


Alzheimer’s Disease: An Updated Overview of Its Genetics
 , ,
, ,  ,
,  , , ,
, , ,  , ,
, ,  and
and 
Abstract
:1. Introduction
2. The Four Classical Genes Associated with Alzheimer’s Disease
3. Unraveling the Genetics of Alzheimer’s Disease: What Is New?

{kind=link}
{kind=link}
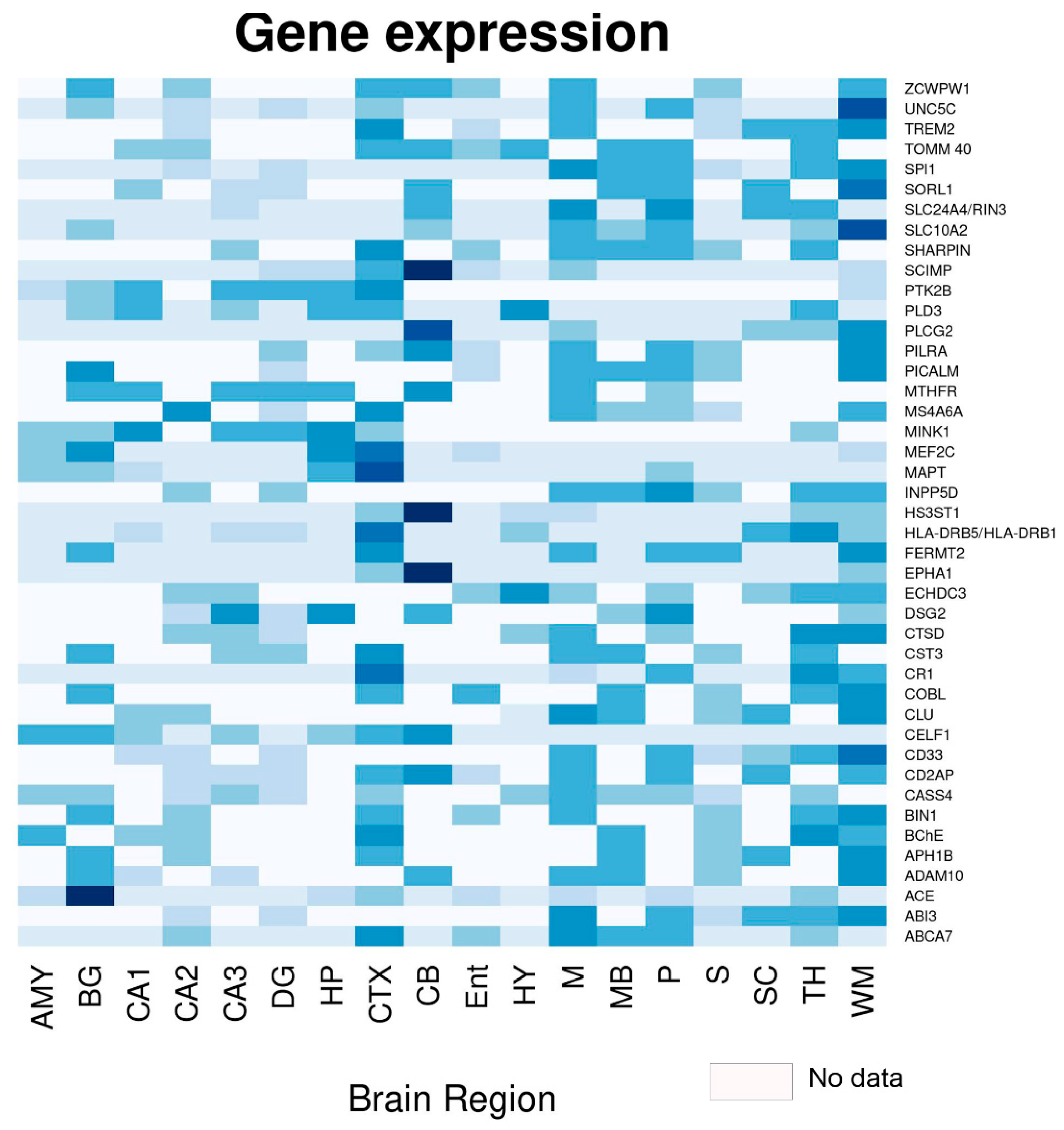{kind=link}
| Gene/Location | Physiological Role * | Molecular Mechanisms Implicated in AD | Ref. |
|---|---|---|---|
| ADAM10 (15q21.3) | It is the most important α-secretase in the brain and contributes to the non-amyloidogenic pathway of APP metabolism. | Alteration in APP metabolism (through non-amyloidogenic pathway), synaptic plasticity, and hippocampal neurogenesis. | [33,34,35] |
| ABCA7 (19p13.3) | Modulates lipid metabolism, phagocytosis, apoptosis, and phagocytic of Aβ by microglia. | Alteration in Aβ processing and regulation of APP by β-secretase. | [64,65,66] |
| BIN1 (2q14.3) | Participates in immune response, calcium homeostasis, apoptosis, endocytosis of synaptic vesicles, and plasma membrane dynamic. | Contributes to Amyloid (through β-secretase activity) and Tau pathology and is related to inflammation, apoptosis, and calcium homeostasis. | [67,68,69] |
| CD2AP (6p12.3) | Regulates actin cytoskeleton and membrane trafficking through endocytosis and cytokinesis. | Associated with increased Aβ production, Tau neurotoxicity, abnormal modulation of the neurite structure, and altered integrity of the BBB. | [52,70,71] |
| EPHA1 (7q34) | Plays a role in synaptic development and plasticity, modulating cell migration, angiogenesis, cell proliferation, and apoptosis. | Alterations in immune response and endocytosis, as well as disruption of BBB integrity. | [29,72] |
| PICALM (11q14.2) | Modulates autophagy, membrane metabolism, internalization of cell receptors, synaptic transmission, removal of apoptotic cells, and endocytic pathways for APP processing. | Dysfunction of Aβ metabolism and APP processing. Possible association with tauopathy, synaptic dysfunction, and altered lipid metabolism. | [36,37,38,73,74] |
| SORL1 (11q24.1) | Plays a role in endocytosis. | Restricts delivery of precursors to endocytic compartments that promote amyloidogenic clearance. | [75,76] |
| MS4A6A (11q12.2) | Regulates Ca2+ entry and signal transduction of several proteins. | Its variants affect the processing of the TREM2 microglial receptor increasing the risk of AD. | [71,77] |
| CR1 (1q32.2) | Facilitates the capture and clearance of complement-opsonized pathogens by erythrocytes, monocytes, macrophages, and microglia. | Decreases complement-mediated clearance of Aβ42. Metabolism and clearance of Aβ and its interaction with APOE-4 are related to cognitive impairment and the appearance of LOAD. | [78,79,80] |
| CLU (8p21.1) | Participates in lipid metabolism, cell proliferation, apoptosis, immune response, and neuronal differentiation. | Alteration in Aβ aggregation, lipid metabolism, regulation of cell cycle and neuronal apoptosis, and neuroinflammation. | [40,41,81] |
| CD33 (19q13.3) | Involved in the inhibition of immune cell function and cytokine production. | Modulates microglial activation (neuroinflammation) and Aβ clearance through microglial cells. | [49,50,82] |
| TREM2 (6p21.1) | This microglia receptor regulates proliferation, survival, phagocytosis, and inflammation. | Related to microglial dysfunctions, stress of the endoplasmic reticulum, Tau and amyloid pathology, and neuroinflammation. | [83,84,85] |
| TOMM40 (19q13.32) | Plays a role in the stabilization of the mitochondrial membrane respiratory chain (Complex I). | Dysfunction of the mitochondrial membrane and subsequent oxidative stress. | [86,87,88] |
| MAPT (17q21.21) | Related to the assembly and stability of microtubules and neuronal polarity. | Encoding for the Tau protein that is involved in the microtubule disassembly during AD. | [89,90] |
| FERMT2 (14q22.1) | Participates in cell differentiation, biogenesis, and connection between extracellular matrix adhesion sites and the actin cytoskeleton. | Associated with increasing levels of mature APP at the cell surface, resulting in increased Aβ-peptide production. | [91,92] |
| CASS4 (20q13.31) | Associated with cell adhesion, cell spreading, calcium signaling, and microtubule stabilization. | Involved in the formation of neuritic plaques and NFT, neuroinflammation, and dysfunction in synapsis, calcium signaling, and microtubule stabilization. | [93,94] |
| PTK2B (8p21.2) | Involved in the regulation of calcium flux, LTP, neurite growth, synapsis, and angiogenesis. | Contributes to hypoperfusion, vascular permeability, and Tau toxicity. | [93,94,95] |
| INPP5D (2q37.1) | Participates in the regulation of microglia gene expression. | Involved in microglia with deficient phagocytic capacity, resulting in increased Aβ deposition. | [96,97] |
| SLC10A2 (13q33.1) | Has an important role in encoding the sodium/bile acid cotransporter, as well as in cholesterol metabolism. | Associated with LOAD by dysfunctional cholesterol metabolism, neuronal death, memory impairment, and increased Aβ generation. | [58,59] |
| COBL (7p12.1) | Regulates actin cytoskeleton reorganization and neuron morphogenesis and increases the branching of axons and dendrites. | Reduction in the number of dendritic branch points and neurites. | [59] |
| UNC5C (4q22.3) | Favors apoptosis and directs axon extension and cell migration during neural development. | Contributes to neuronal death, Tau pathology, and Aβ-associated pathways. | [98,99] |
| PLD3 (19q13.2) | Hydrolysis of membrane phospholipids, which influences the processing of APP. | Promotes the generation of amyloid plaques and cognitive decline. | [100,101] |
| SLC24A4/RIN3 (14q32.12) | Plays a role in calcium transport and lipid and glucose metabolism. | Associated with Aβ loading and Tau pathology. | [102] |
| HLA-DRB5/DRB1 (6p21.32) | Encodes proteins for MHC and plays an important role in the immune response, including antigen processing and presentation, and self-recognition by immune cells. | It is associated with the presence of capillary β-amyloid and the development of cerebral Amyloid angiopathy. It also induces microglial activation. | [103,104] |
| DSG2 (18ql2.1) | Encodes adhesion molecule proteins to promote contact between epithelial cells and other cells. | Mediates APOE-associated Aβ aggregation in neurons. | [105] |
| MTHFR (1p36.3) | The regulatory connection between the folate and methionine cycles. | Associated with high levels of homocysteine and subsequent vascular damage and neuroinflammation. | [63,106] |
| CST3 (20p11.21) | Promotes neurogenesis, reduces Aβ deposition, and inhibits fibril formation. | Involved with amyloid angiopathy and cell death induced by oligomeric and fibrillar Aβ. | [107,108,109] |
| BCHE (3q26.1) | Contributes to acetylcholine inactivation and hydrolysis of neurotoxic organophosphate esters. | Related to NFT and neuritic plaque formation. | [16,110,111] |
| CTSD (11p15.5) | Favors the activation/degradation of polypeptide hormones and growth factors. | Implicated in the processing of APP/Aβ deposition and autophagy dysfunction. | [112,113] |
| ZCWPW1 (7q22.1) | Regulation of the DNA metabolic process. Additionally, it is involved in epigenetic modulation. | Suppresses insulin resistance. It may activate the PI3K signaling pathway in neurons. | [60] |
| MEF2C (5q14.3) | Involved in vascular development, neurogenesis, inflammatory processes, and hippocampal-dependent learning and memory. | Promotes neuroinflammation, cell apoptosis, Aβ aggregates, synaptic plasticity dysfunction, and increases the oxidative stress level. | [114,115,116] |
| ABI3 (17q21.32) | Regulates actin cytoskeleton organization, cytokinesis, migration, endocytosis, and phagocytosis. | Associated with alterations in microglial migration and phagocytosis, Aβ accumulation, and neuroinflammation. | [117,118] |
| PLCG2 (16q24.1) | Regulates divergent microglial functions through TREM2 signaling and is involved in the transition to a microglial state. | Correlation with amyloid plaque density, expression levels of microglial marker genes, and neuroinflammation. | [119,120,121,122] |
| SCIMP (17p13.2) | Regulates MHC-II signaling in B cells and the host’s innate immune responses to danger signals. | Associated with alteration in TLR-mediated microglial phagocytosis via MHC II. | [123] |
| SHARPIN (8q24.3) | Induces NF-kB activation and regulation of inflammation, and cell death. Furthermore, it regulates angiogenesis and NLRP3 activation. | Associated with neuroinflammation, and defective Aβ clearance that leads to pathogenic Aβ accumulation. | [124,125,126,127,128] |
| MINK1 (17p13.2) | Controls glutamate receptor signaling, synaptic density, dendrite complexity, actin cytoskeleton reorganization, cell-matrix adhesion, cell–cell adhesion, and cell migration. | It is related to glutamatergic synapse impairment, dysfunction of axon regeneration, neuronal degeneration, neuroinflammation, and increased ROS levels. | [129,130,131] |
| APH1B (15q22.2) | Serves as a scaffold for the assembly of γ-secretase complex; therefore, it cleaves APP. It also favors excitatory synaptic transmission and plasticity. | Decreases excitatory synapsis and promotes Aβ aggregation and spine formation. | [132,133,134] |
| HS3ST1 (4p15.33) | Modulates stem cell differentiation and neuronal targeting. | Promotes Tau spreading mediated by Tau binding to the cell surface heparan sulfate. | [135,136] |
| ECHDC3 (10p14) | Involved in fatty acid biosynthesis in mitochondria and insulin sensitivity. | Alterations in lipid and cholesterol metabolism, such as cognitive functions. | [137,138] |
| ACE (17q23.3) | Regulates blood pressure, electrolyte homeostasis, synaptic plasticity, and Aβ metabolism. | Associated with cognitive decline, oxidative stress, neuroinflammation, and higher levels of Aβ and Tau load. | [139,140,141] |
| PILRA (7q22.1) | Involved in immune system regulation and plays a key role in the life cycle of HSV-1. | Correlation between AD and HSV-1. In addition, it is related to a decrease in the inhibition of microglial activation. | [54,55] |
| SPI1 (11p11.2) | Regulates the immune response and learning-related neuronal activity in the cerebral cortex. | Alters the microglial phenotype and transcriptome, involving interferon pathways. | [47,48] |
| IGF1 (12q23.2) | Inhibits abnormal Tau phosphorylation and Aβ deposition. Stimulates neurogenesis and prevents apoptosis in the hippocampus. | Associated with Tau and Aβ pathology. | [142,143] |
| INSR (19p13.2) | The insulin-INSR signaling pathway regulates glucose uptake and release, as well as the synthesis and storage of carbohydrates, lipids, and proteins. | Can activate GSK-3β by initiating the PI3K-AKT signaling pathway and lead to Aβ accumulation and Tau phosphorylation. | [57] |
| LKB1 (19p13.3) | Plays a role in cell metabolism, cell polarity, apoptosis, and the response to DNA damage by regulating AMPK activity. | Autophagy dysfunction, Aβ accumulation, and Tau phosphorylation. | [57] |
4. Impact of GWAS on Understanding Alzheimer’s Disease
| Year | Population (Ethnicity/Race) | New Genes Found * | Ref. |
|---|---|---|---|
| 2007 | British and Americans (n = 3870); LOAD = 1808; Ctrl = 2062 | APOE | [156] |
| 2008 | European ancestry (n = 1376). | CD33 | [157] |
| 2009 | 1. French (n = 7360); LOAD = 2032; Ctrl = 5238 2. Belgians, Finns, Italians, Spanish (n = 7275); LOAD = 3978; Ctrl = 3297 | CR1, CLU | [158] |
| 2009 | 1. British, Germans, and Americans (n = 11,789); LOAD = 3941; Ctrl = 7848 2. Europeans (n = 4372); LOAD = 2032; Ctrl = 2340. | PICALM | [41] |
| 2010 | Spanish (n = 2349); LOAD = 1140; Ctrl = 1209 | EXOC3L2, BIN1 | [159] |
| 2010 | Caucasians of German extraction (n = 970); LOAD = 491; Ctrl = 479 | TOMM40 | [160] |
| 2011 | 1. Europeans (n = 9799); LOAD = 4896; Ctrl = 4903 2. Europeans (n = 29,544); LOAD = 8286; Ctrl = 21,258 | ABCA7, MS4A6, EPHA1, CD2AP | [71] |
| 2011 | European Americans (n = 3839); LOAD = 1848; Ctrl = 1991 | CUGBP2 | [161] |
| 2011 | 1. African Americans (n = 1009); LOAD = 513; Ctrl = 496 2. White (n = 9773); LOAD = 3568; Ctrl = 6205 | PVRL2 | [162] |
| 2012 | Caribbean Hispanics (n = 1093); LOAD = 549; Ctrl = 544 | DGKB, GWA-10q23.1 (PCDH21, LRIT1, RGR), HPCAL1 | [163] |
| 2012 | Polish (n = 282); Cases = 141 (94 LOAD, and 47 MCI); Ctrl = 141 | GWA-9q21.33 | [164] |
| 2012 | 1. European Americans (n = 2440); LOAD = 1440; Ctrl = 1000 2. European Americans (n = 6063); LOAD = 2727; Ctrl = 3336 | PPP1R3B | [165] |
| 2013 | European ancestry and African Americans (n = 4689); LOAD = 2493; Ctrl = 2196 | PLD3 | [166] |
| 2013 | 1. Icelanders (n = 4786); LOAD = 3550; Ctrl = 1236. 2. Americans, Norwegians, Germans, Dutch (n = 11,764); LOAD = 2037; Ctrl = 9727 | TREM2 | [167] |
| 2014 | European Americans (n = 3656); LOAD = 2151; Ctrl = 1505 | APOC1, CAMK1D, FBXL13 | [168] |
| 2014 | Multiethnic (mostly Caucasians; n = 32,346); LOAD = 14,967; Ctrl = 17,379 | PLXNA4 | [169] |
| 2015 | 1. Caribbean Hispanics (n = 4514); LOAD = 2451; Ctrl = 2063 2. Caribbean Hispanics (n = 5300); LOAD = 3001; Ctrl = 2299 | FBXL7, FRMD4A, CELF1, FERMT2, SLC24A4-RIN3. | [170] |
| 2015 | Japanese (n = 8808); LOAD = 816; Ctrl = 7992 | CNTNAP2, GWA-18p11.32, GWA-12q24.23 | [171] |
| 2016 | European ancestry (n = 3467) LOAD = 2488; Ctrl = 979 | OSBPL6, PTPRG, PDCL3, | [172] |
| 2017 | African Americans (n = 5609); LOAD = 1825; Ctrl = 3784 | COBL, SLC10A2 | [59] |
| 2018 | Multiethnic (n = 54,162); LOAD = 17,008; Ctrl = 37,154 | MLH3, FNBP4, CEACAM19, CLPTM1. | [173] |
| 2018 | British. Maternal AD = 27,696; Ctrl = 260,980 Paternal AD= 14,338; Ctrl = 245,941 | ADAM10, BCKDK/KAT8 (VKORC1), ACE, TREML2, PLCG2, IL-34. | [154] |
| 2019 | European ancestry (n = 455,258); LOAD = 71,880; Controls = 383,378 | ADAMTS4, INPP5D, HESX1, CLNK, HS3ST1, HLA-DRB1, ZCWPW1 (SPDYE3), CNTNAP2, CLU/PTK2B, ECHDC3, APH1B, SCIMP, AB13, BZRAP1-AS1, SUZ12P1, ALPK2, AC074212.3. | [42] |
| 2019 | Caucasian ancestry (n = 17,480); LOAD = 2741; Ctrl = 14,739 | CEACAM16, BCL3, MIR8085, CBLC, BCAM, PVRL2, APOC1, APOC1P1, APOC4, APOC2, LINC00158, MIR155HG, MIR155, LINC00515, MRPL39, JAM2, C2orf74, ATG10, MS4A6A, ABCB9, ZNF815, TRA2A, MED30, LPXN, IRAK3, N4BP2L2, UQCC, APOBEC3F, SFNa, ESPN, GNAI3, C9orf72, MTMR3 | [150] |
| 2019 | Non-Hispanic Whites (n = 94,437); LOAD = 35,274; Ctrl = 59,163 | NYAP1 (SPDYE3), ECHDC3 (USP6NL), IQCK, WWOX. | [174] |
| 2020 | African Americans (n = 8006); LOAD = 2784; Ctrl = 5222 | TRANK1, FABP2, LARP1B, TSRM, ARAP1, STARD10, SPHK1, SERPINB13, EDEM1, ALCAM, GPC6, VRK3, SIPA1L2, WDR70, API5, ACER3, PIK3C2G, ARRDC4, IGF1R, RBFOX1, MSX2, AKAP9. | [175] |
| 2021 | British (n = 495,000); LOAD = 75,000; Ctrl = 420,000 | PILRA, NCK2, SPI1, TSPAN14, SPPL2A, ACE, CCDC6, ADAMTS1, SHARPIN, GRN, SPRED2, ADAMTS4, TMEM163, SIGLEC11, PLCG2, IGHG1, IKZF1, TSPOAP1. | [147] |
| 2022 | Japanese (n = 3777); LOAD = 1744; Ctrl = 2033 | FAM126A, ZFHX4, LGR5, ZFC3H1, OR51G1, OR4X2, ARHGEF4, PRUNE2, MLKL, NCOR2, DMD, NEDD4, PLEC | [176] |
| 2022 | European (n = 788,989); LOAD = 111,326; Ctrl = 677,663 | UNC5CL, EPDR1, WNT3, SORT1, ADAM17, PRKD3, MME, IDUA, RHOH, ANKH, COX7C, TNIP1, RASGEF1C, HS3STS, UMAD1, ICA1, TMEM106B, JAZF1, SEC61G, CTSB, ABCA1, ANK3, BLNK, PLEKHA1, TPCN1, SNX1, DOC2A, MAF, FOXF1, PRDM7, WDR91, MYO15A, KLF16, LILRB2, RBCK1, SLC2A4RG, APP. | [14] |
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- 2022 Alzheimer’s disease facts and figures. Alzheimers Dement. 2022, 18, 700–789.
- GBD 2016 Neurology Collaborators. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 88–106. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 3 November 2022).
- Matthews, K.A.; Xu, W.; Gaglioti, A.H.; Holt, J.B.; Croft, J.B.; Mack, D.; McGuire, L.C. Racial and ethnic estimates of Alzheimer’s disease and related dementias in the United States (2015–2060) in adults aged >/=65 years. Alzheimers Dement. 2019, 15, 17–24. [Google Scholar] [CrossRef]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Maccioni, R.B.; Tapia, J.P.; Guzman-Martinez, L. Pathway to Tau Modifications and the Origins of Alzheimer’s Disease. Arch. Med. Res. 2018, 49, 130–131. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.; Singh, S.K.; Churruca, M.; Maccioni, R.B. Alzheimer’s Disease and Tau Self-Assembly: In the Search of the Missing Link. Int. J. Mol. Sci. 2022, 23, 4192. [Google Scholar] [CrossRef]
- Silva, M.V.F.; Loures, C.M.G.; Alves, L.C.V.; de Souza, L.C.; Borges, K.B.G.; Carvalho, M.D.G. Alzheimer’s disease: Risk factors and potentially protective measures. J. Biomed. Sci. 2019, 26, 33. [Google Scholar] [CrossRef]
- Reiss, A.B.; Arain, H.A.; Stecker, M.M.; Siegart, N.M.; Kasselman, L.J. Amyloid toxicity in Alzheimer’s disease. Rev. Neurosci. 2018, 29, 613–627. [Google Scholar] [CrossRef]
- Soto-Rojas, L.O.; Pacheco-Herrero, M.; Martinez-Gomez, P.A.; Campa-Cordoba, B.B.; Apatiga-Perez, R.; Villegas-Rojas, M.M.; Harrington, C.R.; de la Cruz, F.; Garces-Ramirez, L.; Luna-Munoz, J. The Neurovascular Unit Dysfunction in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 2022. [Google Scholar] [CrossRef]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef] [PubMed]
- Livingston, G.; Sommerlad, A.; Orgeta, V.; Costafreda, S.G.; Huntley, J.; Ames, D.; Ballard, C.; Banerjee, S.; Burns, A.; Cohen-Mansfield, J.; et al. Dementia prevention, intervention, and care. Lancet 2017, 390, 2673–2734. [Google Scholar] [CrossRef] [PubMed]
- Bellenguez, C.; Kucukali, F.; Jansen, I.E.; Kleineidam, L.; Moreno-Grau, S.; Amin, N.; Naj, A.C.; Campos-Martin, R.; Grenier-Boley, B.; Andrade, V.; et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 2022, 54, 412–436. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Wang, X.; Geng, M. Alzheimer’s disease hypothesis and related therapies. Transl. Neurodegener. 2018, 7, 2. [Google Scholar] [CrossRef]
- Sharma, P.; Srivastava, P.; Seth, A.; Tripathi, P.N.; Banerjee, A.G.; Shrivastava, S.K. Comprehensive review of mechanisms of pathogenesis involved in Alzheimer’s disease and potential therapeutic strategies. Prog. Neurobiol. 2019, 174, 53–89. [Google Scholar] [CrossRef]
- Wingo, A.P.; Liu, Y.; Gerasimov, E.S.; Gockley, J.; Logsdon, B.A.; Duong, D.M.; Dammer, E.B.; Robins, C.; Beach, T.G.; Reiman, E.M.; et al. Integrating human brain proteomes with genome-wide association data implicates new proteins in Alzheimer’s disease pathogenesis. Nat. Genet. 2021, 53, 143–146. [Google Scholar] [CrossRef]
- Cacace, R.; Sleegers, K.; Van Broeckhoven, C. Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimers Dement. 2016, 12, 733–748. [Google Scholar] [CrossRef]
- Lanoiselee, H.M.; Nicolas, G.; Wallon, D.; Rovelet-Lecrux, A.; Lacour, M.; Rousseau, S.; Richard, A.C.; Pasquier, F.; Rollin-Sillaire, A.; Martinaud, O.; et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med. 2017, 14, e1002270. [Google Scholar] [CrossRef]
- Alzforum. Available online: https://www.alzforum.org/mutations (accessed on 3 November 2022).
- Van Cauwenberghe, C.; Van Broeckhoven, C.; Sleegers, K. The genetic landscape of Alzheimer disease: Clinical implications and perspectives. Genet. Med. 2016, 18, 421–430. [Google Scholar] [CrossRef]
- Nalivaeva, N.N.; Turner, A.J. The amyloid precursor protein: A biochemical enigma in brain development, function and disease. FEBS Lett. 2013, 587, 2046–2054. [Google Scholar] [CrossRef]
- Belyaev, N.D.; Kellett, K.A.; Beckett, C.; Makova, N.Z.; Revett, T.J.; Nalivaeva, N.N.; Hooper, N.M.; Turner, A.J. The transcriptionally active amyloid precursor protein (APP) intracellular domain is preferentially produced from the 695 isoform of APP in a β-secretase-dependent pathway. J. Biol. Chem. 2010, 285, 41443–41454. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhang, C.M.; Prokopenko, D.; Liang, Y.; Zhen, S.Y.; Weigle, I.Q.; Han, W.; Aryal, M.; Tanzi, R.E.; Sisodia, S.S. An APP ectodomain mutation outside of the Abeta domain promotes Abeta production in vitro and deposition in vivo. J. Exp. Med. 2021, 218, e20210313. [Google Scholar] [CrossRef] [PubMed]
- Muratore, C.R.; Rice, H.C.; Srikanth, P.; Callahan, D.G.; Shin, T.; Benjamin, L.N.; Walsh, D.M.; Selkoe, D.J.; Young-Pearse, T.L. The familial Alzheimer’s disease APPV717I mutation alters APP processing and Tau expression in iPSC-derived neurons. Hum. Mol. Genet. 2014, 23, 3523–3536. [Google Scholar] [CrossRef]
- Doran, E.; Keator, D.; Head, E.; Phelan, M.J.; Kim, R.; Totoiu, M.; Barrio, J.R.; Small, G.W.; Potkin, S.G.; Lott, I.T. Down Syndrome, Partial Trisomy 21, and Absence of Alzheimer’s Disease: The Role of APP. J. Alzheimers Dis. 2017, 56, 459–470. [Google Scholar] [CrossRef]
- Kasuga, K.; Shimohata, T.; Nishimura, A.; Shiga, A.; Mizuguchi, T.; Tokunaga, J.; Ohno, T.; Miyashita, A.; Kuwano, R.; Matsumoto, N.; et al. Identification of independent APP locus duplication in Japanese patients with early-onset Alzheimer disease. J. Neurol. Neurosurg. Psychiatry 2009, 80, 1050–1052. [Google Scholar] [CrossRef] [PubMed]
- Sleegers, K.; Brouwers, N.; Gijselinck, I.; Theuns, J.; Goossens, D.; Wauters, J.; Del-Favero, J.; Cruts, M.; van Duijn, C.M.; Van Broeckhoven, C. APP duplication is sufficient to cause early onset Alzheimer’s dementia with cerebral amyloid angiopathy. Brain 2006, 129 Pt 11, 2977–2983. [Google Scholar] [CrossRef] [PubMed]
- Karch, C.M.; Goate, A.M. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 2015, 77, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Zhao, N.; Caulfield, T.R.; Liu, C.C.; Bu, G. Apolipoprotein E and Alzheimer disease: Pathobiology and targeting strategies. Nat. Rev. Neurol. 2019, 15, 501–518. [Google Scholar] [CrossRef] [PubMed]
- Safieh, M.; Korczyn, A.D.; Michaelson, D.M. ApoE4: An emerging therapeutic target for Alzheimer’s disease. BMC Med. 2019, 17, 64. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda-Falla, D.; Sanchez, J.S.; Almeida, M.C.; Boassa, D.; Acosta-Uribe, J.; Vila-Castelar, C.; Ramirez-Gomez, L.; Baena, A.; Aguillon, D.; Villalba-Moreno, N.D.; et al. Distinct tau neuropathology and cellular profiles of an APOE3 Christchurch homozygote protected against autosomal dominant Alzheimer’s dementia. Acta Neuropathol. 2022, 144, 589–601. [Google Scholar] [CrossRef] [PubMed]
- Manzine, P.R.; Ettcheto, M.; Cano, A.; Busquets, O.; Marcello, E.; Pelucchi, S.; Di Luca, M.; Endres, K.; Olloquequi, J.; Camins, A.; et al. ADAM10 in Alzheimer’s disease: Pharmacological modulation by natural compounds and its role as a peripheral marker. Biomed. Pharmacother. 2019, 113, 108661. [Google Scholar] [CrossRef] [PubMed]
- Peron, R.; Vatanabe, I.P.; Manzine, P.R.; Camins, A.; Cominetti, M.R. Alpha-Secretase ADAM10 Regulation: Insights into Alzheimer’s Disease Treatment. Pharmaceuticals 2018, 11, 12. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.Z.; Sun, S.; Tan, C.C.; Yu, J.T.; Tan, L. The Role of ADAM10 in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 58, 303–322. [Google Scholar] [CrossRef]
- Narayan, P.; Sienski, G.; Bonner, J.M.; Lin, Y.T.; Seo, J.; Baru, V.; Haque, A.; Milo, B.; Akay, L.A.; Graziosi, A.; et al. PICALM Rescues Endocytic Defects Caused by the Alzheimer’s Disease Risk Factor APOE4. Cell Rep. 2020, 33, 108224. [Google Scholar] [CrossRef] [PubMed]
- Ponomareva, N.; Andreeva, T.; Protasova, M.; Konovalov, R.; Krotenkova, M.; Malina, D.; Mitrofanov, A.; Fokin, V.; Illarioshkin, S.; Rogaev, E. Genetic Association Between Alzheimer’s Disease Risk Variant of the PICALM Gene and EEG Functional Connectivity in Non-demented Adults. Front. Neurosci. 2020, 14, 324. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Tan, L.; Yu, J.T. The Role of PICALM in Alzheimer’s Disease. Mol. Neurobiol. 2015, 52, 399–413. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.P.; Ravichandran, J.; Carkaci-Salli, N. Neural regeneration therapies for Alzheimer’s and Parkinson’s disease-related disorders. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165506. [Google Scholar] [CrossRef]
- Uddin, M.S.; Kabir, M.T.; Begum, M.M.; Islam, M.S.; Behl, T.; Ashraf, G.M. Exploring the Role of CLU in the Pathogenesis of Alzheimer’s Disease. Neurotox. Res. 2021, 39, 2108–2119. [Google Scholar] [CrossRef]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1088–1093. [Google Scholar] [CrossRef] [PubMed]
- Jansen, I.E.; Savage, J.E.; Watanabe, K.; Bryois, J.; Williams, D.M.; Steinberg, S.; Sealock, J.; Karlsson, I.K.; Hagg, S.; Athanasiu, L.; et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 2019, 51, 404–413. [Google Scholar] [CrossRef]
- Maccioni, R.B.; Gonzalez, A.; Andrade, V.; Cortes, N.; Tapia, J.P.; Guzman-Martinez, L. Alzheimer s Disease in the Perspective of Neuroimmunology. Open Neurol. J. 2018, 12, 50–56. [Google Scholar] [CrossRef]
- Cortes, N.; Andrade, V.; Guzman-Martinez, L.; Estrella, M.; Maccioni, R.B. Neuroimmune Tau Mechanisms: Their Role in the Progression of Neuronal Degeneration. Int. J. Mol. Sci. 2018, 19, 956. [Google Scholar] [CrossRef]
- Maccioni, R.B.; Farias, G.; Morales, I.; Navarrete, L. The revitalized tau hypothesis on Alzheimer’s disease. Arch. Med. Res. 2010, 41, 226–231. [Google Scholar] [CrossRef]
- Morales, I.; Farias, G.; Maccioni, R.B. Neuroimmunomodulation in the pathogenesis of Alzheimer’s disease. Neuroimmunomodulation 2010, 17, 202–204. [Google Scholar] [CrossRef]
- Cao, H.; Zhou, X.; Chen, Y.; Ip, F.C.F.; Chen, Y.; Lai, N.C.H.; Lo, R.M.N.; Tong, E.P.S.; Mok, V.C.T.; Kwok, T.C.Y.; et al. Association of SPI1 Haplotypes with Altered SPI1 Gene Expression and Alzheimer’s Disease Risk. J. Alzheimers Dis. 2022, 86, 1861–1873. [Google Scholar] [CrossRef]
- Jones, R.E.; Andrews, R.; Holmans, P.; Hill, M.; Taylor, P.R. Modest changes in Spi1 dosage reveal the potential for altered microglial function as seen in Alzheimer’s disease. Sci. Rep. 2021, 11, 14935. [Google Scholar] [CrossRef]
- Wissfeld, J.; Nozaki, I.; Mathews, M.; Raschka, T.; Ebeling, C.; Hornung, V.; Brustle, O.; Neumann, H. Deletion of Alzheimer’s disease-associated CD33 results in an inflammatory human microglia phenotype. Glia 2021, 69, 1393–1412. [Google Scholar] [CrossRef]
- Griciuc, A.; Federico, A.N.; Natasan, J.; Forte, A.M.; McGinty, D.; Nguyen, H.; Volak, A.; LeRoy, S.; Gandhi, S.; Lerner, E.P.; et al. Gene therapy for Alzheimer’s disease targeting CD33 reduces amyloid beta accumulation and neuroinflammation. Hum. Mol. Genet. 2020, 29, 2920–2935. [Google Scholar] [CrossRef]
- Cruchaga, C.; Kauwe, J.S.; Harari, O.; Jin, S.C.; Cai, Y.; Karch, C.M.; Benitez, B.A.; Jeng, A.T.; Skorupa, T.; Carrell, D.; et al. GWAS of cerebrospinal fluid tau levels identifies risk variants for Alzheimer’s disease. Neuron 2013, 78, 256–268. [Google Scholar] [CrossRef]
- Tao, Q.Q.; Chen, Y.C.; Wu, Z.Y. The role of CD2AP in the Pathogenesis of Alzheimer’s Disease. Aging Dis. 2019, 10, 901–907. [Google Scholar] [CrossRef]
- Sochocka, M.; Zwolinska, K.; Leszek, J. The Infectious Etiology of Alzheimer’s Disease. Curr. Neuropharmacol. 2017, 15, 996–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopatko Lindman, K.; Jonsson, C.; Weidung, B.; Olsson, J.; Pandey, J.P.; Prokopenko, D.; Tanzi, R.E.; Hallmans, G.; Eriksson, S.; Elgh, F.; et al. PILRA polymorphism modifies the effect of APOE4 and GM17 on Alzheimer’s disease risk. Sci. Rep. 2022, 12, 13264. [Google Scholar] [CrossRef] [PubMed]
- Agostini, S.; Costa, A.S.; Mancuso, R.; Guerini, F.R.; Nemni, R.; Clerici, M. The PILRA G78R Variant Correlates with Higher HSV-1-Specific IgG Titers in Alzheimer’s Disease. Cell Mol. Neurobiol. 2019, 39, 1217–1221. [Google Scholar] [CrossRef]
- Linard, M.; Letenneur, L.; Garrigue, I.; Doize, A.; Dartigues, J.F.; Helmer, C. Interaction between APOE4 and herpes simplex virus type 1 in Alzheimer’s disease. Alzheimers Dement. 2020, 16, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Wang, H.; Zhang, F.; Zhang, M.; Wang, Q.; Wang, J. The common genes involved in the pathogenesis of Alzheimer’s disease and type 2 diabetes and their implication for drug repositioning. Neuropharmacology 2023, 223, 109327. [Google Scholar] [CrossRef] [PubMed]
- Loera-Valencia, R.; Goikolea, J.; Parrado-Fernandez, C.; Merino-Serrais, P.; Maioli, S. Alterations in cholesterol metabolism as a risk factor for developing Alzheimer’s disease: Potential novel targets for treatment. J. Steroid Biochem. Mol. Biol. 2019, 190, 104–114. [Google Scholar] [CrossRef]
- Mez, J.; Chung, J.; Jun, G.; Kriegel, J.; Bourlas, A.P.; Sherva, R.; Logue, M.W.; Barnes, L.L.; Bennett, D.A.; Buxbaum, J.D.; et al. Two novel loci, COBL and SLC10A2, for Alzheimer’s disease in African Americans. Alzheimers Dement. 2017, 13, 119–129. [Google Scholar] [CrossRef]
- Gao, Y.; Tan, M.S.; Wang, H.F.; Zhang, W.; Wang, Z.X.; Jiang, T.; Yu, J.T.; Tan, L. ZCWPW1 is associated with late-onset Alzheimer’s disease in Han Chinese: A replication study and meta-analyses. Oncotarget 2016, 7, 20305–20311. [Google Scholar] [CrossRef]
- Chen, M.; Huang, N.; Liu, J.; Huang, J.; Shi, J.; Jin, F. AMPK: A bridge between diabetes mellitus and Alzheimer’s disease. Behav. Brain Res. 2021, 400, 113043. [Google Scholar] [CrossRef]
- Pathak, G.A.; Silzer, T.K.; Sun, J.; Zhou, Z.; Daniel, A.A.; Johnson, L.; O’Bryant, S.; Phillips, N.R.; Barber, R.C. Genome-Wide Methylation of Mild Cognitive Impairment in Mexican Americans Highlights Genes Involved in Synaptic Transport, Alzheimer’s Disease-Precursor Phenotypes, and Metabolic Morbidities. J. Alzheimers Dis. 2019, 72, 733–749. [Google Scholar] [CrossRef]
- Reagan, A.M.; Christensen, K.E.; Graham, L.C.; Bedwell, A.A.; Eldridge, K.; Speedy, R.; Figueiredo, L.L.; Persohn, S.C.; Bottiglieri, T.; Nho, K.; et al. The 677C > T variant in methylenetetrahydrofolate reductase causes morphological and functional cerebrovascular deficits in mice. J. Cereb. Blood Flow Metab. 2022, 42, 2333–2350. [Google Scholar] [CrossRef] [PubMed]
- De Roeck, A.; Van Broeckhoven, C.; Sleegers, K. The role of ABCA7 in Alzheimer’s disease: Evidence from genomics, transcriptomics and methylomics. Acta Neuropathol. 2019, 138, 201–220. [Google Scholar] [CrossRef] [PubMed]
- Aikawa, T.; Holm, M.L.; Kanekiyo, T. ABCA7 and Pathogenic Pathways of Alzheimer’s Disease. Brain Sci. 2018, 8, 27. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.F.; Yu, J.T.; Tan, M.S.; Tan, L. ABCA7 in Alzheimer’s Disease. Mol. Neurobiol. 2015, 51, 1008–1016. [Google Scholar] [CrossRef]
- Lambert, E.; Saha, O.; Soares Landeira, B.; Melo de Farias, A.R.; Hermant, X.; Carrier, A.; Pelletier, A.; Gadaut, J.; Davoine, L.; Dupont, C.; et al. The Alzheimer susceptibility gene BIN1 induces isoform-dependent neurotoxicity through early endosome defects. Acta Neuropathol. Commun. 2022, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Perdigao, C.; Barata, M.A.; Burrinha, T.; Guimas Almeida, C. Alzheimer’s disease BIN1 coding variants increase intracellular Abeta levels by interfering with BACE1 recycling. J. Biol. Chem. 2021, 297, 101056. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Ye, L.; Cheng, H.; Li, H. The Mechanistic Role of Bridging Integrator 1 (BIN1) in Alzheimer’s Disease. Cell Mol. Neurobiol. 2021, 41, 1431–1440. [Google Scholar] [CrossRef] [PubMed]
- Naj, A.C.; Jun, G.; Beecham, G.W.; Wang, L.S.; Vardarajan, B.N.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Jarvik, G.P.; Crane, P.K.; et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet. 2011, 43, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 2011, 43, 429–435. [Google Scholar] [CrossRef]
- Talebi, M.; Delpak, A.; Khalaj-Kondori, M.; Sadigh-Eteghad, S.; Talebi, M.; Mehdizadeh, E.; Majdi, A. ABCA7 and EphA1 Genes Polymorphisms in Late-Onset Alzheimer’s Disease. J. Mol. Neurosci. 2020, 70, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Moreau, K.; Fleming, A.; Imarisio, S.; Lopez Ramirez, A.; Mercer, J.L.; Jimenez-Sanchez, M.; Bento, C.F.; Puri, C.; Zavodszky, E.; Siddiqi, F.; et al. PICALM modulates autophagy activity and tau accumulation. Nat. Commun. 2014, 5, 4998. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Chang, J.C.; Fan, E.Y.; Flajolet, M.; Greengard, P. Adaptor complex AP2/PICALM, through interaction with LC3, targets Alzheimer’s APP-CTF for terminal degradation via autophagy. Proc. Natl. Acad. Sci. USA 2013, 110, 17071–17076. [Google Scholar] [CrossRef]
- Campion, D.; Charbonnier, C.; Nicolas, G. SORL1 genetic variants and Alzheimer disease risk: A literature review and meta-analysis of sequencing data. Acta Neuropathol. 2019, 138, 173–186. [Google Scholar] [CrossRef]
- Yin, R.H.; Yu, J.T.; Tan, L. The Role of SORL1 in Alzheimer’s Disease. Mol. Neurobiol. 2015, 51, 909–918. [Google Scholar] [CrossRef]
- Kunkle, B.W.; Jaworski, J.; Barral, S.; Vardarajan, B.; Beecham, G.W.; Martin, E.R.; Cantwell, L.S.; Partch, A.; Bird, T.D.; Raskind, W.H.; et al. Genome-wide linkage analyses of non-Hispanic white families identify novel loci for familial late-onset Alzheimer’s disease. Alzheimers Dement. 2016, 12, 2–10. [Google Scholar] [CrossRef]
- Lu, L.; Yao, Q.Y.; Ruan, S.S.; Hu, J.W.; Long, W.J.; Dai, W.Z.; Ma, T.; Zhu, X.C. Explore the role of CR1 genetic variants in late-onset Alzheimer’s disease susceptibility. Psychiatr. Genet. 2021, 31, 216–229. [Google Scholar] [CrossRef]
- Zhu, X.C.; Yu, J.T.; Jiang, T.; Wang, P.; Cao, L.; Tan, L. CR1 in Alzheimer’s disease. Mol. Neurobiol. 2015, 51, 753–765. [Google Scholar] [CrossRef]
- Crehan, H.; Holton, P.; Wray, S.; Pocock, J.; Guerreiro, R.; Hardy, J. Complement receptor 1 (CR1) and Alzheimer’s disease. Immunobiology 2012, 217, 244–250. [Google Scholar] [CrossRef]
- Han, S.; Nho, K.; Lee, Y. Alternative Splicing Regulation of an Alzheimer’s Risk Variant in CLU. Int. J. Mol. Sci. 2020, 21, 7079. [Google Scholar] [CrossRef]
- Griciuc, A.; Serrano-Pozo, A.; Parrado, A.R.; Lesinski, A.N.; Asselin, C.N.; Mullin, K.; Hooli, B.; Choi, S.H.; Hyman, B.T.; Tanzi, R.E. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 2013, 78, 631–643. [Google Scholar] [CrossRef]
- Qin, Q.; Teng, Z.; Liu, C.; Li, Q.; Yin, Y.; Tang, Y. TREM2, microglia, and Alzheimer’s disease. Mech. Ageing Dev. 2021, 195, 111438. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Holtzman, D.M. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat. Rev. Immunol. 2018, 18, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Ulland, T.K.; Song, W.M.; Huang, S.C.; Ulrich, J.D.; Sergushichev, A.; Beatty, W.L.; Loboda, A.A.; Zhou, Y.; Cairns, N.J.; Kambal, A.; et al. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell 2017, 170, 649–663.e13. [Google Scholar] [CrossRef]
- Lee, E.G.; Chen, S.; Leong, L.; Tulloch, J.; Yu, C.E. TOMM40 RNA Transcription in Alzheimer’s Disease Brain and Its Implication in Mitochondrial Dysfunction. Genes 2021, 12, 871. [Google Scholar] [CrossRef]
- Bezuch, N.; Bradburn, S.; Robinson, A.C.; Pendleton, N.; Payton, A.; Murgatroyd, C. Superior Frontal Gyrus TOMM40-APOE Locus DNA Methylation in Alzheimer’s Disease. J. Alzheimers Dis. Rep. 2021, 5, 275–282. [Google Scholar] [CrossRef]
- Zhu, Z.; Yang, Y.; Xiao, Z.; Zhao, Q.; Wu, W.; Liang, X.; Luo, J.; Cao, Y.; Shao, M.; Guo, Q.; et al. TOMM40 and APOE variants synergistically increase the risk of Alzheimer’s disease in a Chinese population. Aging Clin. Exp. Res. 2021, 33, 1667–1675. [Google Scholar] [CrossRef]
- Sengoku, R. Aging and Alzheimer’s disease pathology. Neuropathology 2020, 40, 22–29. [Google Scholar] [CrossRef]
- Zhu, J.B.; Tan, C.C.; Tan, L.; Yu, J.T. State of Play in Alzheimer’s Disease Genetics. J. Alzheimers Dis. 2017, 58, 631–659. [Google Scholar] [CrossRef]
- Eysert, F.; Coulon, A.; Boscher, E.; Vreulx, A.C.; Flaig, A.; Mendes, T.; Hughes, S.; Grenier-Boley, B.; Hanoulle, X.; Demiautte, F.; et al. Alzheimer’s genetic risk factor FERMT2 (Kindlin-2) controls axonal growth and synaptic plasticity in an APP-dependent manner. Mol. Psychiatry 2021, 26, 5592–5607. [Google Scholar] [CrossRef]
- Chapuis, J.; Flaig, A.; Grenier-Boley, B.; Eysert, F.; Pottiez, V.; Deloison, G.; Vandeputte, A.; Ayral, A.M.; Mendes, T.; Desai, S.; et al. Genome-wide, high-content siRNA screening identifies the Alzheimer’s genetic risk factor FERMT2 as a major modulator of APP metabolism. Acta Neuropathol. 2017, 133, 955–966. [Google Scholar] [CrossRef]
- Hassan, M.; Shahzadi, S.; Alashwal, H.; Zaki, N.; Seo, S.Y.; Moustafa, A.A. Exploring the mechanistic insights of Cas scaffolding protein family member 4 with protein tyrosine kinase 2 in Alzheimer’s disease by evaluating protein interactions through molecular docking and dynamic simulations. Neurol. Sci. 2018, 39, 1361–1374. [Google Scholar] [CrossRef]
- Beck, T.N.; Nicolas, E.; Kopp, M.C.; Golemis, E.A. Adaptors for disorders of the brain? The cancer signaling proteins NEDD9, CASS4, and PTK2B in Alzheimer’s disease. Oncoscience 2014, 1, 486–503. [Google Scholar] [CrossRef] [PubMed]
- Dourlen, P.; Fernandez-Gomez, F.J.; Dupont, C.; Grenier-Boley, B.; Bellenguez, C.; Obriot, H.; Caillierez, R.; Sottejeau, Y.; Chapuis, J.; Bretteville, A.; et al. Functional screening of Alzheimer risk loci identifies PTK2B as an in vivo modulator and early marker of Tau pathology. Mol. Psychiatry 2017, 22, 874–883. [Google Scholar] [CrossRef] [PubMed]
- Tsai, A.P.; Lin, P.B.; Dong, C.; Moutinho, M.; Casali, B.T.; Liu, Y.; Lamb, B.T.; Landreth, G.E.; Oblak, A.L.; Nho, K. INPP5D expression is associated with risk for Alzheimer’s disease and induced by plaque-associated microglia. Neurobiol. Dis. 2021, 153, 105303. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, Y.; Yamazaki, K.; Ozaki, Y.; Sao, T.; Yoshida, T.; Mori, T.; Mori, Y.; Ochi, S.; Iga, J.I.; Ueno, S.I. INPP5D mRNA Expression and Cognitive Decline in Japanese Alzheimer’s Disease Subjects. J. Alzheimers Dis. 2017, 58, 687–694. [Google Scholar] [CrossRef]
- Chen, G.; