M.V.; Feldmann, G.; Habbe, N.; Jensen, J.N.; Dor, Y.; Stoffers, D.; Jensen, J.; Leach, S.D.; et al. Hedgehog signaling is required for effective regeneration of exocrine pancreas. Gastroenterology. 2008, 135, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lipinski, R.J.; Gipp, J.J.; Shaw, A.K.; Bushman, W. Hedgehog pathway responsiveness correlates with the presence of primary cilia on prostate stromal cells. BMC. Dev. Biol. 2009, 9, 50. [Google Scholar] [CrossRef] [PubMed]
- Watkins, D.N.; Berman, D.M.; Burkholder, S.G.; Wang, B.; Beachy, P.A.; Baylin, S.B. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature 2003, 422, 313–317. [Google Scholar] [CrossRef]
- Le, H.; Kleinerman, R.; Lerman, O.Z.; Brown, D.; Galiano, R.; Gurtner, G.C.; Warren, S.M.; Levine, J.P.; Saadeh, P.B. Hedgehog signaling is essential for normal wound healing. Wound. Repair. Regen. 2008, 16, 768–773. [Google Scholar] [CrossRef]
- Tesanovic, S.; Krenn, P.W.; Aberger, F. Hedgehog/GLI signaling in hematopoietic development and acute myeloid leukemia-From bench to bedside. Front. Cell Dev. Biol. 2022, 10, 944760. [Google Scholar] [CrossRef]
- Ryan, K.E.; Chiang, C. Hedgehog secretion and signal transduction in vertebrates. J. Biol. Chem. 2012, 287, 17905–17913. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, I.; Liang, J.; Hedeen, D.; Roberts, K.J.; Zhang, Y.; Ha, B.; Latorraca, N.R.; Faust, B.; Dror, R.O.; Beachy, P.A.; et al. Smoothened stimulation by membrane sterols drives Hedgehog pathway activity. Nature 2019, 571, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Higgins, M.; Obaidi, I.; McMorrow, T. Primary cilia and their role in cancer. Oncol. Lett. 2019, 17, 3041–3047. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Chaudhry, P.; Merchant, A.A. Primary cilia are present on human blood and bone marrow cells and mediate Hedgehog signaling. Exp. Hematol. 2016, 44, 1181–1187.e1182. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Li, X. Mechanistic Insights into the Generation and Transduction of Hedgehog Signaling. Trends Biochem. Sci. 2020, 45, 397–410. [Google Scholar] [CrossRef]
- Kogerman, P.; Grimm, T.; Kogerman, L.; Krause, D.; Unden, A.B.; Sandstedt, B.; Toftgard, R.; Zaphiropoulos, P.G. Mammalian suppressor-of-fused modulates nuclear-cytoplasmic shuttling of Gli-1. Nat. Cell Biol. 1999, 1, 312–319. [Google Scholar] [CrossRef]
- Tukachinsky, H.; Lopez, L.V.; Salic, A. A mechanism for vertebrate Hedgehog signaling: Recruitment to cilia and dissociation of SuFu-Gli protein complexes. J. Cell Biol. 2010, 191, 415–428. [Google Scholar] [CrossRef]
- Kasper, M.; Regl, G.; Frischauf, A.M.; Aberger, F. GLI transcription factors: Mediators of oncogenic Hedgehog signalling. Eur. J. Cancer 2006, 42, 437–445. [Google Scholar] [CrossRef]
- Pan, Y.; Wang, B. A novel protein-processing domain in Gli2 and Gli3 differentially blocks complete protein degradation by the proteasome. J. Biol. Chem. 2007, 282, 10846–10852. [Google Scholar] [CrossRef]
- Niewiadomski, P.; Kong, J.H.; Ahrends, R.; Ma, Y.; Humke, E.W.; Khan, S.; Teruel, M.N.; Novitch, B.G.; Rohatgi, R. Gli protein activity is controlled by multisite phosphorylation in vertebrate Hedgehog signaling. Cell Rep. 2014, 6, 168–181. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, K.; Fukushima, H.; Ogura, K.; Lien, E.C.; Nihira, N.T.; Zhang, J.; North, B.J.; Guo, A.; Nagashima, K.; Nakagawa, T.; et al. The SCFbeta-TRCP E3 ubiquitin ligase complex targets Lipin1 for ubiquitination and degradation to promote hepatic lipogenesis. Sci. Signal 2017, 10, eaah4117. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Bai, C.B.; Joyner, A.L.; Wang, B. Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol. Cell Biol. 2006, 26, 3365–3377. [Google Scholar] [CrossRef] [PubMed]
- Stone, D.M.; Hynes, M.; Armanini, M.; Swanson, T.A.; Gu, Q.; Johnson, R.L.; Scott, M.P.; Pennica, D.; Goddard, A.; Phillips, H.; et al. The tumour-suppressor gene patched encodes a candidate receptor for Sonic hedgehog. Nature 1996, 384, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Taipale, J.; Beachy, P.A. The Hedgehog and Wnt signalling pathways in cancer. Nature 2001, 411, 349–354. [Google Scholar] [CrossRef]
- Corbit, K.C.; Aanstad, P.; Singla, V.; Norman, A.R.; Stainier, D.Y.; Reiter, J.F. Vertebrate Smoothened functions at the primary cilium. Nature 2005, 437, 1018–1021. [Google Scholar] [CrossRef]
- Rohatgi, R.; Milenkovic, L.; Scott, M.P. Patched1 regulates hedgehog signaling at the primary cilium. Science. 2007, 317, 372–376. [Google Scholar] [CrossRef]
- Qian, H.; Cao, P.; Hu, M.; Gao, S.; Yan, N.; Gong, X. Inhibition of tetrameric Patched1 by Sonic Hedgehog through an asymmetric paradigm. Nat. Commun. 2019, 10, 2320. [Google Scholar] [CrossRef]
- Chen, Y.; Sasai, N.; Ma, G.; Yue, T.; Jia, J.; Briscoe, J.; Jiang, J. Sonic Hedgehog dependent phosphorylation by CK1alpha and GRK2 is required for ciliary accumulation and activation of smoothened. PLoS Biol. 2011, 9, e1001083. [Google Scholar] [CrossRef]
- Kovacs, J.J.; Whalen, E.J.; Liu, R.; Xiao, K.; Kim, J.; Chen, M.; Wang, J.; Chen, W.; Lefkowitz, R.J. Beta-arrestin-mediated localization of smoothened to the primary cilium. Science. 2008, 320, 1777–1781. [Google Scholar] [CrossRef]
- Milenkovic, L.; Scott, M.P.; Rohatgi, R. Lateral transport of Smoothened from the plasma membrane to the membrane of the cilium. J. Cell Biol. 2009, 187, 365–374. [Google Scholar] [CrossRef] [Green Version]
- Dorn, K.V.; Hughes, C.E.; Rohatgi, R. A Smoothened-Evc2 complex transduces the Hedgehog signal at primary cilia. Dev. Cell 2012, 23, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Chen, W.; Chen, Y.; Jiang, J. Smoothened transduces Hedgehog signal by forming a complex with Evc/Evc2. Cell Res. 2012, 22, 1593–1604. [Google Scholar] [CrossRef] [PubMed]
- Caparros-Martin, J.A.; Valencia, M.; Reytor, E.; Pacheco, M.; Fernandez, M.; Perez-Aytes, A.; Gean, E.; Lapunzina, P.; Peters, H.; Goodship, J.A.; et al. The ciliary Evc/Evc2 complex interacts with Smo and controls Hedgehog pathway activity in chondrocytes by regulating Sufu/Gli3 dissociation and Gli3 trafficking in primary cilia. Hum. Mol. Genet. 2013, 22, 124–139. [Google Scholar] [CrossRef]
- Humke, E.W.; Dorn, K.V.; Milenkovic, L.; Scott, M.P.; Rohatgi, R. The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes. Dev. 2010, 24, 670–682. [Google Scholar] [CrossRef] [PubMed]
- Pietrobono, S.; Gagliardi, S.; Stecca, B. Non-canonical Hedgehog Signaling Pathway in Cancer: Activation of GLI Transcription Factors Beyond Smoothened. Front. Genet. 2019, 10, 556. [Google Scholar] [CrossRef] [PubMed]
- Belloni, E.; Muenke, M.; Roessler, E.; Traverso, G.; Siegel-Bartelt, J.; Frumkin, A.; Mitchell, H.F.; Donis-Keller, H.; Helms, C.; Hing, A.V.; et al. Identification of Sonic hedgehog as a candidate gene responsible for holoprosencephaly. Nat. Genet. 1996, 14, 353–356. [Google Scholar] [CrossRef]
- Chiang, C.; Litingtung, Y.; Lee, E.; Young, K.E.; Corden, J.L.; Westphal, H.; Beachy, P.A. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature 1996, 383, 407–413. [Google Scholar] [CrossRef]
- Yang, Y.; Guillot, P.; Boyd, Y.; Lyon, M.F.; McMahon, A.P. Evidence that preaxial polydactyly in the Doublefoot mutant is due to ectopic Indian Hedgehog signaling. Development 1998, 125, 3123–3132. [Google Scholar] [CrossRef]
- Hahn, H.; Wicking, C.; Zaphiropoulous, P.G.; Gailani, M.R.; Shanley, S.; Chidambaram, A.; Vorechovsky, I.; Holmberg, E.; Unden, A.B.; Gillies, S.; et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 1996, 85, 841–851. [Google Scholar] [CrossRef]
