
Deletion of Annexin A1 in Mice Upregulates the Expression of Its Receptor, Fpr2/3, and Reactivity to the AnxA1 Mimetic Peptide in Platelets
 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
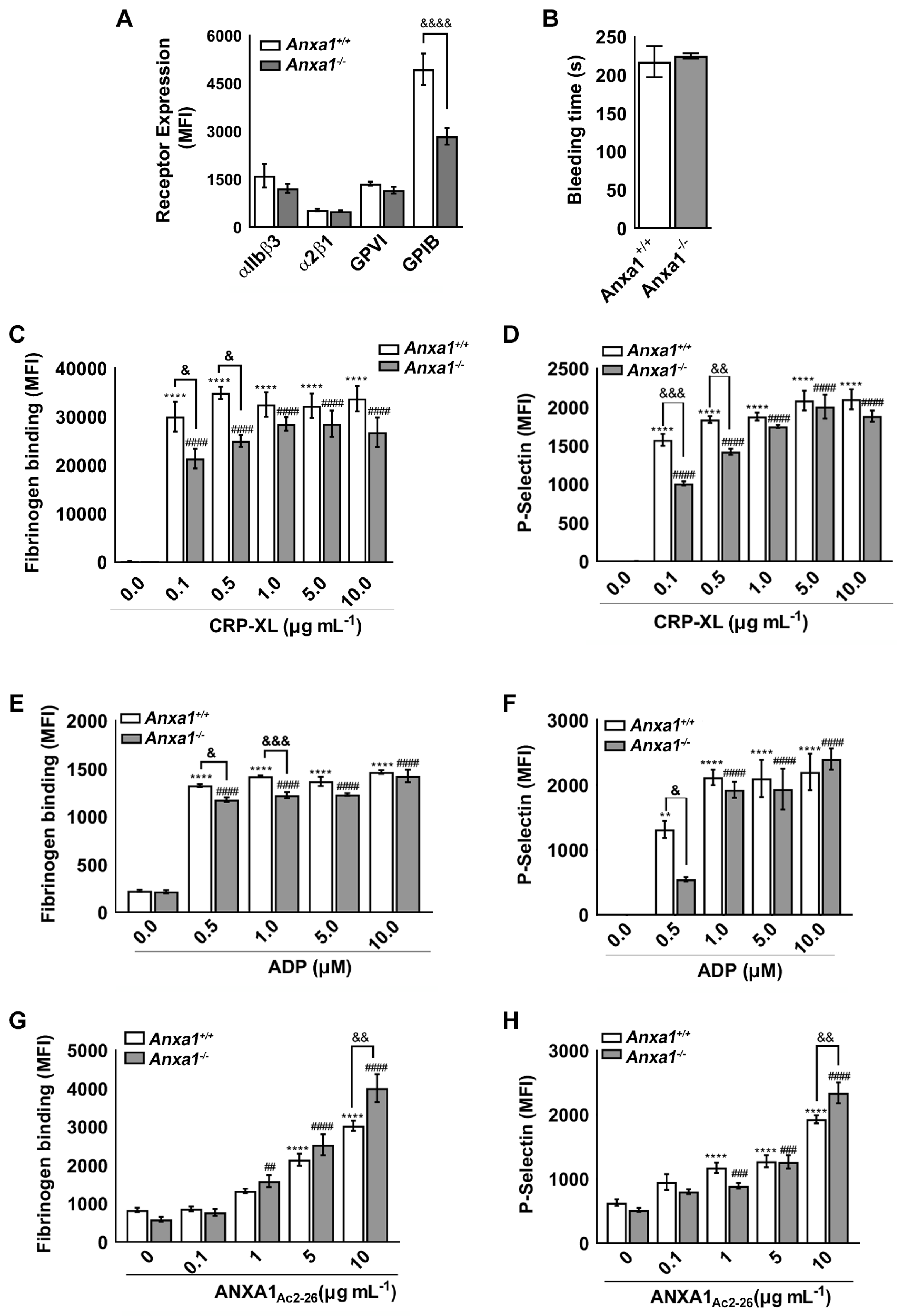
2.1. Anxa1−/− Mouse Platelets Exhibit Reduced GPIbα Level
2.2. Haemostasis Is Unaffected in Anxa1−/− Mice
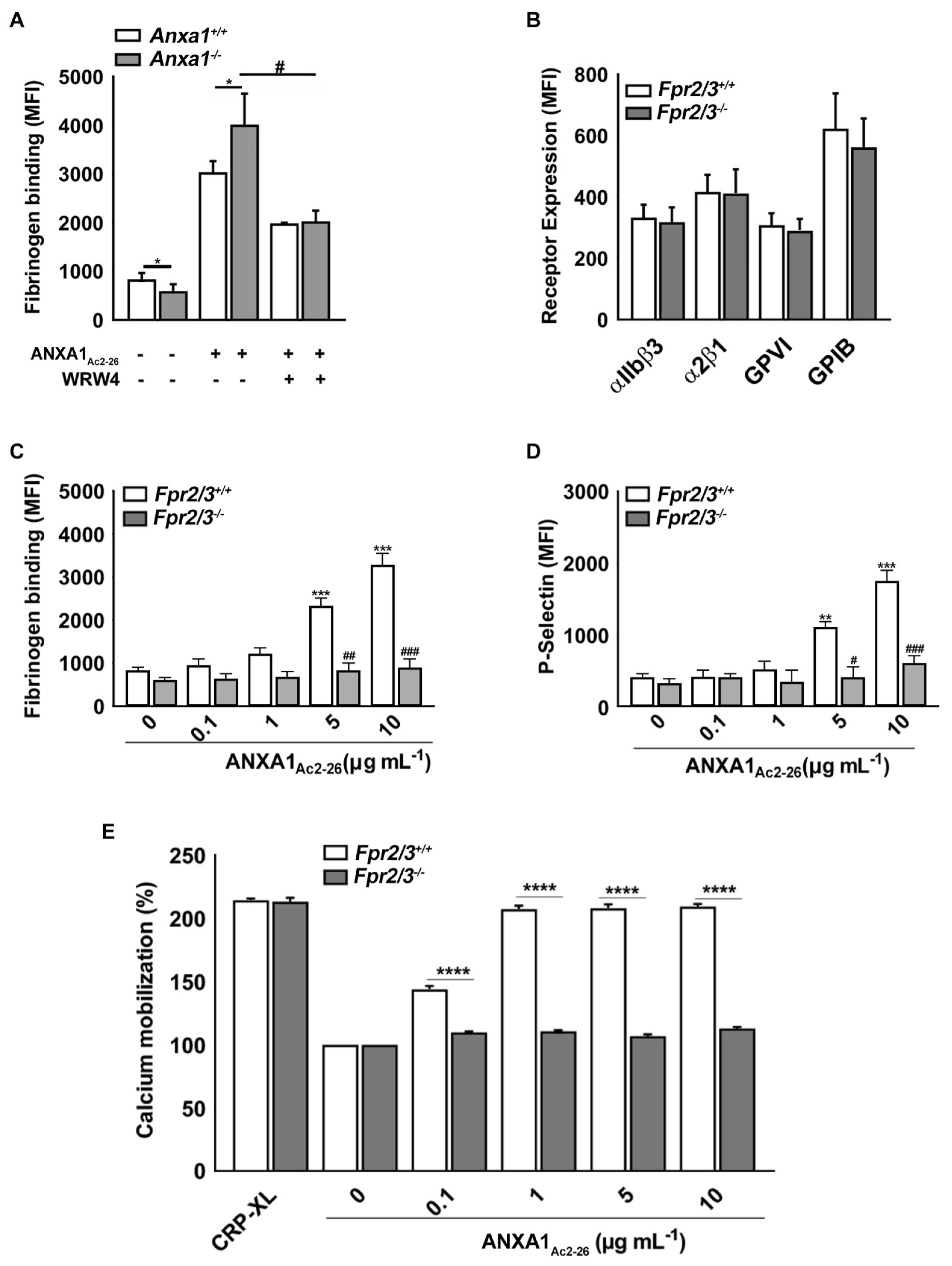
2.3. ANXA1Ac2-26 Exhibits Increased Platelet Reactivity in Anxa1−/− Mouse Platelets
2.4. ANXA1Ac2-26 Induces Platelet Activation through FPR2/ALX
2.5. Fpr2/3 Is Overexpressed in Anxa1−/− Mouse Platelets
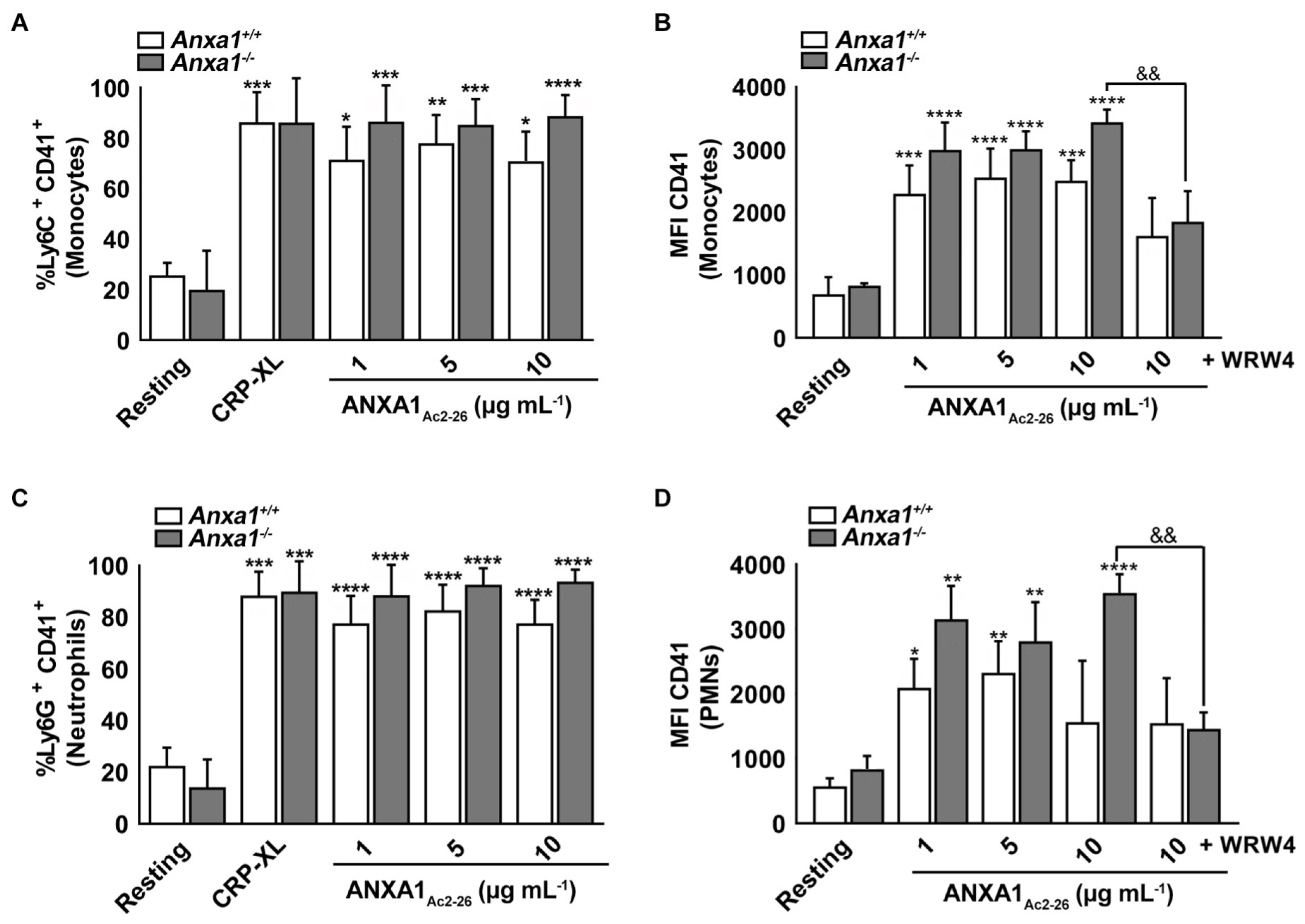
2.6. ANXA1Ac2-26 Induces Platelet–Leukocyte Aggregates in the Whole Blood of Anxa1−/− Mice
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Preparation of Mouse Platelets
4.3. Tail Bleeding Assay in Mice
4.4. Intracellular Calcium Mobilisation Assay
4.5. Electron Microscopy Analysis
4.6. Flow Cytometry-Based Assays
4.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Duhamel, T.A.; Xu, Y.J.; Arneja, A.S.; Dhalla, N.S. Targeting platelets for prevention and treatment of cardiovascular disease. Expert Opin. Ther. Targets 2007, 11, 1523–1533. [Google Scholar] [CrossRef]
- Lam, F.W.; Vijayan, K.V.; Rumbaut, R.E. Platelets and Their Interactions with Other Immune Cells. Compr. Physiol. 2015, 5, 1265–1280. [Google Scholar] [CrossRef]
- Gros, A.; Ollivier, V.; Ho-Tin-Noé, B. Platelets in inflammation: Regulation of leukocyte activities and vascular repair. Front. Immunol. 2014, 5, 678. [Google Scholar] [CrossRef] [PubMed]
- Headland, S.E.; Norling, L.V. The resolution of inflammation: Principles and challenges. Semin. Immunol. 2015, 27, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Rossaint, J.; Margraf, A.; Zarbock, A. Role of Platelets in Leukocyte Recruitment and Resolution of Inflammation. Front. Immunol. 2018, 9, 2712. [Google Scholar] [CrossRef] [PubMed]
