
Studies on the Oxidation of Aromatic Amines Catalyzed by Trametes versicolor Laccase


1
Istituto di Scienze e Tecnologie Chimiche-SCITEC, Consiglio Nazionale delle Ricerche, Via Mario Bianco 9, 20131 Milan, Italy
2
Dipartimento di Scienze Farmaceutiche, Università degli Studi di Milano, Via Mangiagalli 25, 20133 Milan, Italy
3
Istituto di Scienze e Tecnologie Chimiche-SCITEC, Consiglio Nazionale delle Ricerche, Via Luigi Mancinelli 7, 20131 Milan, Italy
*
Authors to whom correspondence should be addressed.
Int. J. Mol. Sci. 2023, 24(4), 3524; https://doi.org/10.3390/ijms24043524
Submission received: 16 January 2023
/
Revised: 3 February 2023
/
Accepted: 6 February 2023
/
Published: 9 February 2023
(This article belongs to the Special Issue Biotechnological Applications of Oxidoreductases)
Abstract
:The bio-oxidation of a series of aromatic amines catalyzed by T. versicolor laccase has been investigated exploiting either commercially available nitrogenous substrates [(E)-4-vinyl aniline and diphenyl amine] or ad hoc synthetized ones [(E)-4-styrylaniline, (E)-4-(prop-1-en-1-yl)aniline and (E)-4-(((4-methoxyphenyl)imino)methyl)phenol]. At variance to their phenolic equivalents, the investigated aromatic amines were not converted into the expected cyclic dimeric structures under T. versicolor catalysis. The formation of complex oligomeric/polymeric or decomposition by-products was mainly observed, with the exception of the isolation of two interesting but unexpected chemical skeletons. Specifically, the biooxidation of diphenylamine resulted in an oxygenated quinone-like product, while, to our surprise, in the presence of T. versicolor laccase (E)-4-vinyl aniline was converted into a 1,2-substited cyclobutane ring. To the best of our knowledge, this is the first example of an enzymatically triggered [2 + 2] olefin cycloaddition. Possible reaction mechanisms to explain the formation of these products are also reported.
1. Introduction
Laccases, blue copper oxidases [1], are enzymes whose natural function(s) are related to their ability to catalyze polymerization and depolymerization processes. As examples, fungal laccases are involved in lignin degradation, while in plants they are key players in lignification processes and cell wall formation [2].
Generally speaking, laccases are regarded as “green tools” enabling the employment of biocatalyzed processes in different fields of industrial interest [3,4]. While laccases’ main (bio)technological applications are usually found in the textile, pulp, paper, and food industries [1,3,5], according also to the large number of patents filed during the last years [6], the versatility and the surprisingly wide substrate scope of these enzymes makes them appealing for the synthesis of fine chemical and for the development of novel and green organic transformations, as we recently reviewed [7].
Radical chemistry is a trivial synthetic tool that, under the proper choice of reactants and reaction media engineering, allows to obtain complex molecular skeletons in simple one-pot processes. In this context, laccase–catalysis represents a convenient activation protocol of a normally inert Csp2-H bond, requiring only molecular oxygen and ad hoc designed aromatic substrates. Specifically, the laccase-mediated generation of reactive radical intermediates can be efficiently exploited to build domino, cascade, and/or one-pot ring closure processes to be applied to the preparation of heterocyclic compounds. It is noteworthy that no toxic, hazardous, and expensive metal-based chemical catalysts are needed to perform these biocatalytic processes.
Multi-step sequences composed by, e.g., a series of (pseudo)quinones formations, nucleophilic aromatic substitutions, and C-C and C-N radical couplings can be merged to synthesize nitrogenous heterocycles starting from aromatic amines. It has been shown that these transformations result in formal oxidative homo- and/or hetero-couplings involving two molecules of the same substrate or of two different partners [8]. Scheme 1 summarizes some of the results obtained by Sousa et al. in the biocatalytic oxidation of differently substituted anilines using the bacterial CotA-laccase (spore coat protein A, CotA) from Bacillus subtilis as the biocatalyst for the facile synthesis of phenazines and phenoxazinones [9,10,11]. These elegant biocatalytic syntheses could represent a convenient entry to analogues of bioactive phenazines [12,13,14].
Among the different species of laccases, the enzyme from T. versicolor, a common polypore mushroom, has been widely exploited and characterized as a biocatalyst for the development of synthetic processes based on radical chemistry and differently substituted natural or ad hoc designed phenol substrates [7,15,16]. As for most of these enzymes, the catalytic cycle of T. versicolor laccase involves the mono-electronic oxidation of four equivalent of an organic (reducing) substrate forming radicals at the expense of molecular oxygen, which is eventually reduced to two molecules of water. The core of the catalytic machinery is represented by a four-membered copper cluster, which is the site of oxygen coordination and reduction, water formation and release, as well as of substrate oxidation [1,3,17].
Besides being largely exploited in the (bio)technological applications mentioned above, T. versicolor laccase has been extensively employed in the development of green and convenient processes to afford oxygen-containing heterocyclic compounds starting from the formal oxidative homocoupling of differently substituted phenols or ad hoc designed phenolic synthetic derivatives [18,19,20]. These biocatalyzed multi-step, one-pot ring closing reactions are usually guided by the structural features of the reacted substrates that can control the profile of the molecular skeletons obtained. When novel stereocenters are formed, no control of their absolute configuration is achieved, while steric hindrance and thermodynamics drive their relative configuration.
Specifically, when vinyl phenols and stilbenoids, molecules structurally related to the laccases’ natural substrates mono-lignols, are reacted in the presence of T. versicolor laccase, three different groups of oxygenated heterocycles can generally be obtained, as trans-racemate, as described in Scheme 2: in the presence of an allylic alcohol (R = OH) and of an alkyl substituent such as R’ bicyclic hexahydrofuro [3,4-c]furans, the core of the natural product pinoresinol [21,22,23,24], are preferentially formed (a) while benzodioxanes (b) and 2,3-dihydrobenzofurans (2,3-DHBs) (c) are obtained starting from vinyl catechols and phenols, respectively.
As we extensively reported [25,26,27,28,29,30,31,32,33], 2,3-DHBs are obtained as the main products when the designed substrate (Scheme 2) is characterized by an R1 ‘spectator group’ (i.e., hydrogen, alkyl chains, substituted phenols) and by an R substituent that is either an alkyl or an aryl group. From a mechanistic point of view (Figure 1), the ring closure occurs via a sequence of phenol oxidation, C-C/O radical coupling, and 1,4-conjugate addition forming two novel stereocenters. In particular, we exploited T. versicolor laccases for the convenient chemo-enzymatic preparation of libraries of 2,3-DHB-based bioactive compounds or for the one-pot, selective manipulation of valuable natural compounds [26,31,34].
