
Supt16 Haploinsufficiency Impairs PI3K/AKT/mTOR/Autophagy Pathway in Human Pluripotent Stem Cells Derived Neural Stem Cells
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Supt16 Haploinsufficiency Disrupts the Self-Renewal of Human iPSCs Derived NSCs
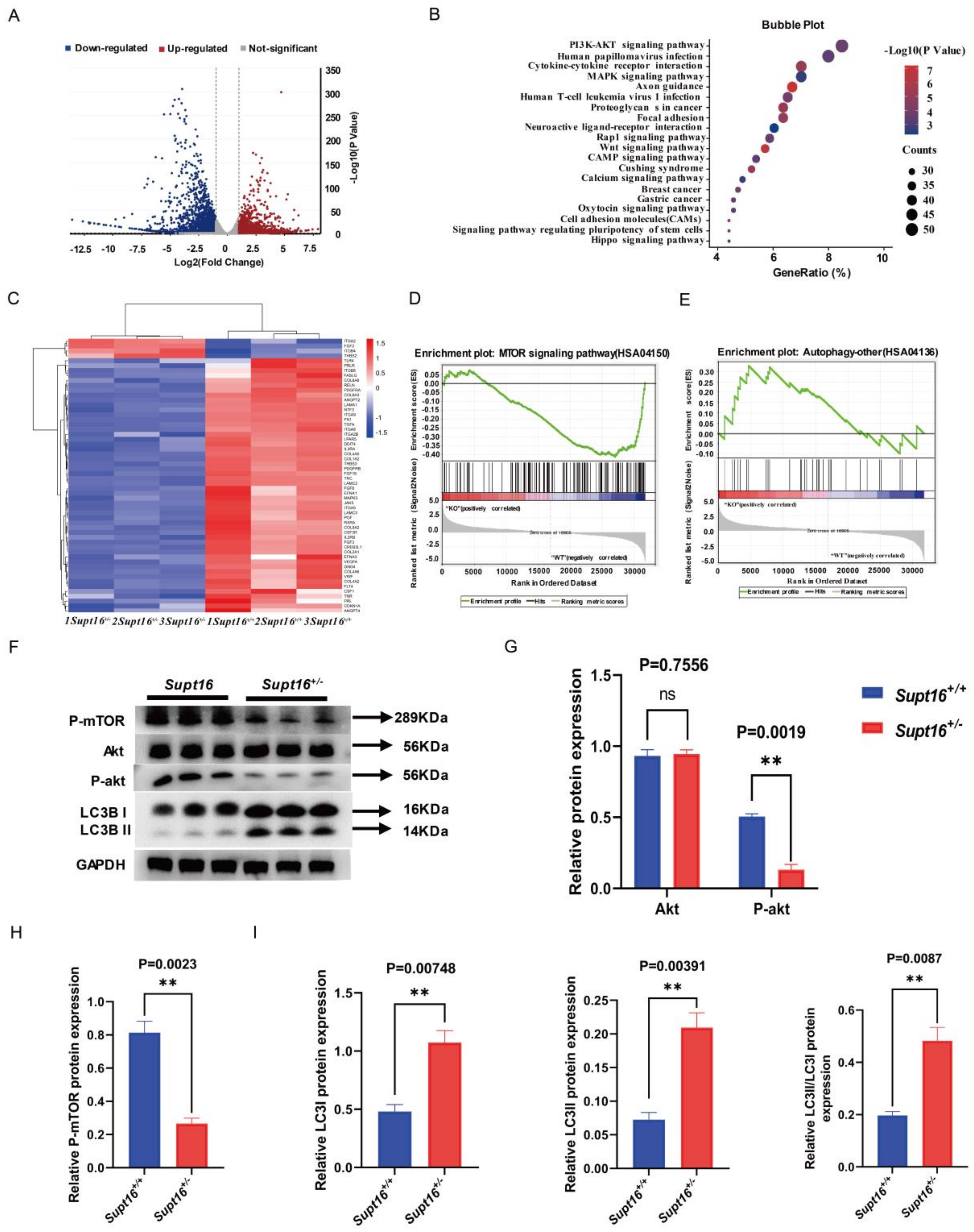
2.2. Supt16 Haploinsufficiency Impaired hNSCs Self-Renewal by Activating PI3K/AKT/mTOR-Mediated Autophagy
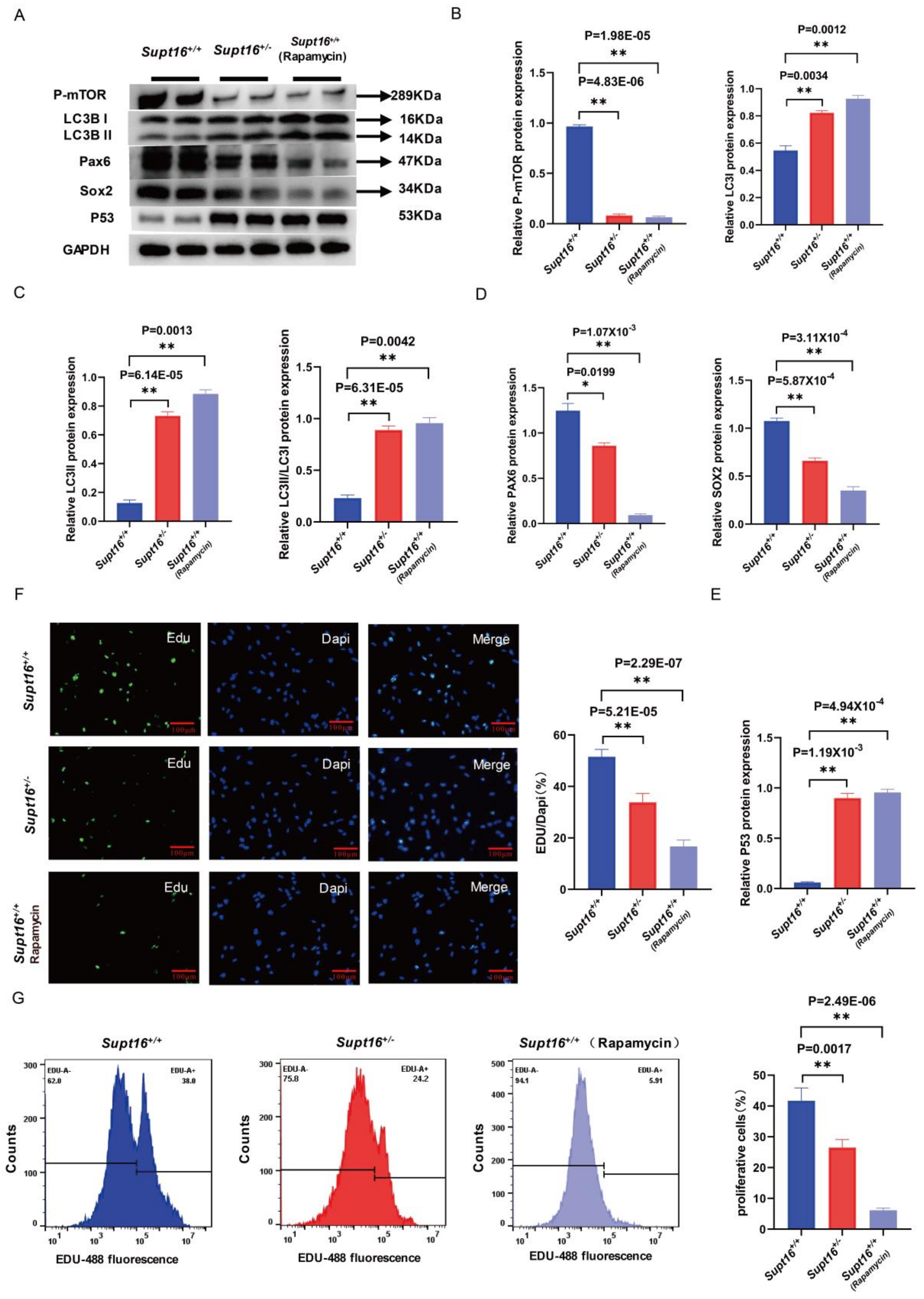
2.3. Wild-Type hNSCs Treated by Rapamycin Reproduced the Phenotype of Supt16 Haploinsufficient hNSCs
2.4. MHY1485 Treatment Rescued the Phenotypes of Supt16 Haploinsufficient hNSCs
3. Discussion
4. Method
4.1. Generation of Supt16+/− hNSCs
4.2. Human Neural Stem Cell Differentiation
4.3. Flow Cytometry Analysis Experiments
4.4. EdU Proliferation Assay
4.5. Western Blot
4.6. RNA-Seq Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gilissen, C.; Hehir-Kwa, J.Y.; Thung, D.T.; van de Vorst, M.; van Bon, B.W.; Willemsen, M.H.; Kwint, M.; Janssen, I.M.; Hoischen, A.; Schenck, A.; et al. Genome sequencing identifies major causes of severe intellectual disability. Nature 2014, 511, 344–347. [Google Scholar] [CrossRef] [PubMed]
- Niemi, M.E.K.; Martin, H.C.; Rice, D.L.; Gallone, G.; Gordon, S.; Kelemen, M.; McAloney, K.; McRae, J.; Radford, E.J.; Yu, S.; et al. Common genetic variants contribute to risk of rare severe neurodevelopmental disorders. Nature 2018, 562, 268–271. [Google Scholar] [CrossRef] [PubMed]
- Tărlungeanu, D.C.; Novarino, G. Genomics in neurodevelopmental disorders: An avenue to personalized medicine. Exp. Mol. Med. 2018, 50, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kriegstein, A.; Alvarez-Buylla, A. The glial nature of embryonic and adult neural stem cells. Annu. Rev. Neurosci. 2009, 32, 149–184. [Google Scholar] [CrossRef] [PubMed]
- Grabel, L. Developmental origin of neural stem cells: The glial cell that could. Stem Cell Rev. Rep. 2012, 8, 577–585. [Google Scholar] [CrossRef]
- Okano, H.; Temple, S. Cell types to order: Temporal specification of CNS stem cells. Curr. Opin. Neurobiol. 2009, 19, 112–119. [Google Scholar] [CrossRef]