- Johnson, R.L.; Rothman, A.L.; Xie, J.; Goodrich, L.V.; Bare, J.W.; Bonifas, J.M.; Quinn, A.G.; Myers, R.M.; Cox, D.R.; Epstein, E.H., Jr.; et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science. 1996, 272, 1668–1671. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, L.T.; Ocampo-Garza, S.S.; Elizondo-Riojas, G.; Ocampo-Candiani, J. Basal cell nevus syndrome: An update on clinical findings. Int. J. Dermatol. 2022, 61, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- Patel, T.N.; Dhanyamraju, P.K. Role of aberrant Sonic hedgehog signaling pathway in cancers and developmental anomalies. J. Biomed. Res. 2021, 36, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bisht, S.; Nigam, M.; Kunjwal, S.S.; Sergey, P.; Mishra, A.P.; Sharifi-Rad, J. Cancer Stem Cells: From an Insight into the Basics to Recent Advances and Therapeutic Targeting. Stem. Cells Int. 2022, 2022, 9653244. [Google Scholar] [CrossRef]
- Katoh, Y.; Katoh, M. Hedgehog target genes: Mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr. Mol. Med. 2009, 9, 873–886. [Google Scholar] [CrossRef]
- Lemos, T.; Merchant, A. The hedgehog pathway in hematopoiesis and hematological malignancy. Front. Oncol. 2022, 12, 960943. [Google Scholar] [CrossRef]
- Jain, R.; Dubey, S.K.; Singhvi, G. The Hedgehog pathway and its inhibitors: Emerging therapeutic approaches for basal cell carcinoma. Drug. Discov. Today 2022, 27, 1176–1183. [Google Scholar] [CrossRef] [PubMed]
- Ramadan, F.; Fahs, A.; Ghayad, S.E.; Saab, R. Signaling pathways in Rhabdomyosarcoma invasion and metastasis. Cancer Metastasis. Rev. 2020, 39, 287–301. [Google Scholar] [CrossRef]
- Wang, W.; Shiraishi, R.; Kawauchi, D. Sonic Hedgehog Signaling in Cerebellar Development and Cancer. Front. Cell Dev. Biol. 2022, 10, 864035. [Google Scholar] [CrossRef]
- Abe, Y.; Tanaka, N. The Hedgehog Signaling Networks in Lung Cancer: The Mechanisms and Roles in Tumor Progression and Implications for Cancer Therapy. Biomed. Res. Int. 2016, 2016, 7969286. [Google Scholar] [CrossRef]
- Clement, V.; Sanchez, P.; de Tribolet, N.; Radovanovic, I.; Ruiz i Altaba, A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 2007, 17, 165–172. [Google Scholar] [CrossRef]
- Kawahira, H.; Scheel, D.W.; Smith, S.B.; German, M.S.; Hebrok, M. Hedgehog signaling regulates expansion of pancreatic epithelial cells. Dev. Biol. 2005, 280, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Berman, D.M.; Karhadkar, S.S.; Maitra, A.; Montes De Oca, R.; Gerstenblith, M.R.; Briggs, K.; Parker, A.R.; Shimada, Y.; Eshleman, J.R.; Watkins, D.N.; et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 2003, 425, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Yauch, R.L.; Gould, S.E.; Scales, S.J.; Tang, T.; Tian, H.; Ahn, C.P.; Marshall, D.; Fu, L.; Januario, T.; Kallop, D.; et al. A paracrine requirement for hedgehog signalling in cancer. Nature 2008, 455, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Callahan, C.A.; DuPree, K.J.; Darbonne, W.C.; Ahn, C.P.; Scales, S.J.; de Sauvage, F.J. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 4254–4259. [Google Scholar] [CrossRef]
- Lindemann, R.K. Stroma-initiated hedgehog signaling takes center stage in B-cell lymphoma. Cancer Res. 2008, 68, 961–964. [Google Scholar] [CrossRef]
- Peacock, C.D.; Wang, Q.; Gesell, G.S.; Corcoran-Schwartz, I.M.; Jones, E.; Kim, J.; Devereux, W.L.; Rhodes, J.T.; Huff, C.A.; Beachy, P.A.; et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc. Natl. Acad. Sci. USA 2007, 104, 4048–4053. [Google Scholar] [CrossRef]
- Becher, O.J.; Hambardzumyan, D.; Fomchenko, E.I.; Momota, H.; Mainwaring, L.; Bleau, A.M.; Katz, A.M.; Edgar, M.; Kenney, A.M.; Cordon-Cardo, C.; et al. Gli activity correlates with tumor grade in platelet-derived growth factor-induced gliomas. Cancer Res. 2008, 68, 2241–2249. [Google Scholar] [CrossRef]
- Suchors, C.; Kim, J. Canonical Hedgehog Pathway and Noncanonical GLI Transcription Factor Activation in Cancer. Cells 2022, 11, 2523. [Google Scholar] [CrossRef]
- Whisenant, T.C.; Ho, D.T.; Benz, R.W.; Rogers, J.S.; Kaake, R.M.; Gordon, E.A.; Huang, L.; Baldi, P.; Bardwell, L. Computational prediction and experimental verification of new MAP kinase docking sites and substrates including Gli transcription factors. PLoS Comput. Biol. 2010, 6. [Google Scholar] [CrossRef]
- Bardwell, A.J.; Wu, B.; Sarin, K.Y.; Waterman, M.L.; Atwood, S.X.; Bardwell, L. ERK2 MAP kinase regulates SUFU binding by multisite phosphorylation of GLI1. Life Sci. Alliance 2022, 5. [Google Scholar] [CrossRef]
- Peng, Z.; Ji, Z.; Mei, F.; Lu, M.; Ou, Y.; Cheng, X. Lithium inhibits tumorigenic potential of PDA cells through targeting hedgehog-GLI signaling pathway. PLoS ONE 2013, 8, e61457. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ding, Q.; Yen, C.J.; Xia, W.; Izzo, J.G.; Lang, J.Y.; Li, C.W.; Hsu, J.L.; Miller, S.A.; Wang, X.; et al. The crosstalk of mTOR/S6K1 and Hedgehog pathways. Cancer Cell 2012, 21, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Varjosalo, M.; Bjorklund, M.; Cheng, F.; Syvanen, H.; Kivioja, T.; Kilpinen, S.; Sun, Z.; Kallioniemi, O.; Stunnenberg, H.G.; He, W.W.; et al. Application of active and kinase-deficient kinome collection for identification of kinases regulating hedgehog signaling. Cell 2008, 133, 537–548. [Google Scholar] [CrossRef]
- Gruber, W.; Hutzinger, M.; Elmer, D.P.; Parigger, T.; Sternberg, C.; Cegielkowski, L.; Zaja, M.; Leban, J.; Michel, S.; Hamm, S.; et al. DYRK1B as therapeutic target in Hedgehog/GLI-dependent cancer cells with Smoothened inhibitor resistance. Oncotarget 2016, 7, 7134–7148. [Google Scholar] [CrossRef] [PubMed]
- Zwerner, J.P.; Joo, J.; Warner, K.L.; Christensen, L.; Hu-Lieskovan, S.; Triche, T.J.; May, W.A. The EWS/FLI1 oncogenic transcription factor deregulates GLI1. Oncogene 2008, 27, 3282–3291. [Google Scholar] [CrossRef] [PubMed]
- Coni, S.; Di Magno, L.; Canettieri, G. Determination of Acetylation of the Gli Transcription Factors. Methods. Mol. Biol. 2015, 1322, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Canettieri, G.; Di Marcotullio, L.; Greco, A.; Coni, S.; Antonucci, L.; Infante, P.; Pietrosanti, L.; De Smaele, E.; Ferretti, E.; Miele, E.; et al. Histone deacetylase and Cullin3-REN(KCTD11) ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation. Nat. Cell Biol. 2010, 12, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Li, B.; Rodriguez-Blanco, J.; Pastori, C.; Volmar, C.H.; Wahlestedt, C.; Capobianco, A.; Bai, F.; Pei, X.H.; Ayad, N.G.; et al. The BET bromodomain inhibitor I-BET151 acts downstream of smoothened protein to abrogate the growth of hedgehog protein-driven cancers. J. Biol. Chem. 2014, 289, 35494–35502. [Google Scholar] [CrossRef]
- Degenhardt, K.; Singh, M.K.; Aghajanian, H.; Massera, D.; Wang, Q.; Li, J.; Li, L.; Choi, C.; Yzaguirre, A.D.; Francey, L.J.; et al. Semaphorin 3d signaling defects are associated with anomalous pulmonary venous connections. Nat. Med. 2013, 19, 760–765. [Google Scholar] [CrossRef]
- Kasiri, S.; Shao, C.; Chen, B.; Wilson, A.N.; Yenerall, P.; Timmons, B.C.; Girard, L.; Tian, H.; Behrens, C.; Wistuba, I.I.; et al. GLI1 Blockade Potentiates the Antitumor Activity of PI3K Antagonists in Lung Squamous Cell Carcinoma. Cancer Res. 2017, 77, 4448–4459. [Google Scholar] [CrossRef] [Green Version]
- Buonamici, S.; Williams, J.; Morrissey, M.; Wang, A.; Guo, R.; Vattay, A.; Hsiao, K.; Yuan, J.; Green, J.; Ospina, B.; et al. Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci. Transl. Med. 2010, 2, 51ra70. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; Bayo-Fina, J.M.; Singh, R.; Kumar Dhanyamraju, P.; Holz, P.; Baier, A.; Fendrich, V.; Ramaswamy, A.; Baumeister, S.; Martinez, E.D.; et al. Identification of a novel actin-dependent signal transducing module allows for the targeted degradation of GLI1. Nat. Commun. 2015, 6, 8023. [Google Scholar] [CrossRef]
- Quatannens, D.; Verhoeven, Y.; Van Dam, P.; Lardon, F.; Prenen, H.; Roeyen, G.; Peeters, M.; Smits, E.L.J.; Van Audenaerde, J. Targeting hedgehog signaling in pancreatic ductal adenocarcinoma. Pharmacol. Ther. 2022, 236, 108107. [Google Scholar] [CrossRef]
- Chen, J.K.; Taipale, J.; Cooper, M.K.; Beachy, P.A. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes. Dev. 2002, 16, 2743–2748. [Google Scholar] [CrossRef] [PubMed]
- Robarge, K.D.; Brunton, S.A.; Castanedo, G.M.; Cui, Y.; Dina, M.S.; Goldsmith, R.; Gould, S.E.; Guichert, O.; Gunzner, J.L.; Halladay, J.; et al. GDC-0449-a potent inhibitor of the hedgehog pathway. Bioorg. Med. Chem. Lett. 2009, 19, 5576–5581. [Google Scholar] [CrossRef] [PubMed]
- Axelson, M.; Liu, K.; Jiang, X.; He, K.; Wang, J.; Zhao, H.; Kufrin, D.; Palmby, T.; Dong, Z.; Russell, A.M.; et al. Food and Drug Administration approval: Vismodegib for recurrent, locally advanced, or metastatic basal cell carcinoma. Clin. Cancer Res. 2013, 19, 2289–2293. [Google Scholar] [CrossRef]
- Pan, S.; Wu, X.; Jiang, J.; Gao, W.; Wan, Y.; Cheng, D.; Han, D.; Liu, J.; Englund, N.P.; Wang, Y.; et al. Discovery of NVP-LDE225, a Potent and Selective Smoothened Antagonist. ACS. Med. Chem. Lett. 2010, 1, 130–134. [Google Scholar] [CrossRef]
- Casey, D.; Demko, S.; Shord, S.; Zhao, H.; Chen, H.; He, K.; Putman, A.; Helms, W.; Keegan, P.; Pazdur, R. FDA Approval Summary: Sonidegib for Locally Advanced Basal Cell Carcinoma. Clin. Cancer Res. 2017, 23, 2377–2381. [Google Scholar] [CrossRef]
- Munchhof, M.J.; Li, Q.; Shavnya, A.; Borzillo, G.V.; Boyden, T.L.; Jones, C.S.; LaGreca, S.D.; Martinez-Alsina, L.; Patel, N.; Pelletier, K.; et al. Discovery of PF-04449913, a Potent and Orally Bioavailable Inhibitor of Smoothened. ACS. Med. Chem. Lett. 2012, 3, 106–111. [Google Scholar] [CrossRef]
- Norsworthy, K.J.; By, K.; Subramaniam, S.; Zhuang, L.; Del Valle, P.L.; Przepiorka, D.; Shen, Y.L.; Sheth, C.M.; Liu, C.; Leong, R.; et al. FDA Approval Summary: Glasdegib for Newly Diagnosed Acute Myeloid Leukemia. Clin. Cancer Res. 2019, 25, 6021–6025. [Google Scholar] [CrossRef] [Green Version]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.; Sivaraman, A.; Lee, K. Development of taladegib as a sonic hedgehog signaling pathway inhibitor. Arch. Pharm. Res. 2017, 40, 1390–1393. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, T.; Oguro, Y.; Tanaka, T.; Shiokawa, Z.; Tanaka, Y.; Shibata, S.; Sato, Y.; Yamakawa, H.; Hattori, H.; Yamamoto, Y.; et al. Discovery of the investigational drug TAK-441, a pyrrolo[3,2-c]pyridine derivative, as a highly potent and orally active hedgehog signaling inhibitor: Modification of the core skeleton for improved solubility. Bioorg. Med. Chem. 2012, 20, 5507–5517. [Google Scholar] [CrossRef] [PubMed]
- Sandhiya, S.; Melvin, G.; Kumar, S.S.; Dkhar, S.A. The dawn of hedgehog inhibitors: Vismodegib. J. Pharmacol. Pharmacother. 2013, 4, 4–7. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; LoRusso, P.M.; Rudin, C.M.; Reddy, J.C.; Yauch, R.L.; Tibes, R.; Weiss, G.J.; Borad, M.J.; Hann, C.L.; Brahmer, J.R.; et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N. Engl. J. Med. 2009, 361, 1164–1172. [Google Scholar] [CrossRef]
- Dlugosz, A.; Agrawal, S.; Kirkpatrick, P. Vismodegib. Nat. Rev. Drug. Discov. 2012, 11, 437–438. [Google Scholar] [CrossRef]
- Sekulic, A.; Migden, M.R.; Oro, A.E.; Dirix, L.; Lewis, K.D.; Hainsworth, J.D.; Solomon, J.A.; Yoo, S.; Arron, S.T.; Friedlander, P.A.; et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N. Engl. J. Med. 2012, 366, 2171–2179. [Google Scholar] [CrossRef]
- LoRusso, P.M.; Rudin, C.M.; Reddy, J.C.; Tibes, R.; Weiss, G.J.; Borad, M.J.; Hann, C.L.; Brahmer, J.R.; Chang, I.; Darbonne, W.C.; et al. Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced or metastatic solid tumors. Clin. Cancer Res. 2011, 17, 2502–2511. [Google Scholar] [CrossRef]
- Cortes, J.E.; Heidel, F.H.; Fiedler, W.; Smith, B.D.; Robak, T.; Montesinos, P.; Candoni, A.; Leber, B.; Sekeres, M.A.; Pollyea, D.A.; et al. Survival outcomes and clinical benefit in patients with acute myeloid leukemia treated with glasdegib and low-dose cytarabine according to response to therapy. J. Hematol. Oncol. 2020, 13, 92. [Google Scholar] [CrossRef]
- Cortes, J.E.; Douglas Smith, B.; Wang, E.S.; Merchant, A.; Oehler, V.G.; Arellano, M.; DeAngelo, D.J.; Pollyea, D.A.; Sekeres, M.A.; Robak, T.; et al. Glasdegib in combination with cytarabine and daunorubicin in patients with AML or high-risk MDS: Phase 2 study results. Am. J. Hematol. 2018, 93, 1301–1310. [Google Scholar] [CrossRef] [Green Version]
- Chai, J.Y.; Sugumar, V.; Alshawsh, M.A.; Wong, W.F.; Arya, A.; Chong, P.P.; Looi, C.Y. The Role of Smoothened-Dependent and -Independent Hedgehog Signaling Pathway in Tumorigenesis. Biomedicines 2021, 9, 1188. [Google Scholar] [CrossRef] [PubMed]
- Lauth, M.; Bergstrom, A.; Shimokawa, T.; Toftgard, R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc. Natl. Acad. Sci. USA 2007, 104, 8455–8460. [Google Scholar] [CrossRef] [PubMed]
- Chenna, V.; Hu, C.; Khan, S.R. Synthesis and cytotoxicity studies of Hedgehog enzyme inhibitors SANT-1 and GANT-61 as anticancer agents. J. Environ. Sci. Health A Tox. Hazard. Subst. Environ. Eng. 2014, 49, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Mullard, M.; Cade, M.; Morice, S.; Dupuy, M.; Danieau, G.; Amiaud, J.; Renault, S.; Lezot, F.; Brion, R.; Thepault, R.A.; et al. Sonic Hedgehog Signature in Pediatric Primary Bone Tumors: Effects of the GLI Antagonist GANT61 on Ewing’s Sarcoma Tumor Growth. Cancers 2020, 12, 3438. [Google Scholar] [CrossRef]
- Rajurkar, M.; De Jesus-Monge, W.E.; Driscoll, D.R.; Appleman, V.A.; Huang, H.; Cotton, J.L.; Klimstra, D.S.; Zhu, L.J.; Simin, K.; Xu, L.; et al. The activity of Gli transcription factors is essential for Kras-induced pancreatic tumorigenesis. Proc. Natl. Acad. Sci. USA 2012, 109, E1038–E1047. [Google Scholar] [CrossRef]
- Sneha, S.; Nagare, R.P.; Sidhanth, C.; Krishnapriya, S.; Garg, M.; Ramachandran, B.; Murhekar, K.; Sundersingh, S.; Ganesan, T.S. The hedgehog pathway regulates cancer stem cells in serous adenocarcinoma of the ovary. Cell Oncol. 2020, 43, 601–616. [Google Scholar] [CrossRef]
- Lu, Y.; Zhu, Y.; Deng, S.; Chen, Y.; Li, W.; Sun, J.; Xu, X. Targeting the Sonic Hedgehog Pathway to Suppress the Expression of the Cancer Stem Cell (CSC)-Related Transcription Factors and CSC-Driven Thyroid Tumor Growth. Cancers 2021, 13, 418. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Matsubara, S.; Ding, Q.; Tsukasa, K.; Yoshimitsu, M.; Kosai, K.; Takao, S. Efficient elimination of pancreatic cancer stem cells by hedgehog/GLI inhibitor GANT61 in combination with mTOR inhibition. Mol. Cancer 2016, 15, 49. [Google Scholar] [CrossRef]
- Infante, P.; Mori, M.; Alfonsi, R.; Ghirga, F.; Aiello, F.; Toscano, S.; Ingallina, C.; Siler, M.; Cucchi, D.; Po, A.; et al. Gli1/DNA interaction is a druggable target for Hedgehog-dependent tumors. EMBO J. 2015, 34, 200–217. [Google Scholar] [CrossRef]
- Kim, J.; Lee, J.J.; Kim, J.; Gardner, D.; Beachy, P.A. Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc. Natl. Acad. Sci. USA 2010, 107, 13432–13437. [Google Scholar] [CrossRef] [Green Version]
- Beauchamp, E.M.; Ringer, L.; Bulut, G.; Sajwan, K.P.; Hall, M.D.; Lee, Y.C.; Peaceman, D.; Ozdemirli, M.; Rodriguez, O.; Macdonald, T.J.; et al. Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/GLI pathway. J. Clin. Invest. 2011, 121, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Chin, L.; Kumana, C.R.; Kwong, Y.L.; Gill, H. The Development and Clinical Applications of Oral Arsenic Trioxide for Acute Promyelocytic Leukaemia and Other Diseases. Pharmaceutics 2022, 14, 1945. [Google Scholar] [CrossRef] [PubMed]
- Yousefnia, S. Mechanistic effects of arsenic trioxide on acute promyelocytic leukemia and other types of leukemias. Cell Biol. Int. 2021, 45, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Cao, F.; Ye, X.; Zhao, H.; Liu, X.; Li, Y.; Shi, C.; Wang, H.; Zhou, J. Arsenic trioxide inhibits the Hedgehog pathway which is aberrantly activated in acute promyelocytic leukemia. Acta Haematol. 2013, 130, 260–267. [Google Scholar] [CrossRef]
- Ally, M.S.; Ransohoff, K.; Sarin, K.; Atwood, S.X.; Rezaee, M.; Bailey-Healy, I.; Kim, J.; Beachy, P.A.; Chang, A.L.; Oro, A.; et al. Effects of Combined Treatment With Arsenic Trioxide and Itraconazole in Patients With Refractory Metastatic Basal Cell Carcinoma. JAMA Dermatol. 2016, 152, 452–456. [Google Scholar] [CrossRef]
- Kim, J.; Tang, J.Y.; Gong, R.; Kim, J.; Lee, J.J.; Clemons, K.V.; Chong, C.R.; Chang, K.S.; Fereshteh, M.; Gardner, D.; et al. Itraconazole, a commonly used antifungal that inhibits Hedgehog pathway activity and cancer growth. Cancer Cell 2010, 17, 388–399. [Google Scholar] [CrossRef]
- Kenchegowda, M.; Rahamathulla, M.; Hani, U.; Begum, M.Y.; Guruswamy, S.; Osmani, R.A.M.; Gowrav, M.P.; Alshehri, S.; Ghoneim, M.M.; Alshlowi, A.; et al. Smart Nanocarriers as an Emerging Platform for Cancer Therapy: A Review. Molecules 2021, 27, 146. [Google Scholar] [CrossRef]
- Kumar, V.; Wang, Q.; Sethi, B.; Lin, F.; Kumar, V.; Coulter, D.W.; Dong, Y.; Mahato, R.I. Polymeric nanomedicine for overcoming resistance mechanisms in hedgehog and Myc-amplified medulloblastoma. Biomaterials 2021, 278, 121138. [Google Scholar] [CrossRef]
- Singh, S.K.; Gordetsky, J.B.; Bae, S.; Acosta, E.P.; Lillard, J.W., Jr.; Singh, R. Selective Targeting of the Hedgehog Signaling Pathway by PBM Nanoparticles in Docetaxel-Resistant Prostate Cancer. Cells 2020, 9, 1976. [Google Scholar] [CrossRef]
- Poulaki, A.; Giannouli, S. Metabolic Swifts Govern Normal and Malignant B Cell Lymphopoiesis. Int. J. Mol. Sci. 2021, 22, 8269. [Google Scholar] [CrossRef]
- Montel-Hagen, A.; Crooks, G.M. From pluripotent stem cells to T cells. Exp. Hematol. 2019, 71, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Ran, G.H.; Lin, Y.Q.; Tian, L.; Zhang, T.; Yan, D.M.; Yu, J.H.; Deng, Y.C. Natural killer cell homing and trafficking in tissues and tumors: From biology to application. Signal Transduct. Target Ther. 2022, 7, 205. [Google Scholar] [CrossRef] [PubMed]
- Mar, B.G.; Amakye, D.; Aifantis, I.; Buonamici, S. The controversial role of the Hedgehog pathway in normal and malignant hematopoiesis. Leukemia 2011, 25, 1665–1673. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Graves, S.; Koch, U.; Liu, S.; Jankovic, V.; Buonamici, S.; El Andaloussi, A.; Nimer, S.D.; Kee, B.L.; Taichman, R.; et al. Hedgehog signaling is dispensable for adult hematopoietic stem cell function. Cell Stem. Cell 2009, 4, 548–558. [Google Scholar] [CrossRef]
- Hofmann, I.; Stover, E.H.; Cullen, D.E.; Mao, J.; Morgan, K.J.; Lee, B.H.; Kharas, M.G.; Miller, P.G.; Cornejo, M.G.; Okabe, R.; et al. Hedgehog signaling is dispensable for adult murine hematopoietic stem cell function and hematopoiesis. Cell Stem. Cell 2009, 4, 559–567. [Google Scholar] [CrossRef]
- Gering, M.; Patient, R. Hedgehog signaling is required for adult blood stem cell formation in zebrafish embryos. Dev. Cell 2005, 8, 389–400. [Google Scholar] [CrossRef]
- Merchant, A.; Joseph, G.; Wang, Q.; Brennan, S.; Matsui, W. Gli1 regulates the proliferation and differentiation of HSCs and myeloid progenitors. Blood 2010, 115, 2391–2396. [Google Scholar] [CrossRef]
- Merchant, A.A.; Matsui, W. Smoothening the controversial role of hedgehog in hematopoiesis. Cell Stem. Cell 2009, 4, 470–471. [Google Scholar] [CrossRef]
- Uhmann, A.; Dittmann, K.; Nitzki, F.; Dressel, R.; Koleva, M.; Frommhold, A.; Zibat, A.; Binder, C.; Adham, I.; Nitsche, M.; et al. The Hedgehog receptor Patched controls lymphoid lineage commitment. Blood 2007, 110, 1814–1823. [Google Scholar] [CrossRef]
- Crompton, T.; Outram, S.V.; Hager-Theodorides, A.L. Sonic hedgehog signalling in T-cell development and activation. Nat. Rev. Immunol. 2007, 7, 726–735. [Google Scholar] [CrossRef]
- Barbarulo, A.; Lau, C.I.; Mengrelis, K.; Ross, S.; Solanki, A.; Saldana, J.I.; Crompton, T. Hedgehog Signalling in the Embryonic Mouse Thymus. J. Dev. Biol. 2016, 4, 22. [Google Scholar] [CrossRef] [PubMed]
- Alves, N.L.; Richard-Le Goff, O.; Huntington, N.D.; Sousa, A.P.; Ribeiro, V.S.; Bordack, A.; Vives, F.L.; Peduto, L.; Chidgey, A.; Cumano, A.; et al. Characterization of the thymic IL-7 niche in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 1512–1517. [Google Scholar] [CrossRef]
- Shah, D.K.; Zuniga-Pflucker, J.C. An overview of the intrathymic intricacies of T cell development. J. Immunol. 2014, 192, 4017–4023. [Google Scholar] [CrossRef] [PubMed]
- Ok, C.Y.; Singh, R.R.; Vega, F. Aberrant activation of the hedgehog signaling pathway in malignant hematological neoplasms. Am. J. Pathol. 2012, 180, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Zhao, F.; Chen, X.; Li, J.; Jin, X.; Han, Y.; Wang, L.; Zhou, Y.; Lu, L. NPAT Supports CD8(+) Immature Single-Positive Thymocyte Proliferation and Thymic Development. J. Immunol. 2022, 209, 916–925. [Google Scholar] [CrossRef]
- Godfrey, D.I.; Zlotnik, A. Control points in early T-cell development. Immunol. Today 1993, 14, 547–553. [Google Scholar] [CrossRef]
- von Boehmer, H. Shaping the T cell repertoire. J. Immunol. 2006, 176, 3–4. [Google Scholar] [CrossRef]
- von Boehmer, H.; Kisielow, P. Negative selection of the T-cell repertoire: Where and when does it occur? Immunol. Rev. 2006, 209, 284–289. [Google Scholar] [CrossRef]
- von Boehmer, H.; Aifantis, I.; Feinberg, J.; Lechner, O.; Saint-Ruf, C.; Walter, U.; Buer, J.; Azogui, O. Pleiotropic changes controlled by the pre-T-cell receptor. Curr. Opin. Immunol. 1999, 11, 135–142. [Google Scholar] [CrossRef]
- Aifantis, I.; Buer, J.; von Boehmer, H.; Azogui, O. Essential role of the pre-T cell receptor in allelic exclusion of the T cell receptor beta locus. Immunity 1997, 7, 601–607. [Google Scholar] [CrossRef] [Green Version]
- Aifantis, I.; Azogui, O.; Feinberg, J.; Saint-Ruf, C.; Buer, J.; von Boehmer, H. On the role of the pre-T cell receptor in alphabeta versus gammadelta T lineage commitment. Immunity 1998, 9, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.; Moore, N.C.; Owen, J.J.; Jenkinson, E.J. Cellular interactions in thymocyte development. Annu. Rev. Immunol. 1996, 14, 73–99. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.; Takahama, Y. Thymic epithelial cells: Working class heroes for T cell development and repertoire selection. Trends Immunol. 2012, 33, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Klein, L.; Kyewski, B.; Allen, P.M.; Hogquist, K.A. Positive and negative selection of the T cell repertoire: What thymocytes see (and don’t see). Nat. Rev. Immunol. 2014, 14, 377–391. [Google Scholar] [CrossRef]
- Grabstein, K.H.; Waldschmidt, T.J.; Finkelman, F.D.; Hess, B.W.; Alpert, A.R.; Boiani, N.E.; Namen, A.E.; Morrissey, P.J. Inhibition of murine B and T lymphopoiesis in vivo by an anti-interleukin 7 monoclonal antibody. J. Exp. Med. 1993, 178, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Sudo, T.; Nishikawa, S.; Ohno, N.; Akiyama, N.; Tamakoshi, M.; Yoshida, H.; Nishikawa, S. Expression and function of the interleukin 7 receptor in murine lymphocytes. Proc. Natl. Acad. Sci. USA 1993, 90, 9125–9129. [Google Scholar] [CrossRef]
- Barata, J.T.; Durum, S.K.; Seddon, B. Flip the coin: IL-7 and IL-7R in health and disease. Nat. Immunol. 2019, 20, 1584–1593. [Google Scholar] [CrossRef]
- Rodewald, H.R.; Kretzschmar, K.; Swat, W.; Takeda, S. Intrathymically expressed c-kit ligand (stem cell factor) is a major factor driving expansion of very immature thymocytes in vivo. Immunity 1995, 3, 313–319. [Google Scholar] [CrossRef]
- Koch, U.; Fiorini, E.; Benedito, R.; Besseyrias, V.; Schuster-Gossler, K.; Pierres, M.; Manley, N.R.; Duarte, A.; Macdonald, H.R.; Radtke, F. Delta-like 4 is the essential, nonredundant ligand for Notch1 during thymic T cell lineage commitment. J. Exp. Med. 2008, 205, 2515–2523. [Google Scholar] [CrossRef]
- von Boehmer, H. Unique features of the pre-T-cell receptor alpha-chain: Not just a surrogate. Nat. Rev. Immunol. 2005, 5, 571–577. [Google Scholar] [CrossRef]
- Schilham, M.W.; Wilson, A.; Moerer, P.; Benaissa-Trouw, B.J.; Cumano, A.; Clevers, H.C. Critical involvement of Tcf-1 in expansion of thymocytes. J. Immunol. 1998, 161, 3984–3991. [Google Scholar] [CrossRef] [PubMed]
- Roozen, P.P.; Brugman, M.H.; Staal, F.J. Differential requirements for Wnt and Notch signaling in hematopoietic versus thymic niches. Ann. N. Y. Acad. Sci. 2012, 1266, 78–93. [Google Scholar] [CrossRef] [PubMed]
- Ciofani, M.; Zuniga-Pflucker, J.C. Notch promotes survival of pre-T cells at the beta-selection checkpoint by regulating cellular metabolism. Nat. Immunol. 2005, 6, 881–888. [Google Scholar] [CrossRef]
- Sacedon, R.; Varas, A.; Hernandez-Lopez, C.; Gutierrez-deFrias, C.; Crompton, T.; Zapata, A.G.; Vicente, A. Expression of hedgehog proteins in the human thymus. J. Histochem. Cytochem. 2003, 51, 1557–1566. [Google Scholar] [CrossRef] [PubMed]
- Outram, S.V.; Varas, A.; Pepicelli, C.V.; Crompton, T. Hedgehog signaling regulates differentiation from double-negative to double-positive thymocyte. Immunity 2000, 13, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Greaves, W.O.; Kim, J.E.; Singh, R.R.; Drakos, E.; Kunkalla, K.; Sanchez-Espiridion, B.; Garcia, J.F.; Medeiros, L.J.; Vega, F. Glioma-associated oncogene homologue 3, a hedgehog transcription factor, is highly expressed in Hodgkin and Reed-Sternberg cells of classical Hodgkin lymphoma. Hum. Pathol. 2011, 42, 1643–1652. [Google Scholar] [CrossRef]
- Saldana, J.I.; Solanki, A.; Lau, C.I.; Sahni, H.; Ross, S.; Furmanski, A.L.; Ono, M.; Hollander, G.; Crompton, T. Sonic Hedgehog regulates thymic epithelial cell differentiation. J. Autoimmun. 2016, 68, 86–97. [Google Scholar] [CrossRef]
- Outram, S.V.; Hager-Theodorides, A.L.; Shah, D.K.; Rowbotham, N.J.; Drakopoulou, E.; Ross, S.E.; Lanske, B.; Dessens, J.T.; Crompton, T. Indian hedgehog (Ihh) both promotes and restricts thymocyte differentiation. Blood 2009, 113, 2217–2228. [Google Scholar] [CrossRef]
- El Andaloussi, A.; Graves, S.; Meng, F.; Mandal, M.; Mashayekhi, M.; Aifantis, I. Hedgehog signaling controls thymocyte progenitor homeostasis and differentiation in the thymus. Nat. Immunol. 2006, 7, 418–426. [Google Scholar] [CrossRef]
- Rowbotham, N.J.; Hager-Theodorides, A.L.; Furmanski, A.L.; Ross, S.E.; Outram, S.V.; Dessens, J.T.; Crompton, T. Sonic hedgehog negatively regulates pre-TCR-induced differentiation by a Gli2-dependent mechanism. Blood 2009, 113, 5144–5156. [Google Scholar] [CrossRef] [Green Version]
- Hager-Theodorides, A.L.; Dessens, J.T.; Outram, S.V.; Crompton, T. The transcription factor Gli3 regulates differentiation of fetal CD4- CD8- double-negative thymocytes. Blood 2005, 106, 1296–1304. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.K.; Hager-Theodorides, A.L.; Outram, S.V.; Ross, S.E.; Varas, A.; Crompton, T. Reduced thymocyte development in sonic hedgehog knockout embryos. J. Immunol. 2004, 172, 2296–2306. [Google Scholar] [CrossRef] [PubMed]
- Drakopoulou, E.; Outram, S.V.; Rowbotham, N.J.; Ross, S.E.; Furmanski, A.L.; Saldana, J.I.; Hager-Theodorides, A.L.; Crompton, T. Non-redundant role for the transcription factor Gli1 at multiple stages of thymocyte development. Cell Cycle 2010, 9, 4144–4152. [Google Scholar] [CrossRef]
- Furmanski, A.L.; Saldana, J.I.; Rowbotham, N.J.; Ross, S.E.; Crompton, T. Role of Hedgehog signalling at the transition from double-positive to single-positive thymocyte. Eur. J. Immunol. 2012, 42, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Rowbotham, N.J.; Hager-Theodorides, A.L.; Cebecauer, M.; Shah, D.K.; Drakopoulou, E.; Dyson, J.; Outram, S.V.; Crompton, T. Activation of the Hedgehog signaling pathway in T-lineage cells inhibits TCR repertoire selection in the thymus and peripheral T-cell activation. Blood 2007, 109, 3757–3766. [Google Scholar] [CrossRef] [PubMed]
- Rowbotham, N.J.; Furmanski, A.L.; Hager-Theodorides, A.L.; Ross, S.E.; Drakopoulou, E.; Koufaris, C.; Outram, S.V.; Crompton, T. Repression of hedgehog signal transduction in T-lineage cells increases TCR-induced activation and proliferation. Cell Cycle 2008, 7, 904–908. [Google Scholar] [CrossRef]
- Solanki, A.; Yanez, D.C.; Ross, S.; Lau, C.I.; Papaioannou, E.; Li, J.; Saldana, J.I.; Crompton, T. Gli3 in fetal thymic epithelial cells promotes thymocyte positive selection and differentiation by repression of Shh. Development 2018, 145. [Google Scholar] [CrossRef]
- Neumann, C.J. Hedgehogs as negative regulators of the cell cycle. Cell Cycle 2005, 4, 1139–1140. [Google Scholar] [CrossRef]
- van den Brink, G.R. Hedgehog signaling in development and homeostasis of the gastrointestinal tract. Physiol. Rev. 2007, 87, 1343–1375. [Google Scholar] [CrossRef]
- Gutierrez-Frias, C.; Sacedon, R.; Hernandez-Lopez, C.; Cejalvo, T.; Crompton, T.; Zapata, A.G.; Varas, A.; Vicente, A. Sonic hedgehog regulates early human thymocyte differentiation by counteracting the IL-7-induced development of CD34+ precursor cells. J. Immunol. 2004, 173, 5046–5053. [Google Scholar] [CrossRef] [Green Version]
- Belver, L.; Ferrando, A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat. Rev. Cancer 2016, 16, 494–507. [Google Scholar] [CrossRef] [PubMed]
- Malczewska, M.; Kosmider, K.; Bednarz, K.; Ostapinska, K.; Lejman, M.; Zawitkowska, J. Recent Advances in Treatment Options for Childhood Acute Lymphoblastic Leukemia. Cancers 2022, 14, 2021. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zou, Y.; Zhang, L.; Guo, Y.; Chen, Y.; Yang, W.; Chen, X.; Wang, S.; Zhang, Y.; Ruan, M.; et al. A Novel Risk Defining System for Pediatric T-Cell Acute Lymphoblastic Leukemia From CCCG-ALL-2015 Group. Front. Oncol. 2022, 12, 841179. [Google Scholar] [CrossRef]
- Qi, H.Z.; Xu, J.; Yang, Q.Q.; Lin, R.; Wang, Z.X.; Zhao, K.; Wang, Q.; Zhou, X.; Fan, Z.P.; Huang, F.; et al. Effect of pediatric- versus adult-type chemotherapy regimens on outcomes of allogeneic hematopoietic stem cell transplants for adult T-cell acute lymphoblastic leukemia in first complete remission. Bone Marrow Transplant. 2022. [Google Scholar] [CrossRef]
- Schrappe, M.; Hunger, S.P.; Pui, C.H.; Saha, V.; Gaynon, P.S.; Baruchel, A.; Conter, V.; Otten, J.; Ohara, A.; Versluys, A.B.; et al. Outcomes after induction failure in childhood acute lymphoblastic leukemia. N. Engl. J. Med. 2012, 366, 1371–1381. [Google Scholar] [CrossRef] [PubMed]
- Wayne, N.J.; Li, Y.; Chung, P.; Coffan, K.; Rheingold, S.R. Outcomes of children and young adults with T-cell acute lymphoblastic leukemia/lymphoma who present in critical status. Pediatr. Blood Cancer 2022, 69, e29457. [Google Scholar] [CrossRef]
- Genesca, E.; Gonzalez-Gil, C. Latest Contributions of Genomics to T-Cell Acute Lymphoblastic Leukemia (T-ALL). Cancers 2022, 14, 2474. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yang, K.; Zhang, H. Targeting Leukemia-Initiating Cells and Leukemic Niches: The Next Therapy Station for T-Cell Acute Lymphoblastic Leukemia? Cancers 2022, 14, 5655. [Google Scholar] [CrossRef]
- Genesca, E.; la Starza, R. Early T-Cell Precursor ALL and Beyond: Immature and Ambiguous Lineage T-ALL Subsets. Cancers 2022, 14, 1873. [Google Scholar] [CrossRef]
- Ferrando, A.A.; Neuberg, D.S.; Staunton, J.; Loh, M.L.; Huard, C.; Raimondi, S.C.; Behm, F.G.; Pui, C.H.; Downing, J.R.; Gilliland, D.G.; et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell 2002, 1, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Bardelli, V.; Arniani, S.; Pierini, V.; Di Giacomo, D.; Pierini, T.; Gorello, P.; Mecucci, C.; La Starza, R. T-Cell Acute Lymphoblastic Leukemia: Biomarkers and Their Clinical Usefulness. Genes 2021, 12, 1118. [Google Scholar] [CrossRef]
- Toribio, M.L.; Gonzalez-Garcia, S. Notch Partners in the Long Journey of T-ALL Pathogenesis. Int. J. Mol. Sci. 2023, 24, 1383. [Google Scholar] [CrossRef]
- Coustan-Smith, E.; Mullighan, C.G.; Onciu, M.; Behm, F.G.; Raimondi, S.C.; Pei, D.; Cheng, C.; Su, X.; Rubnitz, J.E.; Basso, G.; et al. Early T-cell precursor leukaemia: A subtype of very high-risk acute lymphoblastic leukaemia. Lancet. Oncol. 2009, 10, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ding, L.; Holmfeldt, L.; Wu, G.; Heatley, S.L.; Payne-Turner, D.; Easton, J.; Chen, X.; Wang, J.; Rusch, M.; et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 2012, 481, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Homminga, I.; Vuerhard, M.J.; Langerak, A.W.; Buijs-Gladdines, J.; Pieters, R.; Meijerink, J.P. Characterization of a pediatric T-cell acute lymphoblastic leukemia patient with simultaneous LYL1 and LMO2 rearrangements. Haematologica. 2012, 97, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Summers, R.J.; Teachey, D.T. SOHO State of the Art Updates and Next Questions | Novel Approaches to Pediatric T-cell ALL and T-Lymphoblastic Lymphoma. Clin. Lymphoma Myeloma Leuk. 2022. [Google Scholar] [CrossRef]
- Qin, L.; Dai, H.; Wang, J. Key Considerations in Targeted Protein Degradation Drug Discovery and Development. Front. Chem. 2022, 10, 934337. [Google Scholar] [CrossRef]
- Buechner, J.; Caruana, I.; Kunkele, A.; Rives, S.; Vettenranta, K.; Bader, P.; Peters, C.; Baruchel, A.; Calkoen, F.G. Chimeric Antigen Receptor T-Cell Therapy in Paediatric B-Cell Precursor Acute Lymphoblastic Leukaemia: Curative Treatment Option or Bridge to Transplant? Front. Pediatr. 2021, 9, 784024. [Google Scholar] [CrossRef] [PubMed]
- Newman, H.; Teachey, D.T. A Bright Horizon: Immunotherapy for Pediatric T-Cell Malignancies. Int. J. Mol. Sci. 2022, 23, 8600. [Google Scholar] [CrossRef]
- Abraham, A.; Matsui, W. Hedgehog Signaling in Myeloid Malignancies. Cancers 2021, 13, 4888. [Google Scholar] [CrossRef]
- Lau, B.W.; Huh, K.; Madero-Marroquin, R.; De Marchi, F.; Lim, Y.; Wang, Q.; Lobo, F.; Marchionni, L.; Smith, D.B.; DeZern, A.; et al. Hedgehog/GLI1 activation leads to leukemic transformation of myelodysplastic syndrome in vivo and GLI1 inhibition results in antitumor activity. Oncogene 2019, 38, 687–698. [Google Scholar] [CrossRef]
- Wellbrock, J.; Latuske, E.; Kohler, J.; Wagner, K.; Stamm, H.; Vettorazzi, E.; Vohwinkel, G.; Klokow, M.; Uibeleisen, R.; Ehm, P.; et al. Expression of Hedgehog Pathway Mediator GLI Represents a Negative Prognostic Marker in Human Acute Myeloid Leukemia and Its Inhibition Exerts Antileukemic Effects. Clin. Cancer Res. 2015, 21, 2388–2398. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, P.; Singh, M.; Triche, T.J.; Guzman, M.; Merchant, A.A. GLI3 repressor determines Hedgehog pathway activation and is required for response to SMO antagonist glasdegib in AML. Blood 2017, 129, 3465–3475. [Google Scholar] [CrossRef]
- Masetti, R.; Bertuccio, S.N.; Astolfi, A.; Chiarini, F.; Lonetti, A.; Indio, V.; De Luca, M.; Bandini, J.; Serravalle, S.; Franzoni, M.; et al. Hh/Gli antagonist in acute myeloid leukemia with CBFA2T3-GLIS2 fusion gene. J. Hematol. Oncol. 2017, 10, 26. [Google Scholar] [CrossRef]
- Zhou, C.; Du, J.; Zhao, L.; Liu, W.; Zhao, T.; Liang, H.; Fang, P.; Zhang, K.; Zeng, H. GLI1 reduces drug sensitivity by regulating cell cycle through PI3K/AKT/GSK3/CDK pathway in acute myeloid leukemia. Cell Death Dis. 2021, 12, 231. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Sun, Z.; Ding, B.; Jiang, X.; Wang, Z.; Zhu, Y.; Meng, F. Suppressing Hedgehog signaling reverses drug resistance of refractory acute myeloid leukemia. Onco. Targets Ther. 2019, 12, 7477–7488. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, S.R.; Lovell, A.R.; Schroeder, M.A. Glasdegib for the treatment of adult patients with newly diagnosed acute myeloid leukemia or high-grade myelodysplastic syndrome who are elderly or otherwise unfit for standard induction chemotherapy. Drugs. Today 2019, 55, 545–562. [Google Scholar] [CrossRef]
- Anusha; Dalal, H.; Subramanian, S.; V. P., S.; Gowda, D.A.; H., K.; Damodar, S.; Vyas, N. Exovesicular-Shh confers Imatinib resistance by upregulating Bcl2 expression in chronic myeloid leukemia with variant chromosomes. Cell Death Dis. 2021, 12, 259. [Google Scholar] [CrossRef]
- Liu, Z.; Xu, J.; He, J.; Zheng, Y.; Li, H.; Lu, Y.; Qian, J.; Lin, P.; Weber, D.M.; Yang, J.; et al. A critical role of autocrine sonic hedgehog signaling in human CD138+ myeloma cell survival and drug resistance. Blood 2014, 124, 2061–2071. [Google Scholar] [CrossRef]
- Ghia, E.M.; Rassenti, L.Z.; Neuberg, D.S.; Blanco, A.; Yousif, F.; Smith, E.N.; McPherson, J.D.; Hudson, T.J.; Consortium, H.P.-L.G.P.; Harismendy, O.; et al. Activation of hedgehog signaling associates with early disease progression in chronic lymphocytic leukemia. Blood 2019, 133, 2651–2663. [Google Scholar] [CrossRef]
- Ramirez, E.; Singh, R.R.; Kunkalla, K.; Liu, Y.; Qu, C.; Cain, C.; Multani, A.S.; Lennon, P.A.; Jackacky, J.; Ho, M.; et al. Defining causative factors contributing in the activation of hedgehog signaling in diffuse large B-cell lymphoma. Leuk. Res. 2012, 36, 1267–1273. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, T.; Kawaguchi-Ihara, N.; Okuhashi, Y.; Itoh, M.; Nara, N.; Tohda, S. Cyclopamine and quercetin suppress the growth of leukemia and lymphoma cells. Anticancer Res. 2009, 29, 4629–4632. [Google Scholar] [PubMed]
- Okuhashi, Y.; Itoh, M.; Nara, N.; Tohda, S. Effects of combination of notch inhibitor plus hedgehog inhibitor or Wnt inhibitor on growth of leukemia cells. Anticancer Res. 2011, 31, 893–896. [Google Scholar] [PubMed]
- Zhao, Y.; Tong, C.; Jiang, J. Hedgehog regulates smoothened activity by inducing a conformational switch. Nature 2007, 450, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishna, U.; Bornholdt, D.; Scott, H.S.; Patel, U.C.; Rossier, C.; Engel, H.; Bottani, A.; Chandal, D.; Blouin, J.L.; Solanki, J.V.; et al. The phenotypic spectrum of GLI3 morphopathies includes autosomal dominant preaxial polydactyly type-IV and postaxial polydactyly type-A/B.; No phenotype prediction from the position of GLI3 mutations. Am. J. Hum. Genet. 1999, 65, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Burns, M.A.; Liao, Z.W.; Yamagata, N.; Pouliot, G.P.; Stevenson, K.E.; Neuberg, D.S.; Thorner, A.R.; Ducar, M.; Silverman, E.A.; Hunger, S.P.; et al. Hedgehog pathway mutations drive oncogenic transformation in high-risk T-cell acute lymphoblastic leukemia. Leukemia 2018, 32, 2126–2137. [Google Scholar] [CrossRef]
- Liu, Y.; Easton, J.; Shao, Y.; Maciaszek, J.; Wang, Z.; Wilkinson, M.R.; McCastlain, K.; Edmonson, M.; Pounds, S.B.; Shi, L.; et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat. Genet. 2017, 49, 1211–1218. [Google Scholar] [CrossRef]
- Martelli, A.M.; Paganelli, F.; Fazio, A.; Bazzichetto, C.; Conciatori, F.; McCubrey, J.A. The Key Roles of PTEN in T-Cell Acute Lymphoblastic Leukemia Development, Progression, and Therapeutic Response. Cancers 2019, 11, 629. [Google Scholar] [CrossRef]
- Kosmider, K.; Karska, K.; Kozakiewicz, A.; Lejman, M.; Zawitkowska, J. Overcoming Steroid Resistance in Pediatric Acute Lymphoblastic Leukemia-The State-of-the-Art Knowledge and Future Prospects. Int. J. Mol. Sci. 2022, 23, 3795. [Google Scholar] [CrossRef]
- Nyakern, M.; Cappellini, A.; Mantovani, I.; Martelli, A.M. Synergistic induction of apoptosis in human leukemia T cells by the Akt inhibitor perifosine and etoposide through activation of intrinsic and Fas-mediated extrinsic cell death pathways. Mol. Cancer Ther. 2006, 5, 1559–1570. [Google Scholar] [CrossRef] [Green Version]
- Chiarini, F.; Del Sole, M.; Mongiorgi, S.; Gaboardi, G.C.; Cappellini, A.; Mantovani, I.; Follo, M.Y.; McCubrey, J.A.; Martelli, A.M. The novel Akt inhibitor, perifosine, induces caspase-dependent apoptosis and downregulates P-glycoprotein expression in multidrug-resistant human T-acute leukemia cells by a JNK-dependent mechanism. Leukemia 2008, 22, 1106–1116. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Chen, X.; Zhang, P.; Fan, Y.; Ma, A.; Pang, T.; Song, Z.; Jin, Y.; Hao, W.; Liu, F.; et al. Inhibition of hedgehog signaling by GANT58 induces apoptosis and shows synergistic antitumor activity with AKT inhibitor in acute T cell leukemia cells. Biochimie 2014, 101, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Filbin, M.G.; Dabral, S.K.; Pazyra-Murphy, M.F.; Ramkissoon, S.; Kung, A.L.; Pak, E.; Chung, J.; Theisen, M.A.; Sun, Y.; Franchetti, Y.; et al. Coordinate activation of Shh and PI3K signaling in PTEN-deficient glioblastoma: New therapeutic opportunities. Nat. Med. 2013, 19, 1518–1523. [Google Scholar] [CrossRef]
- Stecca, B.; Mas, C.; Clement, V.; Zbinden, M.; Correa, R.; Piguet, V.; Beermann, F.; Ruiz i Altaba, A. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 5895–5900. [Google Scholar] [CrossRef]
- Mazumdar, T.; DeVecchio, J.; Agyeman, A.; Shi, T.; Houghton, J.A. The GLI genes as the molecular switch in disrupting Hedgehog signaling in colon cancer. Oncotarget 2011, 2, 638–645. [Google Scholar] [CrossRef]
- Ji, Z.; Mei, F.C.; Xie, J.; Cheng, X. Oncogenic KRAS activates hedgehog signaling pathway in pancreatic cancer cells. J. Biol. Chem. 2007, 282, 14048–14055. [Google Scholar] [CrossRef]
- Singh, R.R.; Cho-Vega, J.H.; Davuluri, Y.; Ma, S.; Kasbidi, F.; Milito, C.; Lennon, P.A.; Drakos, E.; Medeiros, L.J.; Luthra, R.; et al. Sonic hedgehog signaling pathway is activated in ALK-positive anaplastic large cell lymphoma. Cancer Res. 2009, 69, 2550–2558. [Google Scholar] [CrossRef]
- Kaushal, J.B.; Sankhwar, P.; Kumari, S.; Popli, P.; Shukla, V.; Hussain, M.K.; Hajela, K.; Dwivedi, A. The regulation of Hh/Gli1 signaling cascade involves Gsk3beta- mediated mechanism in estrogen-derived endometrial hyperplasia. Sci. Rep. 2017, 7, 6557. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.M.; Paganelli, F.; Evangelisti, C.; Chiarini, F.; McCubrey, J.A. Pathobiology and Therapeutic Relevance of GSK-3 in Chronic Hematological Malignancies. Cells 2022, 11, 1812. [Google Scholar] [CrossRef]
- Wang, J.; Li, W.; Zheng, X.; Pang, X.; Du, G. Research progress on the forkhead box C1. Oncotarget 2018, 9, 12471–12478. [Google Scholar] [CrossRef] [Green Version]
- Ray, T.; Ryusaki, T.; Ray, P.S. Therapeutically Targeting Cancers That Overexpress FOXC1: A Transcriptional Driver of Cell Plasticity, Partial EMT, and Cancer Metastasis. Front. Oncol. 2021, 11, 721959. [Google Scholar] [CrossRef] [PubMed]
- Tosello, V.; Bongiovanni, D.; Liu, J.; Pan, Q.; Yan, K.K.; Saccomani, V.; Van Trimpont, M.; Pizzi, M.; Mazzoni, M.; Dei Tos, A.P.; et al. Cross-talk between GLI transcription factors and FOXC1 promotes T-cell acute lymphoblastic leukemia dissemination. Leukemia 2021, 35, 984–1000. [Google Scholar] [CrossRef]
- Liu, Z.; Li, T.; Reinhold, M.I.; Naski, M.C. MEK1-RSK2 contributes to Hedgehog signaling by stabilizing GLI2 transcription factor and inhibiting ubiquitination. Oncogene 2014, 33, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Qu, Y.; Jin, Y.; Yu, Y.; Deng, N.; Wawrowsky, K.; Zhang, X.; Li, N.; Bose, S.; Wang, Q.; et al. FOXC1 Activates Smoothened-Independent Hedgehog Signaling in Basal-like Breast Cancer. Cell Rep. 2015, 13, 1046–1058. [Google Scholar] [CrossRef]
- Sanchez-Martin, M.; Ferrando, A. The NOTCH1-MYC highway toward T-cell acute lymphoblastic leukemia. Blood 2017, 129, 1124–1133. [Google Scholar] [CrossRef]
- Papayannidis, C.; DeAngelo, D.J.; Stock, W.; Huang, B.; Shaik, M.N.; Cesari, R.; Zheng, X.; Reynolds, J.M.; English, P.A.; Ozeck, M.; et al. A Phase 1 study of the novel gamma-secretase inhibitor PF-03084014 in patients with T-cell acute lymphoblastic leukemia and T-cell lymphoblastic lymphoma. Blood Cancer J. 2015, 5, e350. [Google Scholar] [CrossRef] [PubMed]
- McCarter, A.C.; Wang, Q.; Chiang, M. Notch in Leukemia. Adv. Exp. Med. Biol. 2018, 1066, 355–394. [Google Scholar] [CrossRef]
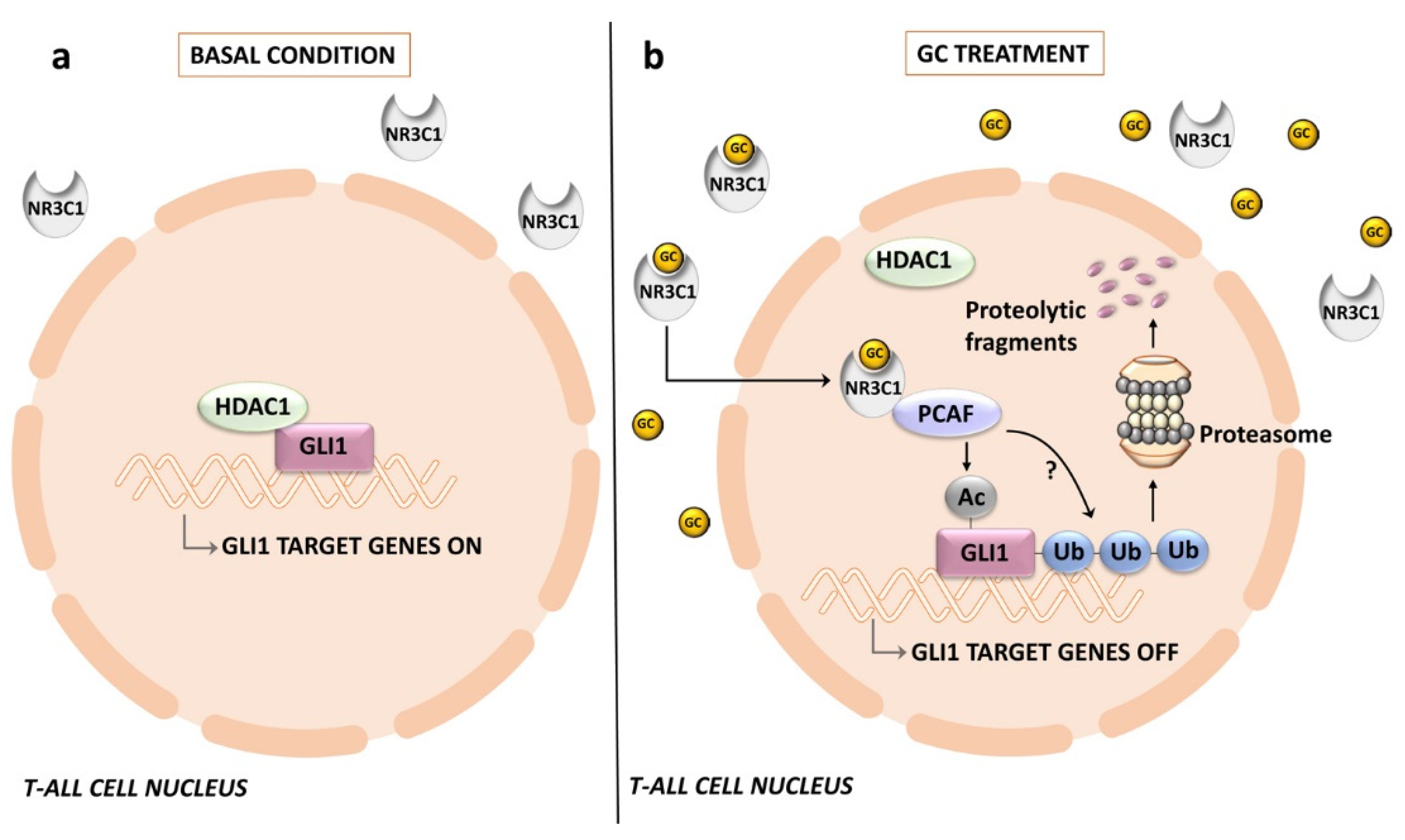
- Bongiovanni, D.; Tosello, V.; Saccomani, V.; Dalla Santa, S.; Amadori, A.; Zanovello, P.; Piovan, E. Crosstalk between Hedgehog pathway and the glucocorticoid receptor pathway as a basis for combination therapy in T-cell acute lymphoblastic leukemia. Oncogene 2020, 39, 6544–6555. [Google Scholar] [CrossRef]
- Schrappe, M.; Valsecchi, M.G.; Bartram, C.R.; Schrauder, A.; Panzer-Grumayer, R.; Moricke, A.; Parasole, R.; Zimmermann, M.; Dworzak, M.; Buldini, B.; et al. Late MRD response determines relapse risk overall and in subsets of childhood T-cell ALL: Results of the AIEOP-BFM-ALL 2000 study. Blood 2011, 118, 2077–2084. [Google Scholar] [CrossRef]
- Coni, S.; Mancuso, A.B.; Di Magno, L.; Sdruscia, G.; Manni, S.; Serrao, S.M.; Rotili, D.; Spiombi, E.; Bufalieri, F.; Petroni, M.; et al. Selective targeting of HDAC1/2 elicits anticancer effects through Gli1 acetylation in preclinical models of SHH Medulloblastoma. Sci. Rep. 2017, 7, 44079. [Google Scholar] [CrossRef] [Green Version]
- Kassel, O.; Herrlich, P. Crosstalk between the glucocorticoid receptor and other transcription factors: Molecular aspects. Mol. Cell Endocrinol. 2007, 275, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Bornhauser, B.C.; Bonapace, L.; Lindholm, D.; Martinez, R.; Cario, G.; Schrappe, M.; Niggli, F.K.; Schafer, B.W.; Bourquin, J.P. Low-dose arsenic trioxide sensitizes glucocorticoid-resistant acute lymphoblastic leukemia cells to dexamethasone via an Akt-dependent pathway. Blood 2007, 110, 2084–2091. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.H.; Luo, J.; Mosley, Y.Y.; Hedrick, V.E.; Paul, L.N.; Chang, J.; Zhang, G.; Wang, Y.K.; Banko, M.R.; Brunet, A.; et al. AMP-Activated Protein Kinase Directly Phosphorylates and Destabilizes Hedgehog Pathway Transcription Factor GLI1 in Medulloblastoma. Cell Rep. 2015, 12, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Huang, S.Y.; Ka-Wai Li, K.; Li, Y.H.; Hsu, W.H.; Zhang, G.J.; Chang, C.J.; Yang, J.Y. Dual degradation signals destruct GLI1: AMPK inhibits GLI1 through beta-TrCP-mediated proteasome degradation. Oncotarget 2017, 8, 49869–49881. [Google Scholar] [CrossRef]
- Penugurti, V.; Mishra, Y.G.; Manavathi, B. AMPK: An odyssey of a metabolic regulator, a tumor suppressor, and now a contextual oncogene. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188785. [Google Scholar] [CrossRef]
- Grimaldi, C.; Chiarini, F.; Tabellini, G.; Ricci, F.; Tazzari, P.L.; Battistelli, M.; Falcieri, E.; Bortul, R.; Melchionda, F.; Iacobucci, I.; et al. AMP-dependent kinase/mammalian target of rapamycin complex 1 signaling in T-cell acute lymphoblastic leukemia: Therapeutic implications. Leukemia 2012, 26, 91–100. [Google Scholar] [CrossRef]