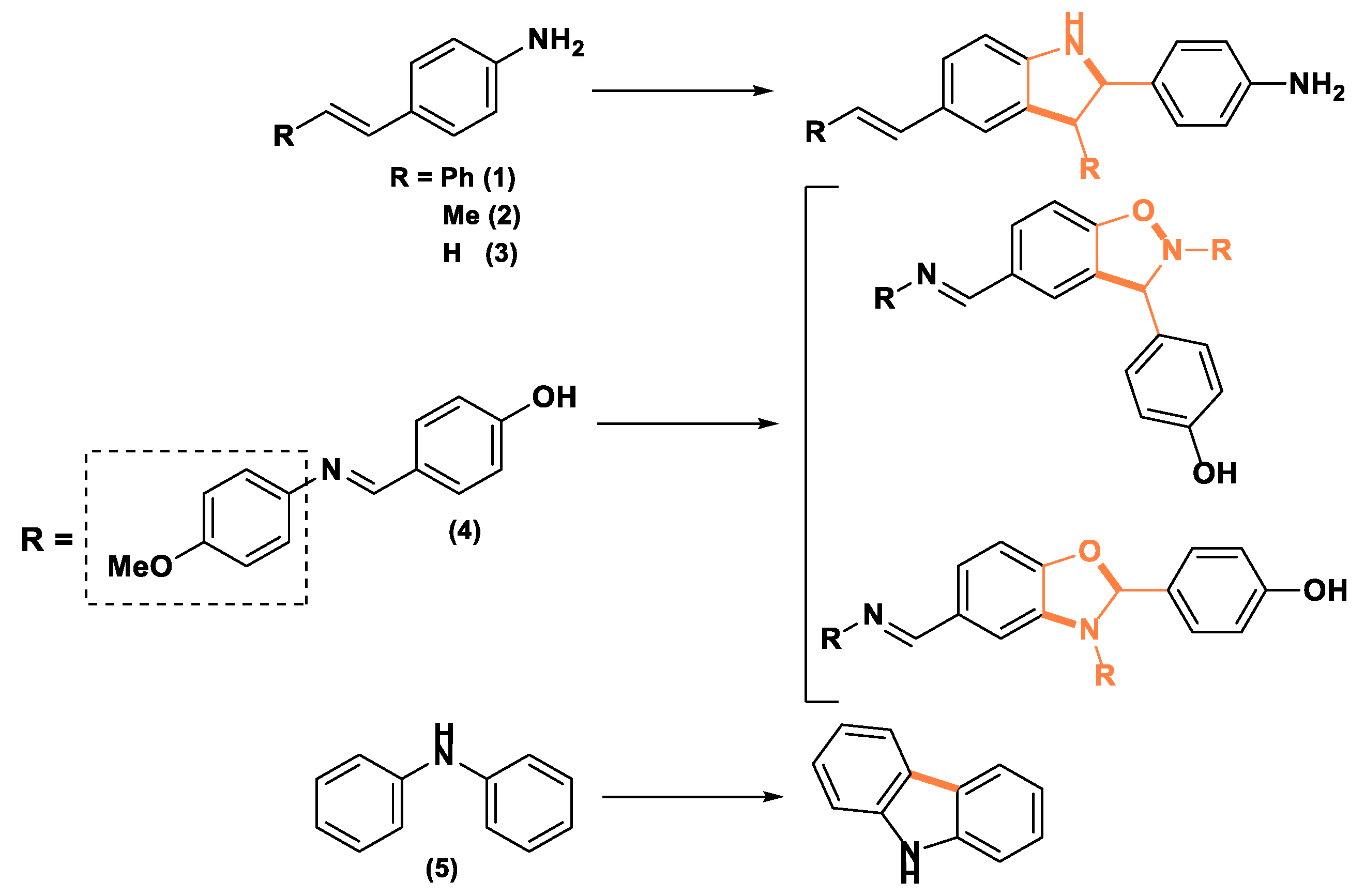
As we previously discussed, while the use of laccases of different origins has been reported for the synthesis of phenazine-like compounds or generic organic dyes possessing the structure of Bandrowski’s base-like trimers (Scheme 1), [9,10,11,35] to the best of our knowledge, laccase-catalysis has not been exploited yet for oxidation of the corresponding vinyl or styryl anilines. Thus, we decided that it was worthy investigating whether T. versicolor laccase catalysis could be exploited to oxidize aromatic amines or imines to build indoline, oxazole/isoxazole, or carbazole skeletons, as described in Scheme 3, using the model substates 1–5, respectively. In the following, we report and discuss the obtained results.
2. Results
2.1. Synthesis of Substrates
Substrates 1, 2, and 4 are not commercially available and therefore they were chemically synthetized for the purpose of this study (Scheme 4).
The styryl aniline 1 was obtained via a sequence of Knoevenagel condensation, involving p-nitro benzoic acid and benzaldehyde working in the presence of piperidine as a base, and the reduction of this nitro group mediated by metallic zinc.
The E-configured methyl vinyl aniline 2 was similarly obtained from the Zn-promoted reduction of the corresponding nitro-derivative which, in turn, was prepared from the decarboxylation of the commercially available (E)-3-(4-nitrophenyl)acrylic acid in the presence of FeCl3.
Finally, imine 3 was easily afforded by the condensation of p-hydroxy benzaldehyde and p-methoxy aniline in pure MeOH at room temperature.
2.2. Laccases-Mediated Biooxidations
2.2.1. Substrate 1, 2, and 4
The biocatalyzed oxidations of aryl and alkyl substituted vinyl anilines 1 and 2 and of imine 4 was explored via a series of small-scale reactions monitored by TLC analysis.
Standard oxidation conditions for T. versicolor laccase catalysis (acid pH, 27 °C) were applied along with modified protocols in which the pH was moved up to neutral (7.0) and slightly basic (up to 8.0) values. In all the cases (data not shown), starting materials and/or a complex mixture of by-products were isolated.
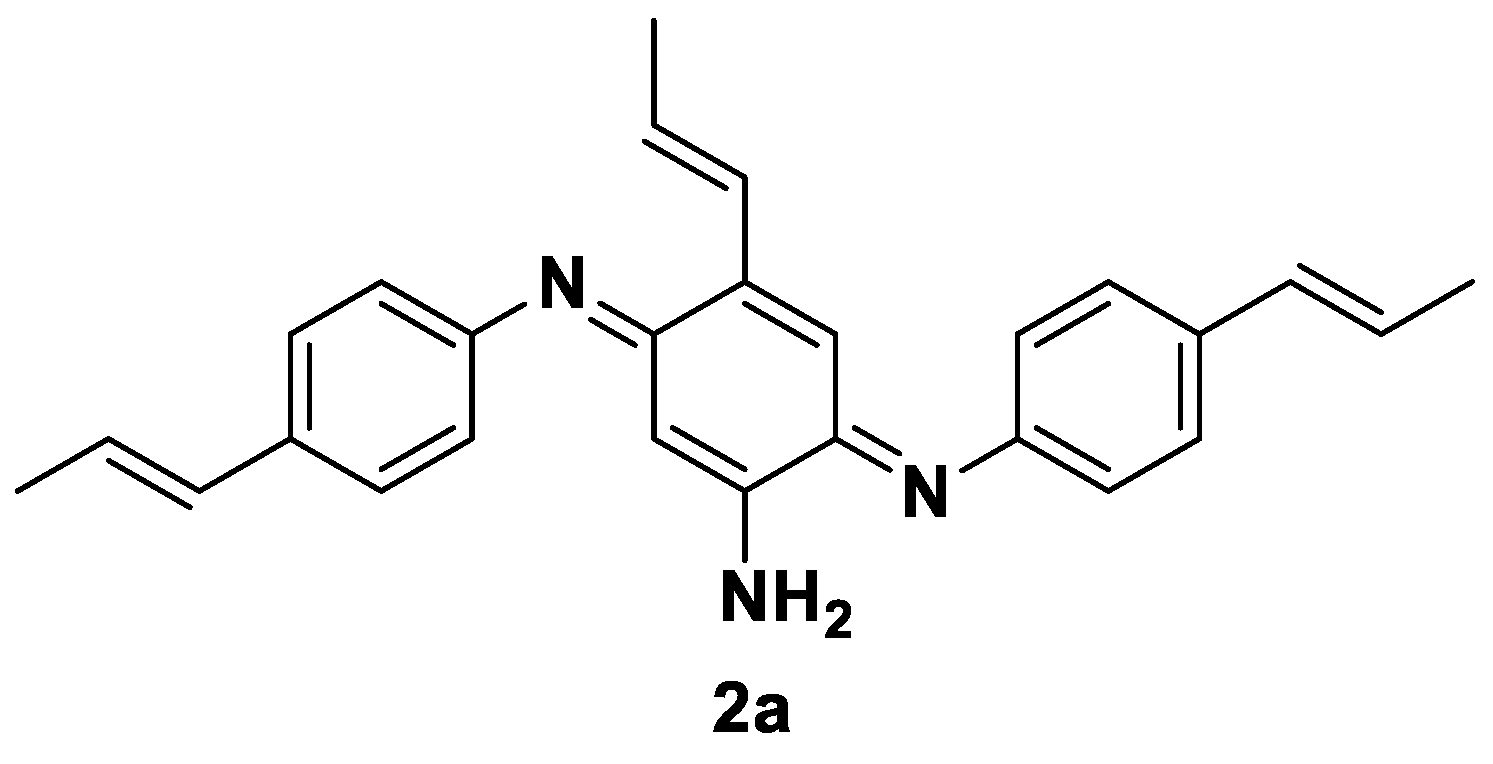
Only in the case of substrate 2, traces of a trimeric structure already reported for the laccase mediated bio-oxidation of similar aryl amines [35,36] were detected via mass spectroscopy, but it was not possible to properly isolated it. According to the literature references, the structure reported in Figure 2 might be hypothesized.
In conclusion, to our disappointment, no evidence of discrete dimeric products could be observed with these three substrates.
2.2.2. Substrate 3
The bio-oxidation of compound 3 was at first investigated by selecting the proper reaction medium and then optimized in terms of enzyme loading and general parameters as reaction time or temperature. Control reactions were also performed to exclude the presence of spontaneous oxidation in the investigated reaction media.