- Ernst, C. Proliferation and Differentiation Deficits are a Major Convergence Point for Neurodevelopmental Disorders. Trends Neurosci. 2016, 39, 290–299. [Google Scholar] [CrossRef]
- Liu, X.; Dai, S.K.; Liu, P.P.; Liu, C.M. Arid1a regulates neural stem/progenitor cell proliferation and differentiation during cortical development. Cell Proliferat. 2021, 54, e13124. [Google Scholar] [CrossRef]
- Gao, Y.; Duque-Wilckens, N.; Aljazi, M.B.; Moeser, A.J.; Mias, G.I.; Robison, A.J.; Zhang, Y.; He, J. Impaired KDM2B-mediated PRC1 recruitment to chromatin causes defective neural stem cell self-renewal and ASD/ID-like behaviors. iScience 2022, 25, 103742. [Google Scholar] [CrossRef]
- Murai, K.; Sun, G.; Ye, P.; Tian, E.; Yang, S.; Cui, Q.; Sun, G.; Trinh, D.; Sun, O.; Hong, T.; et al. The TLX-miR-219 cascade regulates neural stem cell proliferation in neurodevelopment and schizophrenia iPSC model. Nat. Commun. 2016, 7, 10965. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.F.; Zhang, Y.; Wilde, J.; Hansen, K.C.; Lai, F.; Niswander, L. Microcephaly disease gene Wdr62 regulates mitotic progression of embryonic neural stem cells and brain size. Nat. Commun. 2014, 5, 3885. [Google Scholar] [CrossRef]
- Carpentieri, J.A.; Di Cicco, A.; Lampic, M.; Andreau, D.; Del Maestro, L.; El Marjou, F.; Coquand, L.; Bahi-Buisson, N.; Brault, J.B.; Baffet, A.D. Endosomal trafficking defects alter neural progenitor proliferation and cause microcephaly. Nat. Commun. 2022, 13, 16. [Google Scholar] [CrossRef]
- Parchem, R.J.; Moore, N.; Fish, J.L.; Parchem, J.G.; Braga, T.T.; Shenoy, A.; Oldham, M.C.; Rubenstein, J.L.; Schneider, R.A.; Blelloch, R. miR-302 Is Required for Timing of Neural Differentiation, Neural Tube Closure, and Embryonic Viability. Cell Rep. 2015, 12, 760–773. [Google Scholar] [CrossRef]
- Theodosis, D.T.; Fraser, F.C. Early changes in the mouse neuroepithelium preceding exencephaly induced by hypervitaminosis A. Teratology 1978, 18, 219–232. [Google Scholar] [CrossRef]