- Allen, N.; Barrett, T.J.; Guo, Y.; Nardi, M.; Ramkhelawon, B.; Rockman, C.B.; Hochman, J.S.; Berger, J.S. Circulating monocyte-platelet aggregates are a robust marker of platelet activity in cardiovascular disease. Atherosclerosis 2019, 282, 11–18. [Google Scholar] [CrossRef]
- Freedman, J.E.; Loscalzo, J. Platelet-monocyte aggregates: Bridging thrombosis and inflammation. Circulation 2002, 105, 2130–2132. [Google Scholar] [CrossRef]
- Shao, G.; Zhou, H.; Zhang, Q.; Jin, Y.; Fu, C. Advancements of Annexin A1 in inflammation and tumorigenesis. OncoTargets Ther. 2019, 12, 3245–3254. [Google Scholar] [CrossRef]
- Gavins, F.N.; Hickey, M.J. Annexin A1 and the regulation of innate and adaptive immunity. Front. Immunol. 2012, 3, 354. [Google Scholar] [CrossRef]
- Senchenkova, E.Y.; Ansari, J.; Becker, F.; Vital, S.A.; Al-Yafeai, Z.; Sparkenbaugh, E.M.; Pawlinski, R.; Stokes, K.Y.; Carroll, J.L.; Dragoi, A.M.; et al. Novel Role for the AnxA1-Fpr2/ALX Signaling Axis as a Key Regulator of Platelet Function to Promote Resolution of Inflammation. Circulation 2019, 140, 319–335. [Google Scholar] [CrossRef]
- Getting, S.J.; Flower, R.J.; Perretti, M. Inhibition of neutrophil and monocyte recruitment by endogenous and exogenous lipocortin 1. Br. J. Pharmacol. 1997, 120, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.B.; Kornerup, K.N.; Sampaio, A.L.; D’Acquisto, F.; Seed, M.P.; Girol, A.P.; Gray, M.; Pitzalis, C.; Oliani, S.M.; Perretti, M. The impact of endogenous annexin A1 on glucocorticoid control of inflammatory arthritis. Ann. Rheum. Dis. 2012, 71, 1872–1880. [Google Scholar] [CrossRef]
- Babbin, B.A.; Laukoetter, M.G.; Nava, P.; Koch, S.; Lee, W.Y.; Capaldo, C.T.; Peatman, E.; Severson, E.A.; Flower, R.J.; Perretti, M.; et al. Annexin A1 regulates intestinal mucosal injury, inflammation, and repair. J. Immunol. 2008, 181, 5035–5044. [Google Scholar] [CrossRef]