- Kishton, R.J.; Barnes, C.E.; Nichols, A.G.; Cohen, S.; Gerriets, V.A.; Siska, P.J.; Macintyre, A.N.; Goraksha-Hicks, P.; de Cubas, A.A.; Liu, T.; et al. AMPK Is Essential to Balance Glycolysis and Mitochondrial Metabolism to Control T-ALL Cell Stress and Survival. Cell Metab. 2016, 23, 649–662. [Google Scholar] [CrossRef]
- Tosello, V.; Bongiovanni, D.; Di Martino, L.; Franchin, C.; Zanovello, P.; Arrigoni, G.; Piovan, E. Responsiveness to Hedgehog Pathway Inhibitors in T-Cell Acute Lymphoblastic Leukemia Cells Is Highly Dependent on 5’AMP-Activated Kinase Inactivation. Int. J. Mol. Sci. 2021, 22, 6384. [Google Scholar] [CrossRef]
- Di Magno, L.; Basile, A.; Coni, S.; Manni, S.; Sdruscia, G.; D’Amico, D.; Antonucci, L.; Infante, P.; De Smaele, E.; Cucchi, D.; et al. The energy sensor AMPK regulates Hedgehog signaling in human cells through a unique Gli1 metabolic checkpoint. Oncotarget 2016, 7, 9538–9549. [Google Scholar] [CrossRef]
- Gonzalez-Gugel, E.; Villa-Morales, M.; Santos, J.; Bueno, M.J.; Malumbres, M.; Rodriguez-Pinilla, S.M.; Piris, M.A.; Fernandez-Piqueras, J. Down-regulation of specific miRNAs enhances the expression of the gene Smoothened and contributes to T-cell lymphoblastic lymphoma development. Carcinogenesis 2013, 34, 902–908. [Google Scholar] [CrossRef] [Green Version]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Salmeron-Villalobos, J.; Ramis-Zaldivar, J.E.; Balague, O.; Verdu-Amoros, J.; Celis, V.; Sabado, C.; Garrido, M.; Mato, S.; Uriz, J.; Ortega, M.J.; et al. Diverse mutations and structural variations contribute to Notch signaling deregulation in paediatric T-cell lymphoblastic lymphoma. Pediatr. Blood Cancer 2022, 69, e29926. [Google Scholar] [CrossRef] [PubMed]
- Intermesoli, T.; Weber, A.; Leoncin, M.; Frison, L.; Skert, C.; Bassan, R. Lymphoblastic Lymphoma: A Concise Review. Curr. Oncol. Rep. 2022, 24, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kyriakidis, I.; Kyriakidis, K.; Tsezou, A. MicroRNAs and the Diagnosis of Childhood Acute Lymphoblastic Leukemia: Systematic Review, Meta-Analysis and Re-Analysis with Novel Small RNA-Seq Tools. Cancers 2022, 14, 3976. [Google Scholar] [CrossRef] [PubMed]
- Farooqi, A.A.; Naureen, H.; Attar, R. Regulation of cell signaling pathways by circular RNAs and microRNAs in different cancers: Spotlight on Wnt/beta-catenin, JAK/STAT, TGF/SMAD, SHH/GLI, NOTCH and Hippo pathways. Semin Cell Dev. Biol. 2022, 124, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Sargazi, M.L.; Jafarinejad-Farsangi, S.; Moazzam-Jazi, M.; Rostamzadeh, F.; Karam, Z.M. The crosstalk between long non-coding RNAs and the hedgehog signaling pathway in cancer. Med. Oncol. 2022, 39, 127. [Google Scholar] [CrossRef]
- HajiEsmailPoor, Z.; Tabnak, P.; Ahmadzadeh, B.; Ebrahimi, S.S.; Faal, B.; Mashatan, N. Role of hedgehog signaling related non-coding RNAs in developmental and pathological conditions. Biomed. Pharmacother. 2022, 153, 113507. [Google Scholar] [CrossRef]
- Roy, U.; Raghavan, S.C. Deleterious point mutations in T-cell acute lymphoblastic leukemia: Mechanistic insights into leukemogenesis. Int. J. Cancer 2021, 149, 1210–1220. [Google Scholar] [CrossRef]
- Pear, W.S.; Aster, J.C.; Scott, M.L.; Hasserjian, R.P.; Soffer, B.; Sklar, J.; Baltimore, D. Exclusive development of T cell neoplasms in mice transplanted with bone marrow expressing activated Notch alleles. J. Exp. Med. 1996, 183, 2283–2291. [Google Scholar] [CrossRef]
- Radtke, F.; Fasnacht, N.; Macdonald, H.R. Notch signaling in the immune system. Immunity 2010, 32, 14–27. [Google Scholar] [CrossRef] [Green Version]
- Degryse, S.; de Bock, C.E.; Cox, L.; Demeyer, S.; Gielen, O.; Mentens, N.; Jacobs, K.; Geerdens, E.; Gianfelici, V.; Hulselmans, G.; et al. JAK3 mutants transform hematopoietic cells through JAK1 activation, causing T-cell acute lymphoblastic leukemia in a mouse model. Blood 2014, 124, 3092–3100. [Google Scholar] [CrossRef]
- Oliveira, M.L.; Akkapeddi, P.; Alcobia, I.; Almeida, A.R.; Cardoso, B.A.; Fragoso, R.; Serafim, T.L.; Barata, J.T. From the outside, from within: Biological and therapeutic relevance of signal transduction in T-cell acute lymphoblastic leukemia. Cell Signal 2017, 38, 10–25. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Jette, C.; Kanki, J.P.; Aster, J.C.; Look, A.T.; Griffin, J.D. NOTCH1-induced T-cell leukemia in transgenic zebrafish. Leukemia 2007, 21, 462–471. [Google Scholar] [CrossRef]
- Koudijs, M.J.; den Broeder, M.J.; Keijser, A.; Wienholds, E.; Houwing, S.; van Rooijen, E.M.; Geisler, R.; van Eeden, F.J. The zebrafish mutants dre, uki, and lep encode negative regulators of the hedgehog signaling pathway. PLoS Genet. 2005, 1, e19. [Google Scholar] [CrossRef]
- Romer, J.T.; Kimura, H.; Magdaleno, S.; Sasai, K.; Fuller, C.; Baines, H.; Connelly, M.; Stewart, C.F.; Gould, S.; Rubin, L.L.; et al. Suppression of the Shh pathway using a small molecule inhibitor eliminates medulloblastoma in Ptc1(+/-)p53(-/-) mice. Cancer Cell 2004, 6, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Zahreddine, H.A.; Culjkovic-Kraljacic, B.; Assouline, S.; Gendron, P.; Romeo, A.A.; Morris, S.J.; Cormack, G.; Jaquith, J.B.; Cerchietti, L.; Cocolakis, E.; et al. The sonic hedgehog factor GLI1 imparts drug resistance through inducible glucuronidation. Nature 2014, 511, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; Mei, F.C.; Johnson, B.H.; Thompson, E.B.; Cheng, X. Protein kinase A, not Epac, suppresses hedgehog activity and regulates glucocorticoid sensitivity in acute lymphoblastic leukemia cells. J. Biol. Chem. 2007, 282, 37370–37377. [Google Scholar] [CrossRef]
- Pietrobono, S.; Gaudio, E.; Gagliardi, S.; Zitani, M.; Carrassa, L.; Migliorini, F.; Petricci, E.; Manetti, F.; Makukhin, N.; Bond, A.G.; et al. Targeting non-canonical activation of GLI1 by the SOX2-BRD4 transcriptional complex improves the efficacy of HEDGEHOG pathway inhibition in melanoma. Oncogene 2021, 40, 3799–3814. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SMO Inhibitors | National Clinical Trial (NCT) | Tumor Type |
|---|---|---|
| Vismodegib (GDC-0449) | e.g., NCT03035188, NCT02115828 | BCC, prostate cancer |
| Sonidegib (NVP-LDE225) | e.g., NCT02111187, NCT04066504, | Prostate cancer, BCC, |
| Glasdegib (PF-04449913) | e.g., NCT03466450, NCT04231851 | Glioblastoma, AML |
| Saridegib (IPI-926) | e.g., NCT01609179 | BCC |
| Taladegib (LY-2940680) | e.g., NCT01226485 | Advanced cancer |
| TAK-441 | e.g., NCT01204073 | Advanced nonhematologic malignancies |
| BMS-833923 (XL139) | e.g., NCT01413906, NCT00670189 | Solid tumors, BCC |
| Itraconazole | e.g., NCT01791894 | BCC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martelli, A.M.; Paganelli, F.; Truocchio, S.; Palumbo, C.; Chiarini, F.; McCubrey, J.A. Understanding the Roles of the Hedgehog Signaling Pathway during T-Cell Lymphopoiesis and in T-Cell Acute Lymphoblastic Leukemia (T-ALL). Int. J. Mol. Sci. 2023, 24, 2962. https://doi.org/10.3390/ijms24032962
Martelli AM, Paganelli F, Truocchio S, Palumbo C, Chiarini F, McCubrey JA. Understanding the Roles of the Hedgehog Signaling Pathway during T-Cell Lymphopoiesis and in T-Cell Acute Lymphoblastic Leukemia (T-ALL). International Journal of Molecular Sciences. 2023; 24(3):2962. https://doi.org/10.3390/ijms24032962
Chicago/Turabian StyleMartelli, Alberto M., Francesca Paganelli, Serena Truocchio, Carla Palumbo, Francesca Chiarini, and James A. McCubrey. 2023. "Understanding the Roles of the Hedgehog Signaling Pathway during T-Cell Lymphopoiesis and in T-Cell Acute Lymphoblastic Leukemia (T-ALL)" International Journal of Molecular Sciences 24, no. 3: 2962. https://doi.org/10.3390/ijms24032962