- Curtis, M.A.; Penney, E.B.; Pearson, A.G.; van Roon-Mom, W.M.; Butterworth, N.J.; Dragunow, M.; Connor, B.; Faull, R.L. Increased cell proliferation and neurogenesis in the adult human Huntington’s disease brain. Proc. Natl. Acad. Sci. USA 2003, 100, 9023–9027. [Google Scholar] [CrossRef]
- Höglinger, G.U.; Rizk, P.; Muriel, M.P.; Duyckaerts, C.; Oertel, W.H.; Caille, I.; Hirsch, E.C. Dopamine depletion impairs precursor cell proliferation in Parkinson disease. Nat. Neurosci. 2004, 7, 726–735. [Google Scholar] [CrossRef]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. Embo J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef]
- Crutcher, E.; Pal, R.; Naini, F.; Zhang, P.; Laugsch, M.; Kim, J.; Bajic, A.; Schaaf, C.P. mTOR and autophagy pathways are dysregulated in murine and human models of Schaaf-Yang syndrome. Sci. Rep. 2019, 9, 15935. [Google Scholar] [CrossRef]
- Zhu, J.; Dou, S.; Jiang, Y.; Bai, B.; Chen, J.; Wang, C.; Cheng, B. Apelin-36 exerts the cytoprotective effect against MPP(+)-induced cytotoxicity in SH-SY5Y cells through PI3K/Akt/mTOR autophagy pathway. Life Sci. 2019, 224, 95–108. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [Green Version]
- Deng, Z.; Zhou, X.; Lu, J.H.; Yue, Z. Autophagy deficiency in neurodevelopmental disorders. Cell Biosci. 2021, 11, 214. [Google Scholar] [CrossRef] [PubMed]
- Hui, K.K.; Takashima, N.; Watanabe, A.; Chater, T.E.; Matsukawa, H.; Nekooki-Machida, Y.; Nilsson, P.; Endo, R.; Goda, Y.; Saido, T.C.; et al. GABARAPs dysfunction by autophagy deficiency in adolescent brain impairs GABA(A) receptor trafficking and social behavior. Sci. Adv. 2019, 5, eaau8237. [Google Scholar] [CrossRef] [PubMed]
- Dragich, J.M.; Kuwajima, T.; Hirose-Ikeda, M.; Yoon, M.S.; Eenjes, E.; Bosco, J.R.; Fox, L.M.; Lystad, A.H.; Oo, T.F.; Yarygina, O.; et al. Autophagy linked FYVE (Alfy/WDFY3) is required for establishing neuronal connectivity in the mammalian brain. eLife 2016, 5, e14810. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Gudsnuk, K.; Kuo, S.H.; Cotrina, M.; Rosoklija, G.; Sosunov, A.; Sonders, M.; Kanter, E.; Castagna, C.; Ai, Y.J.N. Loss of mTOR-Dependent Macroautophagy Causes Autistic-like Synaptic Pruning Deficits. Neuron 2014, 83, 1131–1143. [Google Scholar] [CrossRef]