- Paschalidis, N.; Iqbal, A.J.; Maione, F.; Wood, E.G.; Perretti, M.; Flower, R.J.; D’Acquisto, F. Modulation of experimental autoimmune encephalomyelitis by endogenous annexin A1. J Neuroinflammation 2009, 6, 33. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.L.; Milne, I.R.; Bagley, C.J.; Gamble, J.R.; Vadas, M.A.; Pitson, S.M.; Khew-Goodall, Y. A proinflammatory role for proteolytically cleaved annexin A1 in neutrophil transendothelial migration. J. Immunol. 2010, 185, 3057–3063. [Google Scholar] [CrossRef]
- Perretti, M.; Getting, S.J.; Solito, E.; Murphy, P.M.; Gao, J.L. Involvement of the receptor for formylated peptides in the in vivo anti-migratory actions of annexin 1 and its mimetics. Am. J. Pathol. 2001, 158, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Cirino, G.; Cicala, C.; Sorrentino, L.; Ciliberto, G.; Arpaia, G.; Perretti, M.; Flower, R.J. Anti-inflammatory actions of an N-terminal peptide from human lipocortin 1. Br. J. Pharmacol. 1993, 108, 573–574. [Google Scholar] [CrossRef]
- Tcherniuk, S.; Cenac, N.; Comte, M.; Frouard, J.; Errazuriz-Cerda, E.; Galabov, A.; Morange, P.E.; Vergnolle, N.; Si-Tahar, M.; Alessi, M.C.; et al. Formyl Peptide Receptor 2 Plays a Deleterious Role During Influenza A Virus Infections. J. Infect. Dis. 2016, 214, 237–247. [Google Scholar] [CrossRef]
- Saito, N.; Qiao, H.; Yanagi, T.; Shinkuma, S.; Nishimura, K.; Suto, A.; Fujita, Y.; Suzuki, S.; Nomura, T.; Nakamura, H.; et al. An annexin A1-FPR1 interaction contributes to necroptosis of keratinocytes in severe cutaneous adverse drug reactions. Sci. Transl. Med. 2014, 6, 245ra95. [Google Scholar] [CrossRef]
- Salamah, M.F.; Ravishankar, D.; Kodji, X.; Moraes, L.A.; Williams, H.F.; Vallance, T.M.; Albadawi, D.A.; Vaiyapuri, R.; Watson, K.; Gibbins, J.M.; et al. The endogenous antimicrobial cathelicidin LL37 induces platelet activation and augments thrombus formation. Blood Adv. 2018, 2, 2973–2985. [Google Scholar] [CrossRef] [Green Version]