The optimized conditions for this biotransformation were identified as adding portion wise T. versicolor laccase, 36 U per mmol of substrate, to an acetate buffer solution of vinyl aniline.
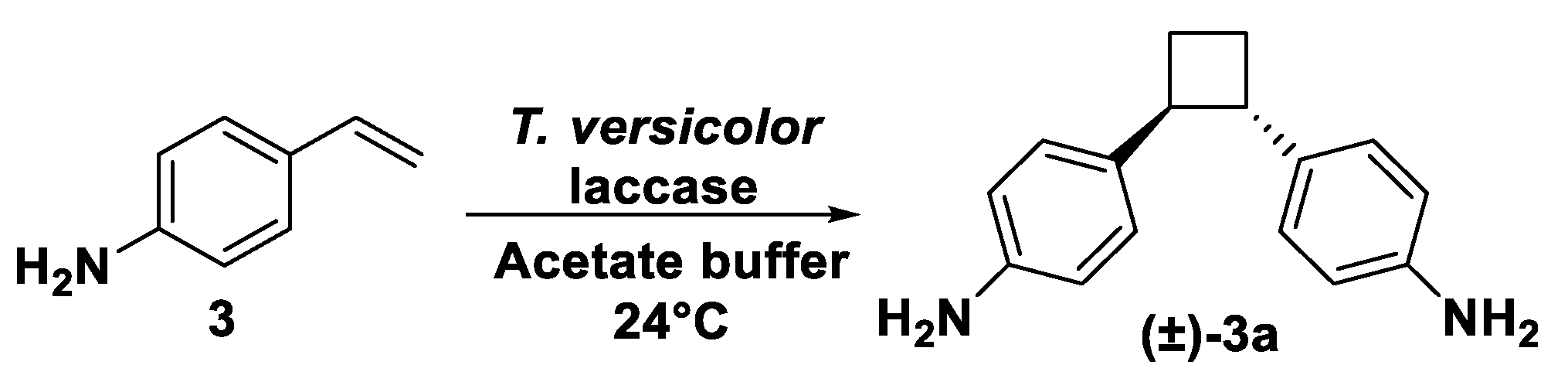
The reaction, shaken at 24 °C, was followed using TLC attesting the disappearing of the starting material and the formation of a main, more polar compound (Rf = 0.25 in petroleum ether–ethyl acetate = 7:3), which was purified by flash column chromatography on silica gel (FC). Product 3a was then characterized by means of NMR spectroscopy and mass spectrometry and was identified as a dimeric structure corresponding to a 4,4′-(cyclobutane-1,2-diyl)dianiline skeleton obtained as a putative trans-isomer (as a racemate) in 40% of isolated yield (3a, Scheme 5).
2.2.3. Substrate 5
As described for 3, a small-preparative scale bio-oxidation of substrate 5 was conducted after a short investigation of the proper reaction medium and enzyme loading.
Specifically, 5 was reacted with T. versicolor laccase in a 30% v/v solution of dioxane in acetate buffer in the presence of 1.5 U mL−1 of biocatalyst. The reaction was incubated for 24 h at 27 °C and followed by TLC analysis until complete disappearance of the starting material. A more polar compound (Rf = 0.34 in petroleum ether–ethyl acetate = 9:1) was formed as a main product along with a complex mixture of apparently indistinguishable spots. The main product was isolated by means of FC and fully characterized via NMR spectroscopy and mass spectrometry. Based on these analyses, the structure of the obtained chemical species (5a, Scheme 6) could be assigned to a 4-(phenylimino)cyclohexa-2,5-dien-1-one skeleton obtained in a 10% of isolated yield.
3. Discussion
As it has been described in the introduction, the CotA-laccase-catalyzed dimerization of substituted anilines to give phenazines is a well-assessed methodology. Moreover, the enzymatic oligomerization and polymerization of anilines is well-documented in the literature, as it has been recently reviewed [36]. However, the structural characterization of dimers and trimers formed during the early stages of the biocatalyzed oxidative polymerization of these compounds is missing in most cases.
In an attempt to tackle this problem, at least with specific aniline derivatives, this research was focused on the π-conjugated vinyl anilines 1–3, with the aim of verifying whether these aromatic amines behaved like their oxygenated cognates, forming indole skeletons under laccase oxidation [25,26,27,28,29,30,31,32,33]. Moreover, we also considered compounds 4 (an easily synthesized imine) and the aromatic secondary amine 5 as possible precursors of cyclic nitrogen-containing heterocycles, respectively of oxazole/isoxazole or carbazole skeletons (Scheme 3). Unfortunately, our approach failed to give the hypothesized results. However, in two cases it was possible to isolate products with defined and unexpected chemical structures.
As described in the previous paragraph, the bio-oxidation of 2 gave only traces of the trimeric compound 2a, previously reported in the literature [36], as attested by the mass spectrum reported in Figure S1 (M* = 293.2 Da).
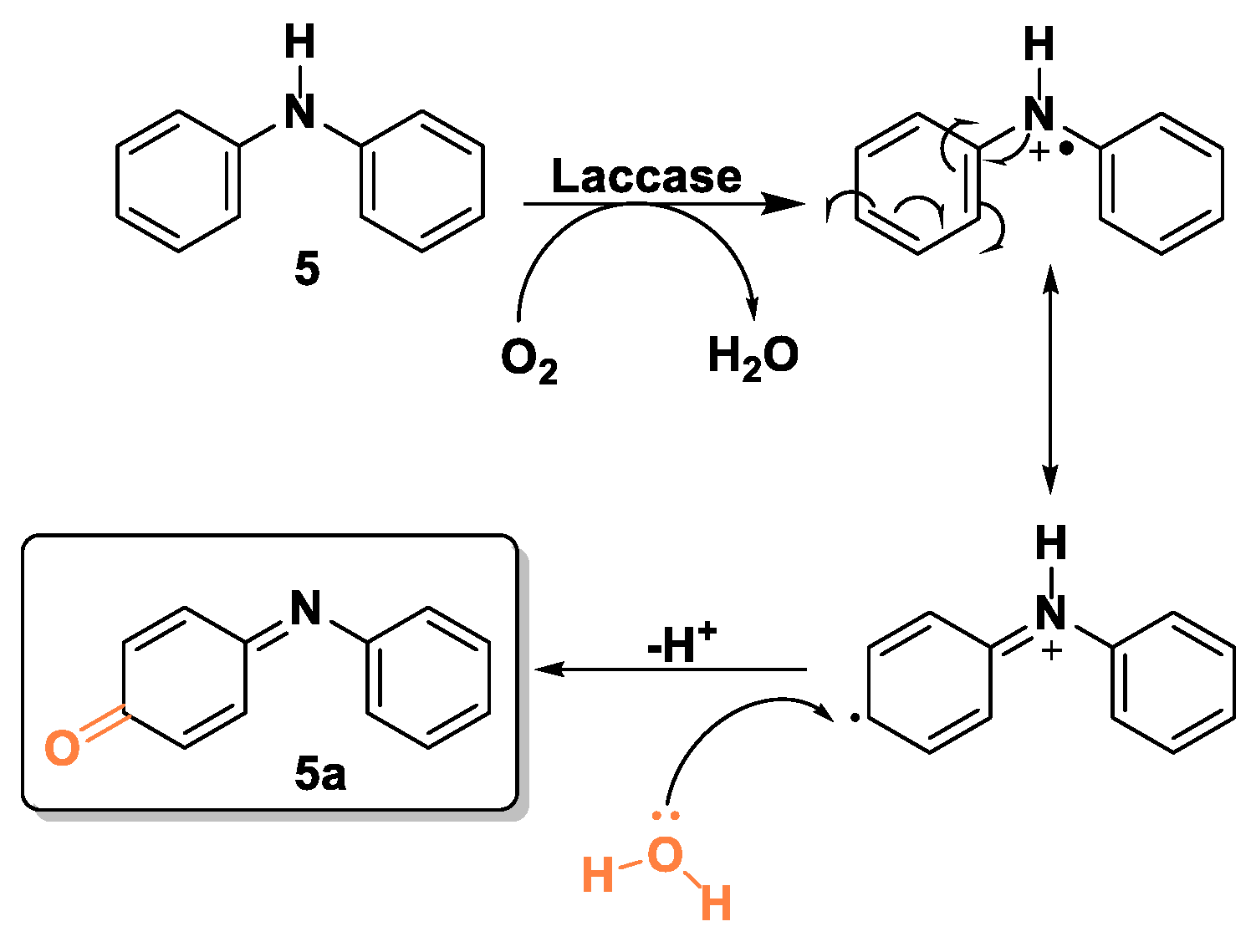
The elucidation of the structure of compound 5a, obtained by the laccase-catalyzed oxidation of diphenyl amine 5, was not straightforward. The mass spectrum (Figure S2), with a molecular value of 184.1 Da, suggested the presence of an oxygenated substituent. A careful inspection of the signals of the 1H NMR spectrum and the exploitation of bidimensional techniques (Figure S2) allowed to identify the structure 5a, formed according to the following hypothesized mechanism (Scheme 7).