- Amegandjin, C.A.; Choudhury, M.; Jadhav, V.; Carriço, J.N.; Quintal, A.; Berryer, M.; Snapyan, M.; Chattopadhyaya, B.; Saghatelyan, A.; Di Cristo, G. Sensitive period for rescuing parvalbumin interneurons connectivity and social behavior deficits caused by TSC1 loss. Nat. Commun. 2021, 12, 3653. [Google Scholar] [CrossRef]
- Schäffner, I.; Minakaki, G.; Khan, M.A.; Balta, E.A.; Schlötzer-Schrehardt, U.; Schwarz, T.J.; Beckervordersandforth, R.; Winner, B.; Webb, A.E.; DePinho, R.A.; et al. FoxO Function Is Essential for Maintenance of Autophagic Flux and Neuronal Morphogenesis in Adult Neurogenesis. Neuron 2018, 99, 1188–1203.e1186. [Google Scholar] [CrossRef]
- Linda, K.; Lewerissa, E.I.; Verboven, A.H.A.; Gabriele, M.; Frega, M.; Klein Gunnewiek, T.M.; Devilee, L.; Ulferts, E.; Hommersom, M.; Oudakker, A.; et al. Imbalanced autophagy causes synaptic deficits in a human model for neurodevelopmental disorders. Autophagy 2022, 18, 423–442. [Google Scholar] [CrossRef]
- Jeronimo, C.; Angel, A.; Nguyen, V.Q.; Kim, J.M.; Poitras, C.; Lambert, E.; Collin, P.; Mellor, J.; Wu, C.; Robert, F. FACT is recruited to the +1 nucleosome of transcribed genes and spreads in a Chd1-dependent manner. Mol. Cell 2021, 81, 3542–3559.e3511. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, X.; Feng, J.; Leng, H.; Li, S.; Xiao, J.; Liu, S.; Xu, Z.; Xu, J.; Li, D.; et al. The Histone Chaperone FACT Contributes to DNA Replication-Coupled Nucleosome Assembly. Cell Rep. 2016, 16, 3414. [Google Scholar] [CrossRef]
- Dinant, C.; Ampatziadis-Michailidis, G.; Lans, H.; Tresini, M.; Lagarou, A.; Grosbart, M.; Theil, A.F.; van Cappellen, W.A.; Kimura, H.; Bartek, J.; et al. Enhanced chromatin dynamics by FACT promotes transcriptional restart after UV-induced DNA damage. Mol. Cell. 2013, 51, 469–479. [Google Scholar] [CrossRef] [Green Version]
- Hossan, T.; Nagarajan, S.; Baumgart, S.J.; Xie, W.; Magallanes, R.T.; Hernandez, C.; Chiaroni, P.M.; Indenbirken, D.; Spitzner, M.; Thomas-Chollier, M.; et al. Histone Chaperone SSRP1 is Essential for Wnt Signaling Pathway Activity During Osteoblast Differentiation. Stem Cells 2016, 34, 1369–1376. [Google Scholar] [CrossRef]
- Shen, Z.; Formosa, T.; Tantin, D. FACT Inhibition Blocks Induction But Not Maintenance of Pluripotency. Stem Cells Dev. 2018, 27, 1693–1701. [Google Scholar] [CrossRef]
- Bina, R.; Matalon, D.; Fregeau, B.; Tarsitano, J.J.; Aukrust, I.; Houge, G.; Bend, R.; Warren, H.; Stevenson, R.E.; Stuurman, K.E.; et al. De novo variants in Supt16H cause neurodevelopmental disorders associated with corpus callosum abnormalities. J. Med Genet. 2020, 57, 461–465. [Google Scholar] [CrossRef]
- Wang, J.; Zhu, X.; Dai, L.; Wang, Z.; Guan, X.; Tan, X.; Li, J.; Zhang, M.; Bai, Y.; Guo, H. Supt16 haploinsufficiency causes neurodevelopment disorder by disrupting MAPK pathway in neural stem cells. Hum. Mol. Genet. 2022. [Google Scholar] [CrossRef]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef]
- Menendez, J.A.; Vellon, L.; Oliveras-Ferraros, C.; Cufí, S.; Vazquez-Martin, A. mTOR-regulated senescence and autophagy during reprogramming of somatic cells to pluripotency: A roadmap from energy metabolism to stem cell renewal and aging. Cell Cycle 2011, 10, 3658–3677. [Google Scholar] [CrossRef]
- Garcia, H.; Fleyshman, D.; Kolesnikova, K.; Safina, A.; Commane, M.; Paszkiewicz, G.; Omelian, A.; Morrison, C.; Gurova, K. Expression of FACT in mammalian tissues suggests its role in maintaining of undifferentiated state of cells. Oncotarget 2011, 2, 783–796. [Google Scholar] [CrossRef]
- Garcia-Luis, J.; Lazar-Stefanita, L.; Gutierrez-Escribano, P.; Thierry, A.; Cournac, A.; García, A.; González, S.; Sánchez, M.; Jarmuz, A.; Montoya, A.; et al. FACT mediates cohesin function on chromatin. Nat. Struct. Mol. Biol. 2019, 26, 970–979. [Google Scholar] [CrossRef]
- Ma, M.; Zhang, X.; Zheng, Y.; Lu, S.; Pan, X.; Mao, X.; Pan, H.; Chung, H.L.; Wang, H.; Guo, H.; et al. The fly homolog of Supt16H, a gene associated with neurodevelopmental disorders, is required in a cell-autonomous fashion for cell survival. Hum. Mol. Genet. 2022. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Boya, P.; Codogno, P.; Rodriguez-Muela, N. Autophagy in stem cells: Repair, remodelling and metabolic reprogramming. Development 2018, 145, dev146506. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.L.; Simon, A.K.; Prescott, M.; Menendez, J.A.; Liu, F.; Wang, F.; Wang, C.; Wolvetang, E.; Vazquez-Martin, A.; Zhang, J. Autophagy in stem cells. Autophagy 2013, 9, 830–849. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Liang, C.C.; Bian, Z.C.; Zhu, Y.; Guan, J.L. FIP200 is required for maintenance and differentiation of postnatal neural stem cells. Nat. Neurosci. 2013, 16, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Choe, S.; Woo, H.; Jeong, H.; An, H.K.; Moon, H.; Ryu, H.Y.; Yeo, B.K.; Lee, Y.W.; Choi, H.; et al. Autophagic death of neural stem cells mediates chronic stress-induced decline of adult hippocampal neurogenesis and cognitive deficits. Autophagy 2020, 16, 512–530. [Google Scholar] [CrossRef]
- Wang, G.; Yang, L.; Grishin, D.; Rios, X.; Ye, L.Y.; Hu, Y.; Li, K.; Zhang, D.; Church, G.M.; Pu, W.T. Efficient, footprint-free human iPSCs genome editing by consolidation of Cas9/CRISPR and piggyBac technologies. Nat. Protoc. 2017, 12, 88–103. [Google Scholar] [CrossRef]
- Ganley, I.G.; Lam du, H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Wang, Z.; Dai, L.; Zhu, X.; Guan, X.; Wang, J.; Li, J.; Zhang, M.; Bai, Y.; Guo, H. Supt16 Haploinsufficiency Impairs PI3K/AKT/mTOR/Autophagy Pathway in Human Pluripotent Stem Cells Derived Neural Stem Cells. Int. J. Mol. Sci. 2023, 24, 3035. https://doi.org/10.3390/ijms24033035
Wang J, Wang Z, Dai L, Zhu X, Guan X, Wang J, Li J, Zhang M, Bai Y, Guo H. Supt16 Haploinsufficiency Impairs PI3K/AKT/mTOR/Autophagy Pathway in Human Pluripotent Stem Cells Derived Neural Stem Cells. International Journal of Molecular Sciences. 2023; 24(3):3035. https://doi.org/10.3390/ijms24033035
Chicago/Turabian StyleWang, Junwen, Ziyi Wang, Limeng Dai, Xintong Zhu, Xingying Guan, Junyi Wang, Jia Li, Mao Zhang, Yun Bai, and Hong Guo. 2023. "Supt16 Haploinsufficiency Impairs PI3K/AKT/mTOR/Autophagy Pathway in Human Pluripotent Stem Cells Derived Neural Stem Cells" International Journal of Molecular Sciences 24, no. 3: 3035. https://doi.org/10.3390/ijms24033035