- Salamah, M.F.; Vallance, T.M.; Kodji, X.; Ravishankar, D.; Williams, H.F.; Brain, S.D.; Vaiyapuri, S. The Antimicrobial Cathelicidin CRAMP Augments Platelet Activation during Psoriasis in Mice. Biomolecules 2020, 10, 1267. [Google Scholar] [CrossRef]
- Salamah, M.F.; Ravishankar, D.; Vaiyapuri, R.; Moraes, L.A.; Patel, K.; Perretti, M.; Gibbins, J.M.; Vaiyapuri, S. The formyl peptide fMLF primes platelet activation and augments thrombus formation. J. Thromb. Haemost. 2019, 17, 1120–1133. [Google Scholar] [CrossRef]
- Rossi, A.; Kontarakis, Z.; Gerri, C.; Nolte, H.; Hölper, S.; Krüger, M.; Stainier, D.Y. Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature 2015, 524, 230–233. [Google Scholar] [CrossRef]
- Aareskjold, E.; Grindheim, A.K.; Hollås, H.; Goris, M.; Lillehaug, J.R.; Vedeler, A. Two tales of Annexin A2 knock-down: One of compensatory effects by antisense RNA and another of a highly active hairpin ribozyme. Biochem. Pharmacol. 2019, 166, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Varga-Szabo, D.; Braun, A.; Nieswandt, B. Calcium signaling in platelets. J. Thromb. Haemost. 2009, 7, 1057–1066. [Google Scholar] [CrossRef] [PubMed]
- Zucoloto, A.Z.; Jenne, C.N. Platelet-Neutrophil Interplay: Insights Into Neutrophil Extracellular Trap (NET)-Driven Coagulation in Infection. Front. Cardiovasc. Med. 2019, 6, 85. [Google Scholar] [CrossRef] [PubMed]
- de Jong, R.J.; Paulin, N.; Lemnitzer, P.; Viola, J.R.; Winter, C.; Ferraro, B.; Grommes, J.; Weber, C.; Reutelingsperger, C.; Drechsler, M.; et al. Protective Aptitude of Annexin A1 in Arterial Neointima Formation in Atherosclerosis-Prone Mice-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 312–315. [Google Scholar] [CrossRef]