Even more puzzling was the characterization of the main product obtained by the laccase-catalyzed oxidation of 4-vinyl aniline (3), isolated in 40% yield. Its exact molecular mass at 239.15410 Da was compatible with a dimeric structure with a brute formula C16H10N2. Its 1H NMR spectrum, far from being trivial (Figure S3), was deeply investigated and, besides aromatic systems, no signs of the original Csp2-H olefinic protons could be identified. In place of those signals, a multiplet centered at 3.36 ppm could be identified. Given the nature of the reacted substrate, the molecular mass of the product obtained, and the identification of novel signals in the region of Csp3-H protons, we hypothesized that 3a could be a disubstituted cyclobutane skeleton. To identify the obtained regioisomer, we carefully looked in the literature for reports of 1H-NMR characterization of 1,2 and 1,3-substituted cyclobutane rings. The 1H spin system characterizing this family of cyclobutane rings can be described as AA’BB’XX’, where X and X’ are the isochronous methine nuclei. In these systems, the width at half height of the signal of the XX’ nuclei depends essentially on the sum of the vicinal coupling constants (J), e.g., J(AX) and J(BX) for X, since the J across the four bonds are small (about 1 Hz) in four membered rings. In cyclobutanes, vicinal J are usually found between 7 and 10 Hz. Thus, the width of the XX’ signal of a 1,2-disubstituted cyclobutane should measure 15–20 Hz (only one adjacent methylene) while 1,3-disubstituted cyclobutanes could reach values greater than 30 Hz (two adjacent methylenes). These considerations allowed us to identify 3a as a 1,2-disubstituted cyclobutane as the width of the signal at 3.36 ppm (corresponding to XX’ nuclei) was found to be 17.0 Hz, in perfect agreement with that reported by Raza et al. in the 1H NMR characterization of diphenyl cyclobutanes [37]. As far as the relative stereochemistry of 3a concerns, we hypothesize a trans-configured cyclobutane due to the similarity of the fine structure of the NMR spectrum with that of the 1,2-trans-diphenylcyclobutane [37] and the fact that generally laccases-mediated dimerization reactions are driven by thermodynamic factors which generally lead to the formation of the more stable and less hindered trans-systems [7].
This structure was quite an unexpected molecular skeleton to be formed under laccase catalysis. As a possible rationale, we propose the following mechanism (Scheme 8) for the formation of 3a, which relies on the well-documented [2 + 2] photochemical cycloadditions of olefins [38].
The isolation of 3a represents, to the best our knowledge, the first example of a biocatalytic [2 + 2] cycloaddition of olefins. In this reaction, T. versicolor laccase is expected to act as an initiator by activating the fully conjugated π-system of vinyl aniline 3 to the radical cation 3*+, an intermediate reported as pivotal in this kind of cyclization reactions [39,40,41].
Interestingly, the same cyclobutane dimeric structures were not isolated with the vinyl anilines 1 and 2. It could be possible to argue that steric hindrance or ring-tension by the additional presence of phenyl and methyl substituents in the position C-2 and C-4 could have prevented the cycloaddition reaction. However, several reports can be found in the literature dealing with the preparation of tri- and tetra-substituted cyclobutanes exploiting protocols of metal-catalyzed [2 + 2] olefin photocycloadditions [38,42,43,44]. For all these reasons, a detailed investigation of this laccase-mediated process will be conducted by us, using different fully conjugated π-systems as substrates.
4. Materials and Methods
All reagents were of the highest purity grade from commercial suppliers: Merck (St Louis, MO, USA) or VWR (Radnor, PA, USA).
Laccase from Trametes versicolor was from Sigma-Aldrich. The enzyme was used based on their respective activities evaluated according to literature assay based on the ABTS (2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid)) as model substrate. [32]
Biotransformations were performed in a G24 Environmental Incubator New Brunswick Scientific Shaker (Edison, USA) or in a Thermomixer Comfort (Eppendorf, DE).
Reactions were monitored by thin-layer chromatography (TLC) (precoated silica gel 60 F254 plates (Merck, DE)); development with UV lamp, Komarovsky reagent (1 mL 50% ethanolic H2SO4 with 10 mL 2% methanolic 4-hydroxybenzaldehyde), a 20% solution of H2SO4 in ethanol or a molybdate reagent ((NH4)6Mo7O24·4H2O, 42 g; Ce(SO4)2, 2 g; H2SO4 conc., 62 mL; made up to 1 L of deionized water). Flash chromatography: silica gel 60 (70–230 mesh, Merck, DE).
NMR spectra were recorded with a Bruker AC spectrometer (400 or 500 MHz) in [D4]MeOH, [D6]DMSO or [D1]CHCl3. Mass spectra were recorded with a Bruker Esquire 3000 Plus spectrometer.
High-resolution mass spectra (HRMS) were conducted on FT-Orbitrap mass spectrometer in positive electrospray ionization (ESI).
4.1. Synthesis of Dubstrates
4.1.1. (E)-1-nitro-4-styrylbenzene
As we previously reported for the synthesis of this compound [31], a solution of benzaldehyde (100 mg, 0.9 mmol, 1 eq) and 4-nitrophenylacetic acid (0.51 g, 2.8 mmol, 3 eq) was prepared with 15 mL of CH2Cl2 at r.t. Piperidine (280 mL, 2.8 mmol, 3 eq) was added and the resulting mixture was gradually heated to 130 °C, distilling the solvent. The resulting neat mixture was left reacting at 130 °C for 24–48 h. After that, the crude residue was analyzed by TLC (CHCl3-acetone = 95:5) and purified by flash column chromatography (petroleum ether-EtOAc = 96:4), obtaining the desired (E)-stilbene as a yellow solid (190 mg, 89% yield). 1H-NMR (400 MHz; [D4]MeOH, r.t.): δ 8.22–8.19 (AA’BB’ system, 2H), 7.74–7.71 (AA’BB’ system, 2H), 7.49–7.47 (AA’BB’ system, 2H), 7.34 (d, J 16.4 Hz, 1H), 7.10 (d, J 16.4 Hz, 1H), 6.84–6.81 (AA’BB’ system, 2H); 13C-NMR (101 MHz; [D4]MeOH, r.t.): δ 146.9, 143–9, 136.3, 133.4, 129.0, 129.9, 127.1, 126.9, 126.4, 124.2. MS, m/z ESI = 264.0 [M + Na]+.
4.1.2. (E)-4-styrylaniline (Substrate 1)
Zinc power (72 mg, 1.1 mmol, 5 eq) and ammonium chloride (60 mg, 1.1 mmol, 5 eq) were added to a stirred solution of p-nitrostilbene (50 mg, 0.2 mmol, 1 eq) in EtOH (0.52 mL). The resulting reaction mixture was heated to 90 °C for 6 h, monitored by TLC (petroleum ether–EtOAc = 9: 1). The reaction mixture was cooled to r.t., filtered through a celite® pad, and volatiles were evaporated under reduced pressure. The crude residue was diluted with EtOAc (20 mL) and washed with a solution of NaHCO3 (3 × 10 mL) and brine (3 × 10 mL), dried over Na2SO4 and the solvent evaporated under reduced pressure to afford the desired product as a white foam (32 mg, 90% yield). 1H-NMR (400 MHz; [D4]MeOH, r.t.): δ 7.48 (AA’BB’ system, 2H), 7.22 (t, J 7.4 Hz, 1H), 7.03 (d, J 16.3, 1H), 6.93 d, J 16.3, 1H), 6.68 (d, J 8.5, 1H), 3.74 (brs, 2H). 13C-NMR (101 MHz; [D4]MeOH, r.t.): δ 146.3, 138.1, 129.0, 128.7, 127.0, 126.2, 125.2, 115.3. MS (ESI): calcd for [C14H14N]+ 196.1126, found 169.1225.
4.1.3. (E)-1-nitro-4-(prop-1-en-1-yl)benzene
A mixture of 4-nitro cinnamic acid (290 mg, 1.5 mmol, 1 eq), DTBP (2-(tert-butylperoxy)-2-methylpropane, 550 μL, 3 mmol, 2 eq), FeCl3 (81 mg, 0.3 mmol, 20 mol%), and DMSO (10 mL) was stirred in a round-bottom flask under nitrogen atmosphere at 130 °C overnight. After that, the mixture was poured into EtOAc (10 mL) and washed with water (25 mL). The aqueous phase was extracted with EtOAc (15 mL). The combined organic layers were dried over Na2SO4 and the solvent evaporated under reduced pressure. The crude product was purified by purified by flash column chromatography (petroleum ether-EtOAc = 97:3), obtaining the desired compound as a sightly yellow solid (43 mg, 17% yield). 1H-NMR (400 MHz; [D4]MeOH, r.t.): δ 8.14 (AA’BB’ system, 2H), 7.43 (AA’BB’ system, 2H), 6.46 (s, 2H), 1.94 (d, J 4.9 Hz, 3H). 13C-NMR (101 MHz; [D4]MeOH, r.t.): δ 146.5, 144.5, 131.4, 129.6, 126.3, 124.0, 18.84. MS, m/z ESI = 186.0 [M + Na]+.
4.1.4. (E)-4-(prop-1-en-1-yl)aniline (Substrate 2)
Following the same reduction protocol applied for the preparation of (1), substrate 2 was obtained as a white foam (93% isolated yield, 80 mg) starting from 106 mg of (E)-1-nitro-4-prop-l-en-1-yl)benzene. 1H-NMR (400 MHz; [D4]MeOH, r.t.): δ 7.14 (AA’BB’ system, 2H), 6.62 (AA’BB’ system, 2H), 6.62 (d, J 8.5, 1H), 6.30 (dd, J 15.7, 1.4 Hz, 1H), 6.30 (dq, J 15.7, 6.6 Hz, 1H), 3.60 (bs, 1H), 1.84 (dd, J 6.6 Hz, 1.4 Hz, 3H). 13C-NMR (101 MHz; [D4]MeOH, r.t.): δ 145.3, 130.8, 128.9, 126.9, 122.0, 115.3, 18.4. MS (ESI): calcd for [C9H12N]+ 133.0891, found 133.0892.
4.1.5. (E)-4-(((4-methoxyphenyl)imino)methyl)phenol (Substrate 4)
p-Hydroxy benzaldehyde (100 mg, 0.8 mmol, 1 eq) and p-methoxy aniline (101 mg, 0.8 mmol, 1 eq) were dissolved in pure EtOH (4.1 mL). The obtained solution was stirred under reflux for 3 h observing the formation of a yellow precipitate, which was isolated by filtration and crystallized from pure EtOH affording the desired imine (80% yield). 1H-NMR (400 MHz; [D6]DMSO, r.t.): δ 10.03 (s, 1H), 8.47 (s, 1H), 7.75 (AA’BB’ system, 2H), 7.33–7.11 (AA’BB’ system, 2H), 7.04–6.89 (AA’BB’ system, 2H), 6.87 (AA’BB’ system, 2H), 3.77 (s, 3H). 13C-NMR (101 MHz; [D4]MeOH, r.t.): δ 160.8, 160.0, 159.0, 144.4, 131.2, 130.7, 129.8, 122.4, 116.2, 116.1, 116.0, 115.6, 55.8. MS (ESI): calcd for [C14H13NO2 + 1]+ 278.0946, found 278.0944.
4.2. Biooxidations with T. versicolor Laccase
4.2.1. Oxidation of Vinyl Aniline (Substate 3)
Vinyl aniline (30 mg, 0.3 mmol) was dissolved in 1.5 mL of sodium acetate buffer (pH 5, 50 mM) and incubated with T. versicolor laccase (36 U mmol−1 substrate) overnight at 27 °C and 180 rpm in an orbital thermoshaker. After attesting the formation of a more polar UV-visible spot in TLC analysis (petroleum ether–EtOAc = 1:1), an additional aliquot of enzyme was added doubling its concentration and the mixture was left reacting for 6 h. The reaction mixture was then extracted with EtOAc and the combined organic layers were dried over Na2SO4 and concentrated in vacuo affording a crude mixture. The more polar spot (compound 3a, Rf = 0.25 in petroleum ether–EtOAc = 7:3) was isolated by means of flash column chromatography on silica gel (petroleum ether–EtOAc = 7:3 → 4:6) and fully characterized (isolated yield = 40%). 1H-NMR (400 MHz; CDCl3, r.t.): δ 7.01 (AA’BB’ system, 2H), 6.61 (AA’BB’ system, 2H), 3.42–3.34 (m, 1H), 2.25–2.19 (m, 1H), 2.06–2.02 (m, 2H). 13C-NMR (101 MHz; CDCl3, r.t.): δ 144.3, 135.1, 127.5, 119.5, 115.1, 47.9, 25.9. MS (ESI): calcd for [C16H10N2]+ 239.15428, found 239.15410.
4.2.2. Oxidation of Diphenyl Amine (Substate 5)
Diphenyl amine (100 mg, 0.6 mmol) was dissolved in 2.5 mL of dioxane and added 5.8 mL of acetate buffer (pH 5, 50 mM) containing T. versicolor laccase (1.5 U mmol−1substrate) and incubated overnight at 27 °C and 180 rpm in an orbital thermoshaker. After attesting the formation of a more polar UV-visible spot in TLC analysis (petroleum ether–EtOAc = 9:1) and the disappearance of the starting material, the reaction mixture was then extracted with EtOAc, and the combined organic layers were dried over Na2SO4 and concentrated in vacuo affording a crude mixture. The polar spot (compound 5a, Rf = 0.34 in petroleum ether–EtOAc = 9:1) was isolated by means of flash column chromatography on silica gel (petroleum ether–EtOAc = from 95:5 to 90:10) and fully characterized (isolated yield = 10%). 1H-NMR (400 MHz; CDCl3, r.t.): δ 7.41 (t, J 7.8 Hz, 1H), 7.31 (dd, J 10.0, 2.6 Hz, 1H), 7.27–7.22 (m, 1H), 7.09 (dd, J 10.3, 2.6 Hz, 1H); 6.89 (d, J 10.1 Hz, 1H), 6.70 (dd, J 10.1, 2.1 Hz, 1H), 6.54 (dd, J 10.3, 2.1 Hz, 1H). 13C-NMR (101 MHz; CDCl3, r.t.): δ 187.7, 157.5, 149.6, 142.0, 133.7, 133.0, 192.2, 128.4, 126.6, 120.8. MS (ESI): calcd for [C12H10NO]+ 184.0762, found 184.0764.
5. Conclusions
The laccase-catalyzed oxidation of a series of aromatic amines has been described. At variance of their phenolic equivalents, it was not possible to isolate the expected nitrogenous cyclic dimeric structures and the oxidation proceeded to give complex oligomeric and polymeric products.
Interestingly, while diphenyl amine gave an unexpected but trivial oxidized product, the laccase-mediated oxidation of vinyl aniline resulted in the isolation a 1,2-trans-disubstituted cyclobutane, possibly via a radical-cationic [2 + 2] olefin cycloaddition. This unexpected but highly valuable result represents the first biocatalyzed example of this reaction, which is presently a hot topic in photo and organometallic catalysis, based on the number of reports published just in the last five years. While this work concludes our long journey on the laccase-catalyzed oxidation of vinyl derivatives to give heterocyclic compounds, at the same time it opens a novel investigation on this laccase-mediated annulation reaction.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms24043524/s1.
Author Contributions
Conceptualization, I.B. and S.R.; methodology: I.B. and S.R, formal analysis, I.B., S.R., and G.F.; investigation, I.B., S.G., and C.T.; data curation, I.B, S.R., and G.F.; writing—original draft preparation, I.B.; writing—review and editing, I.B., S.R., and G.F.; supervision, I.B. and S.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Riva, S. Laccases: Blue Enzymes for Green Chemistry. Trends Biotechnol. 2006, 24, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Mogharabi, M.; Faramarzi, M.A. Laccase and Laccase-Mediated Systems in the Synthesis of Organic Compounds. Adv. Synth. Catal. 2014, 356, 897–927. [Google Scholar] [CrossRef]
- Pezzella, C.; Guarino, L.; Piscitelli, A. How to Enjoy Laccases. Cell. Mol. Life Sci. 2015, 72, 923–940. [Google Scholar] [CrossRef] [PubMed]
- Couto, S.R.; Herrera, J.L.T. Industrial and Biotechnological Applications of Laccases: A Review. Biotechnol. Adv. 2006, 24, 500–513. [Google Scholar] [CrossRef]
- Kunamneni, A.; Plou, F.J.; Ballesteros, A.; Alcalde, M. Laccases and Their Applications: A Patent Review. Recent Pat. Biotechnol. 2008, 2, 10–24. [Google Scholar] [CrossRef]
- Zerva, A.; Simić, S.; Topakas, E.; Nikodinovic-Runic, J. Applications of Microbial Laccases: Patent Review of the Past Decade (2009–2019). Catalysts 2019, 9, 1023. [Google Scholar] [CrossRef]
- Bassanini, I.; Ferrandi, E.E.; Riva, S.; Monti, D. Biocatalysis with Laccases: An Updated Overview. Catalysts 2021, 11, 26. [Google Scholar] [CrossRef]
- Hahn, V.; Davids, T.; Lalk, M.; Schauer, F.; Mikolasch, A. Enzymatic Cyclizations Using Laccases: Multiple Bond Formation between Dihydroxybenzoic Acid Derivatives and Aromatic Amines. Green Chem. 2010, 12, 879. [Google Scholar] [CrossRef]
- Sousa, A.C.; Oliveira, M.C.; Martins, L.O.; Robalo, M.P. Towards the Rational Biosynthesis of Substituted Phenazines and Phenoxazinones by Laccases. Green Chem. 2014, 16, 4127. [Google Scholar] [CrossRef]
- Sousa, A.C.; Piedade, M.F.M.M.; Martins, L.O.; Robalo, M.P. An Enzymatic Route to a Benzocarbazole Framework Using Bacterial CotA Laccase. Green Chem. 2015, 17, 1429–1433. [Google Scholar] [CrossRef]
- Sousa, A.C.; Oliveira, M.C.; Martins, L.O.; Robalo, M.P. A Sustainable Synthesis of Asymmetric Phenazines and Phenoxazinones Mediated by CotA-Laccase. Adv. Synth. Catal. 2018, 360, 575–583. [Google Scholar] [CrossRef]
- Bassanini, I.; Parapini, S.; Basilico, N.; Sparatore, A. Novel Hydrophilic Riminophenazines as Potent Antiprotozoal Agents. ChemMedChem 2019, 14, 1940–1949. [Google Scholar] [CrossRef]
- Koval, A.; Bassanini, I.; Xu, J.; Tonelli, M.; Boido, V.; Sparatore, F.; Amant, F.; Annibali, D.; Leucci, E.; Sparatore, A.; et al. Optimization of the Clofazimine Structure Leads to a Highly Water-Soluble C3-Aminopyridinyl Riminophenazine Endowed with Improved Anti-Wnt and Anti-Cancer Activity in Vitro and in Vivo. Eur. J. Med. Chem. 2021, 222, 113562. [Google Scholar] [CrossRef]
- Barteselli, A.; Casagrande, M.; Basilico, N.; Parapini, S.; Rusconi, C.M.; Tonelli, M.; Boido, V.; Taramelli, D.; Sparatore, F.; Sparatore, A. Clofazimine Analogs with Antileishmanial and Antiplasmodial Activity. Bioorg. Med. Chem. 2015, 23, 55–65. [Google Scholar] [CrossRef]
- Bassanini, I.; Kapešová, J.; Petrásková, L.; Pelantová, H.; Markošová, K.; Rebroš, M.; Valentová, K.; Kotik, M.; Káňová, K.; Bojarová, P.; et al. Glycosidase-Catalyzed Synthesis of Glycosyl Esters and Phenolic Glycosides of Aromatic Acids. Adv. Synth. Catal. 2019, 361, adsc.201900259. [Google Scholar] [CrossRef]
- Bassanini, I.; Krejzova, J.; Panzeri, W.; Monti, D.; Kren, V.; Riva, S. A Sustainable One-Pot Two-Enzymes Synthesis of Naturally Occurring Arylalkyl Glucosides. ChemSusChem 2017, 10, 2040–2045. [Google Scholar] [CrossRef]
- Quintanar, L.; Stoj, C.; Taylor, A.B.; Hart, P.J.; Kosman, D.J.; Solomon, E.I. Shall We Dance? How a Multicopper Oxidase Chooses Its Electron Transfer Partner. Acc. Chem. Res. 2007, 40, 445–452. [Google Scholar] [CrossRef]
- Constantin, M.-A.; Conrad, J.; Beifuss, U. Laccase-Catalyzed Oxidative Phenolic Coupling of Vanillidene Derivatives. Green Chem. 2012, 14, 2375–2379. [Google Scholar] [CrossRef]
- Ricklefs, E.; Girhard, M.; Urlacher, V.B. Three-Steps in One-Pot: Whole-Cell Biocatalytic Synthesis of Enantiopure (+)- and (−)-Pinoresinol via Kinetic Resolution. Microb. Cell Factories 2016, 15, 78. [Google Scholar] [CrossRef]
- Navarra, C.; Goodwin, C.; Burton, S.; Danieli, B.; Riva, S. Laccase-Mediated Oxidation of Phenolic Derivatives. J. Mol. Catal. B Enzym. 2010, 65, 52–57. [Google Scholar] [CrossRef]
- Wan, Y.; Lu, R.; Akiyama, K.; Miyakoshi, T.; Du, Y. Enzymatic Synthesis of Bioactive Compounds by Rhus Laccase from Chinese Rhus Vernicifera. Sci. China Ser. B Chem. 2007, 50, 179–182. [Google Scholar] [CrossRef]
- Ricklefs, E.; Girhard, M.; Koschorreck, K.; Smit, M.S.; Urlacher, V.B. Two-Step One-Pot Synthesis of Pinoresinol from Eugenol in an Enzymatic Cascade. ChemCatChem 2015, 7, 1857–1864. [Google Scholar] [CrossRef]
- Pickel, B.; Constantin, M.A.; Pfannstiel, J.; Conrad, J.; Beifuss, U.; Schaller, A. An Enantiocomplementary Dirigent Protein for the Enantioselective Lacease-Catalyzed Oxidative Coupling of Phenols. Angew Chem. Int. Ed. 2010, 49, 202–204. [Google Scholar] [CrossRef] [PubMed]
- Davin, L.B. Stereoselective Bimolecular Phenoxy Radical Coupling by an Auxiliary (Dirigent) Protein Without an Active Center. Science 1997, 275, 362–367. [Google Scholar] [CrossRef]
- Beneventi, E.; Conte, S.; Cramarossa, M.R.; Riva, S.; Forti, L. Chemo-Enzymatic Synthesis of New Resveratrol-Related Dimers Containing the Benzo[b]Furan Framework and Evaluation of Their Radical Scavenger Activities. Tetrahedron 2015, 71, 3052–3058. [Google Scholar] [CrossRef]
- Gavezzotti, P.; Bertacchi, F.; Fronza, G.; Křen, V.; Monti, D.; Riva, S. Laccase-Catalyzed Dimerization of Piceid, a Resveratrol Glucoside, and Its Further Enzymatic Elaboration. Adv. Synth. Catal. 2015, 357, 1831–1839. [Google Scholar] [CrossRef]
- Bassanini, I.; Gavezzotti, P.; Monti, D.; Krejzová, J.; Křen, V.; Riva, S. Laccase-Catalyzed Dimerization of Glycosylated Lignols. J. Mol. Catal. B Enzym. 2016, 134, 295–301. [Google Scholar] [CrossRef]
- Ponzoni, C.; Beneventi, E.; Cramarossa, M.R.; Raimondi, S.; Trevisi, G.; Pagnoni, U.M.; Riva, S.; Forti, L. Laccase-Catalyzed Dimerization of Hydroxystilbenes. Adv. Synth. Catal. 2007, 349, 1497–1506. [Google Scholar] [CrossRef]
- Gavezzotti, P.; Navarra, C.; Caufin, S.; Danieli, B.; Magrone, P.; Monti, D.; Riva, S. Synthesis of Enantiomerically Enriched Dimers of Vinylphenols by Tandem Action of Laccases and Lipases. Adv. Synth. Catal. 2011, 353, 2421–2430. [Google Scholar] [CrossRef]
- Grosso, S.; Radaelli, F.; Fronza, G.; Passarella, D.; Monti, D.; Riva, S. Studies on the Laccase-Catalyzed Oxidation of 4-Hydroxy-Chalcones. Adv. Synth. Catal. 2019, 361, adsc.201900190. [Google Scholar] [CrossRef]
- Bassanini, I.; D’Annessa, I.; Costa, M.; Monti, D.; Colombo, G.; Riva, S. Chemo-Enzymatic Synthesis of (E)-2,3-Diaryl-5-Styryl- Trans -2,3-Dihydrobenzofuran-Based Scaffolds and Their in Vitro and in Silico Evaluation as a Novel Sub-Family of Potential Allosteric Modulators of the 90 KDa Heat Shock Protein (Hsp90). Org. Biomol. Chem. 2018, 16, 3741–3753. [Google Scholar] [CrossRef]
- Ncanana, S.; Baratto, L.; Roncaglia, L.; Riva, S.; Burton, S.G. Laccase-Mediated Oxidation of Totarol. Adv. Synth. Catal. 2007, 349, 1507–1513. [Google Scholar] [CrossRef]
- Navarra, C.; Gavezzotti, P.; Monti, D.; Panzeri, W.; Riva, S. Biocatalyzed Synthesis of Enantiomerically Enriched β-5-like Dimer of 4-Vinylphenol. J. Mol. Catal. B Enzym. 2012, 84, 115–120. [Google Scholar] [CrossRef]
- Ficarra, S.; Tellone, E.; Pirolli, D.; Russo, A.; Barreca, D.; Galtieri, A.; Giardina, B.; Gavezzotti, P.; Riva, S.; De Rosa, M.C. Insights into the Properties of the Two Enantiomers of Trans-δ-Viniferin, a Resveratrol Derivative: Antioxidant Activity, Biochemical and Molecular Modeling Studies of Its Interactions with Hemoglobin. Mol. Biosyst. 2016, 12, 1276–1286. [Google Scholar] [CrossRef]
- Sousa, A.C.; Martins, L.O.; Robalo, M.P. Laccase-Catalysed Homocoupling of Primary Aromatic Amines towards the Biosynthesis of Dyes. Adv. Synth. Catal. 2013, 355, 2908–2917. [Google Scholar] [CrossRef]
- Ćirić-Marjanović, G.; Milojević-Rakić, M.; Janošević-Ležaić, A.; Luginbühl, S.; Walde, P. Enzymatic Oligomerization and Polymerization of Arylamines: State of the Art and Perspectives. Chem. Zvesti 2017, 71, 199–242. [Google Scholar] [CrossRef]
- Raza, G.; Bella, J.; Segre, A.; Ferrando, A.; Goffredi, G. Structures and NMR Parameters of 1,2-Diphenylcyclobutanes. Struct. Chem. 1998, 9, 419–427. [Google Scholar] [CrossRef]
- Poplata, S.; Tröster, A.; Zou, Y.-Q.; Bach, T. Recent Advances in the Synthesis of Cyclobutanes by Olefin [2 + 2] Photocycloaddition Reactions. Chem. Rev. 2016, 116, 9748–9815. [Google Scholar] [CrossRef]
- Crellin, R.A.; Lamber, C. Photochemical 2 + 2 Cycloaddition via a Cation-Radical Chain Reaction. J. Chem. Soc. D Chem. Commun. 1970, 11, 682–683. [Google Scholar] [CrossRef]
- Horibe, T.; Katagiri, K.; Ishihara, K. Radical-Cation-Induced Crossed [2 + 2] Cycloaddition of Electron-Deficient Anetholes Initiated by Iron(III) Salt. Adv. Synth. Catal. 2020, 362, 960–963. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Horiguchi, G.; Kamiya, H.; Okada, Y. Design of a Photocatalytic [2 + 2] Cycloaddition Reaction Using Redox-Tag Strategy. Chem. Eur. J. 2022, 28, e202202018. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.; Reiser, O.; Díaz, D. Effect of Reaction Media on Photosensitized [2 + 2]-Cycloaddition of Cinnamates. ChemistryOpen 2020, 9, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; López-Arteaga, R.; Weiss, E.A. Quantum Dots Photocatalyze Intermolecular [2 + 2] Cycloadditions of Aromatic Alkenes Adsorbed to Their Surfaces via van Der Waals Interactions. J. Am. Chem. Soc. 2022, 144, 3782–3786. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Wang, C.; Rogers, C.R.; Kodaimati, M.S.; Weiss, E.A. Regio- and Diastereoselective Intermolecular [2 + 2] Cycloadditions Photocatalysed by Quantum Dots. Nat. Chem. 2019, 11, 1034–1040. [Google Scholar] [CrossRef]
Scheme 1.
Mechanistic insights on phenazines formation via radical-cationic oxidation of substituted phenoxazinones (a) and phenazines (b,c) catalyzed by CotA-laccase.
Scheme 1.
Mechanistic insights on phenazines formation via radical-cationic oxidation of substituted phenoxazinones (a) and phenazines (b,c) catalyzed by CotA-laccase.

Scheme 2.
Oxygenated heterocycles synthetized via T. versicolor laccase catalysis: hexahydrofuro [3,4-c]furan (a), benzodioxane (b), and 2,3-dihydrobenzofuran (c) cores.
Scheme 2.
Oxygenated heterocycles synthetized via T. versicolor laccase catalysis: hexahydrofuro [3,4-c]furan (a), benzodioxane (b), and 2,3-dihydrobenzofuran (c) cores.
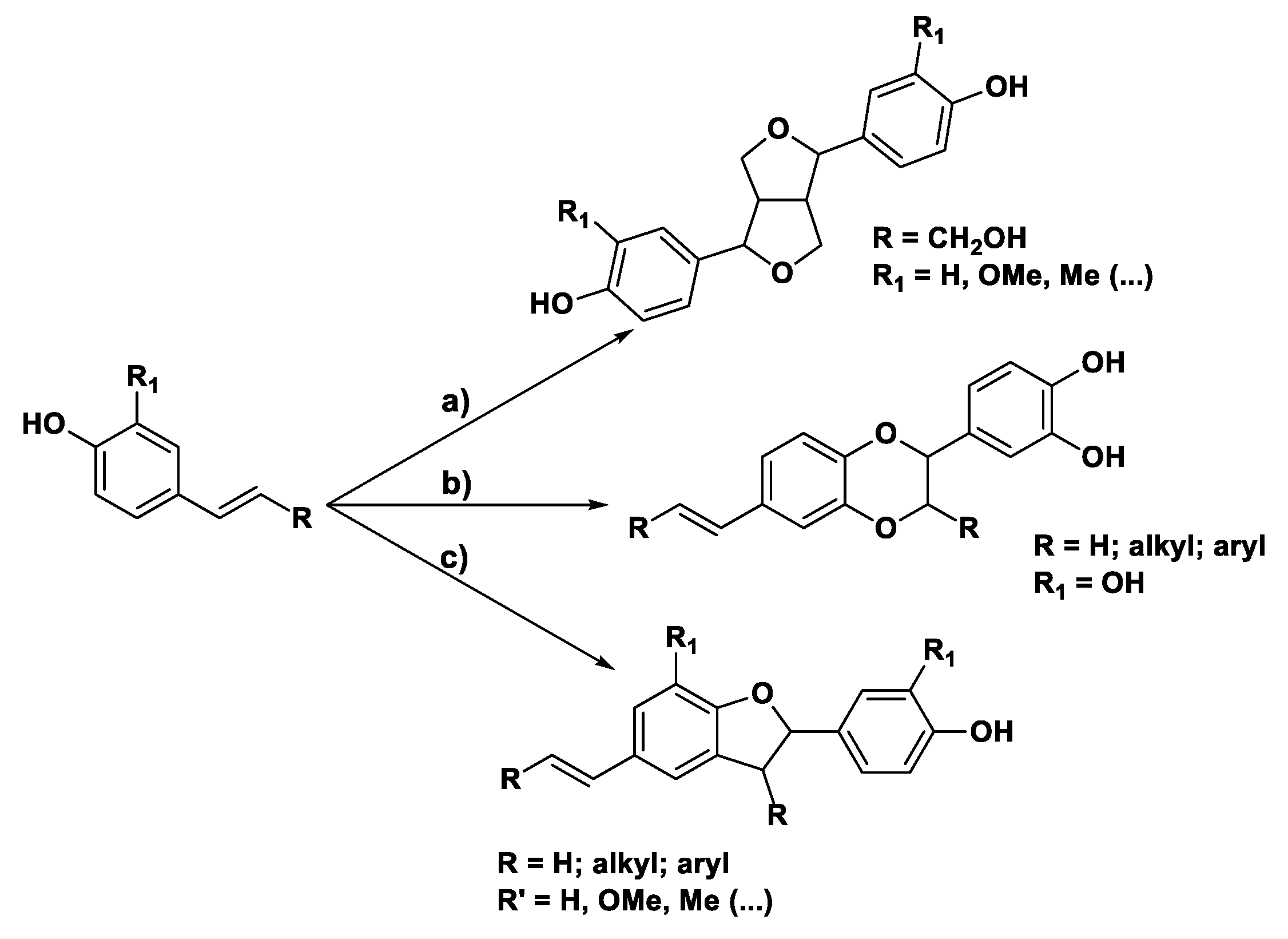
Figure 1.