- De Jong, R.; Leoni, G.; Drechsler, M.; Soehnlein, O. The advantageous role of annexin A1 in cardiovascular disease. Cell Adh. Migr. 2017, 11, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Yang, Y.H.; May, L.; Gao, X.; Stewart, A.G.; Tu, Y.; Woodman, O.L.; Ritchie, R.H. Cardioprotective potential of annexin-A1 mimetics in myocardial infarction. Pharmacol. Ther. 2015, 148, 47–65. [Google Scholar] [CrossRef]
- Cooray, S.N.; Gobbetti, T.; Montero-Melendez, T.; McArthur, S.; Thompson, D.; Clark, A.J.; Flower, R.J.; Perretti, M. Ligand-specific conformational change of the G-protein-coupled receptor ALX/FPR2 determines proresolving functional responses. Proc. Natl. Acad. Sci. USA 2013, 110, 18232–18237. [Google Scholar] [CrossRef] [Green Version]
- Walther, A.; Riehemann, K.; Gerke, V. A novel ligand of the formyl peptide receptor: Annexin I regulates neutrophil extravasation by interacting with the FPR. Mol. Cell 2000, 5, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Ernst, S.; Lange, C.; Wilbers, A.; Goebeler, V.; Gerke, V.; Rescher, U. An annexin 1 N-terminal peptide activates leukocytes by triggering different members of the formyl peptide receptor family. J. Immunol. 2004, 172, 7669–7676. [Google Scholar] [CrossRef]
- Hayhoe, R.P.; Kamal, A.M.; Solito, E.; Flower, R.J.; Cooper, D.; Perretti, M. Annexin 1 and its bioactive peptide inhibit neutrophil-endothelium interactions under flow: Indication of distinct receptor involvement. Blood 2006, 107, 2123–2130. [Google Scholar] [CrossRef]
- Bena, S.; Brancaleone, V.; Wang, J.M.; Perretti, M.; Flower, R.J. Annexin A1 interaction with the FPR2/ALX receptor: Identification of distinct domains and downstream associated signaling. J. Biol. Chem. 2012, 287, 24690–24697. [Google Scholar] [CrossRef]
- Vital, S.A.; Becker, F.; Holloway, P.M.; Russell, J.; Perretti, M.; Granger, D.N.; Gavins, F.N. Formyl-Peptide Receptor 2/3/Lipoxin A4 Receptor Regulates Neutrophil-Platelet Aggregation and Attenuates Cerebral Inflammation: Impact for Therapy in Cardiovascular Disease. Circulation 2016, 133, 2169–2179. [Google Scholar] [CrossRef]
- Manne, B.K.; Denorme, F.; Middleton, E.A.; Portier, I.; Rowley, J.W.; Stubben, C.; Petrey, A.C.; Tolley, N.D.; Guo, L.; Cody, M.; et al. Platelet gene expression and function in patients with COVID-19. Blood 2020, 136, 1317–1329. [Google Scholar] [CrossRef]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef]
- Vaiyapuri, S.; Sage, T.; Rana, R.H.; Schenk, M.P.; Ali, M.S.; Unsworth, A.J.; Jones, C.I.; Stainer, A.R.; Kriek, N.; Moraes, L.A.; et al. EphB2 regulates contact-dependent and contact-independent signaling to control platelet function. Blood 2015, 125, 720–730. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.I.; Sage, T.; Moraes, L.A.; Vaiyapuri, S.; Hussain, U.; Tucker, K.L.; Barrett, N.E.; Gibbins, J.M. Platelet endothelial cell adhesion molecule-1 inhibits platelet response to thrombin and von Willebrand factor by regulating the internalization of glycoprotein Ib via AKT/glycogen synthase kinase-3/dynamin and integrin αIIbβ3. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1968–1976. [Google Scholar] [CrossRef] [PubMed]
- Mazet, F.; Dunster, J.L.; Jones, C.I.; Vaiyapuri, S.; Tindall, M.J.; Fry, M.J.; Gibbins, J.M. A high-density immunoblotting methodology for quantification of total protein levels and phosphorylation modifications. Sci. Rep. 2015, 5, 16995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaiyapuri, S.; Moraes, L.A.; Sage, T.; Ali, M.S.; Lewis, K.R.; Mahaut-Smith, M.P.; Oviedo-Orta, E.; Simon, A.M.; Gibbins, J.M. Connexin40 regulates platelet function. Nat. Commun. 2013, 4, 2564. [Google Scholar] [CrossRef] [PubMed]
- Ravishankar, D.; Salamah, M.; Attina, A.; Pothi, R.; Vallance, T.M.; Javed, M.; Williams, H.F.; Alzahrani, E.M.S.; Kabova, E.; Vaiyapuri, R.; et al. Ruthenium-conjugated chrysin analogues modulate platelet activity, thrombus formation and haemostasis with enhanced efficacy. Sci. Rep. 2017, 7, 5738. [Google Scholar] [CrossRef]