T. versicolor laccase-mediated domino process to 2,3-DHBs via a formal oxidative homocoupling. Examples of preparations of bioactive compounds: (A) δ-viniferin, a natural antioxidant able to bind to human hemoglobin [34] and (B) synthetic inhibitor of Hsp90 variants potentially endowed with antiproliferative activity [31].
Figure 1.
T. versicolor laccase-mediated domino process to 2,3-DHBs via a formal oxidative homocoupling. Examples of preparations of bioactive compounds: (A) δ-viniferin, a natural antioxidant able to bind to human hemoglobin [34] and (B) synthetic inhibitor of Hsp90 variants potentially endowed with antiproliferative activity [31].

Scheme 3.
Substrates 1–5 and their likely expected laccase-catalyzed oxidation products.
Scheme 4.
Synthetic entries to substrates 1, 2 and 4.
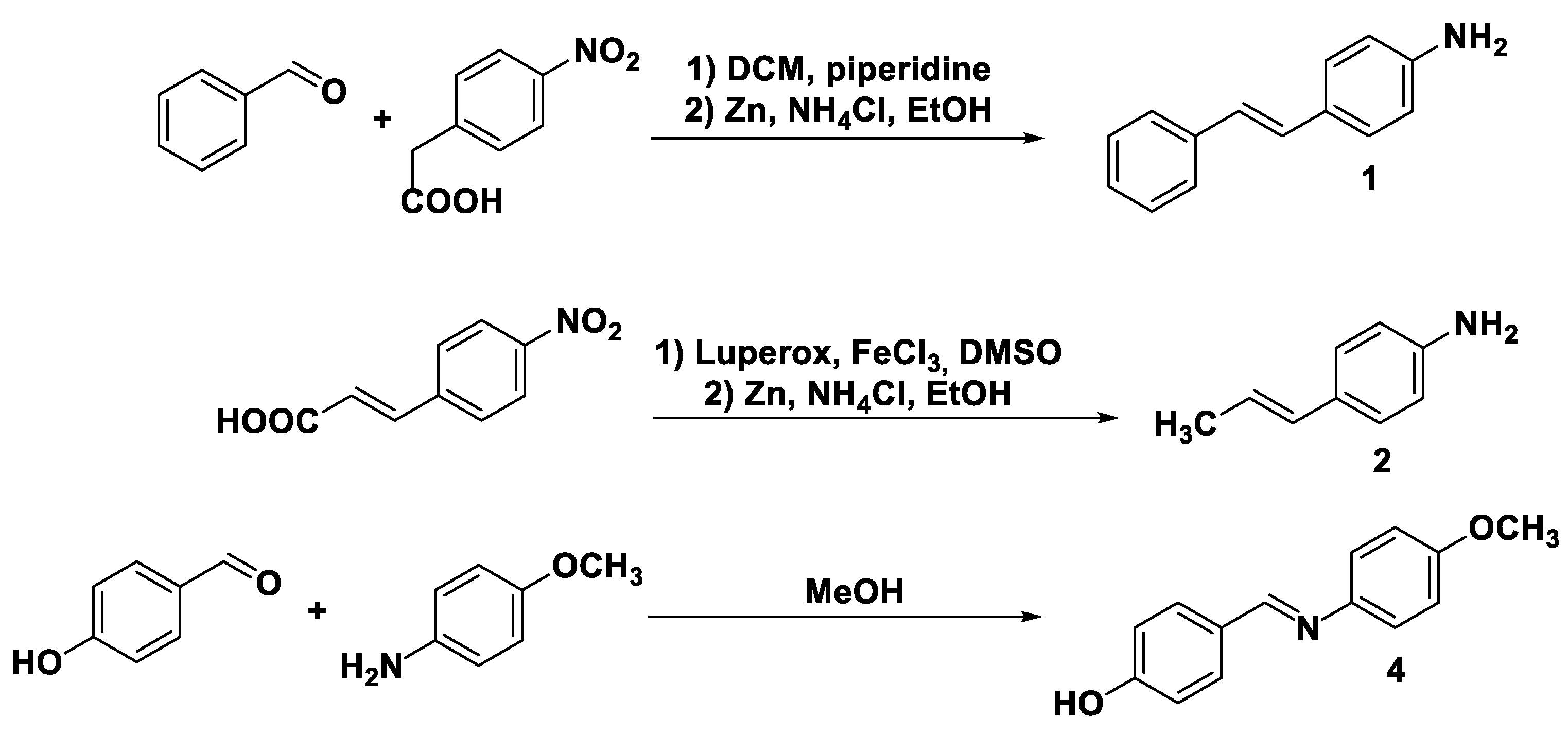
Figure 2.
Hypothesized structure of the trimeric Bandrowski’s base-like product 2a formed in traces from 2 [36].
Figure 2.
Hypothesized structure of the trimeric Bandrowski’s base-like product 2a formed in traces from 2 [36].
Scheme 5.
Reaction of vinyl aniline 3 to give 3a via T. versicolor laccase–catalysis.
Scheme 6.
Reaction of diphenyl amine 5 to give 5a via T. versicolor laccase–catalysis.
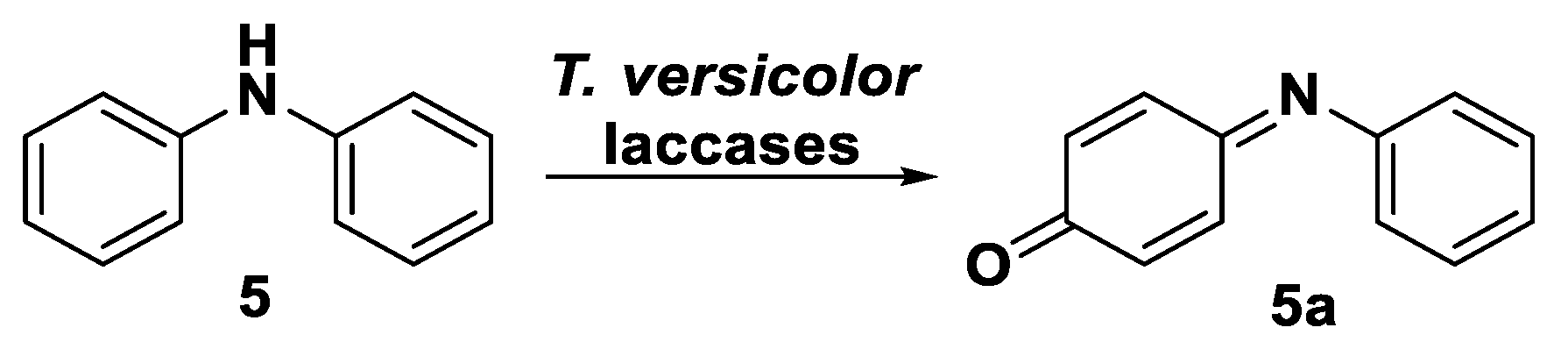
Scheme 7.
Proposed mechanism for the biooxidation of 5.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bassanini, I.; Grosso, S.; Tognoli, C.; Fronza, G.; Riva, S. Studies on the Oxidation of Aromatic Amines Catalyzed by Trametes versicolor Laccase. Int. J. Mol. Sci. 2023, 24, 3524. https://doi.org/10.3390/ijms24043524
AMA Style
Bassanini I, Grosso S, Tognoli C, Fronza G, Riva S. Studies on the Oxidation of Aromatic Amines Catalyzed by Trametes versicolor Laccase. International Journal of Molecular Sciences. 2023; 24(4):3524. https://doi.org/10.3390/ijms24043524
Chicago/Turabian StyleBassanini, Ivan, Simone Grosso, Chiara Tognoli, Giovanni Fronza, and Sergio Riva. 2023. "Studies on the Oxidation of Aromatic Amines Catalyzed by Trametes versicolor Laccase" International Journal of Molecular Sciences 24, no. 4: 3524. https://doi.org/10.3390/ijms24043524
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.