- Vaiyapuri, S.; Jones, C.I.; Sasikumar, P.; Moraes, L.A.; Munger, S.J.; Wright, J.R.; Ali, M.S.; Sage, T.; Kaiser, W.J.; Tucker, K.L.; et al. Gap junctions and connexin hemichannels underpin hemostasis and thrombosis. Circulation 2012, 125, 2479–2491. [Google Scholar] [CrossRef]
- Vaiyapuri, S.; Flora, G.D.; Gibbins, J.M. Gap junctions and connexin hemichannels in the regulation of haemostasis and thrombosis. Biochem. Soc. Trans. 2015, 43, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Elashry, M.I.; Collins-Hooper, H.; Vaiyapuri, S.; Patel, K. Characterisation of connective tissue from the hypertrophic skeletal muscle of myostatin null mice. J. Anat. 2012, 220, 603–611. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zharkova, O.; Salamah, M.F.; Babak, M.V.; Rajan, E.; Lim, L.H.K.; Andrade, F.; Gil, C.D.; Oliani, S.M.; Moraes, L.A.; Vaiyapuri, S. Deletion of Annexin A1 in Mice Upregulates the Expression of Its Receptor, Fpr2/3, and Reactivity to the AnxA1 Mimetic Peptide in Platelets. Int. J. Mol. Sci. 2023, 24, 3424. https://doi.org/10.3390/ijms24043424
Zharkova O, Salamah MF, Babak MV, Rajan E, Lim LHK, Andrade F, Gil CD, Oliani SM, Moraes LA, Vaiyapuri S. Deletion of Annexin A1 in Mice Upregulates the Expression of Its Receptor, Fpr2/3, and Reactivity to the AnxA1 Mimetic Peptide in Platelets. International Journal of Molecular Sciences. 2023; 24(4):3424. https://doi.org/10.3390/ijms24043424
Chicago/Turabian StyleZharkova, Olga, Maryam F. Salamah, Maria V. Babak, Elanchezhian Rajan, Lina H. K. Lim, Frans Andrade, Cristiane D. Gil, Sonia M. Oliani, Leonardo A. Moraes, and Sakthivel Vaiyapuri. 2023. "Deletion of Annexin A1 in Mice Upregulates the Expression of Its Receptor, Fpr2/3, and Reactivity to the AnxA1 Mimetic Peptide in Platelets" International Journal of Molecular Sciences 24, no. 4: 3424. https://doi.org/10.3390/ijms24043424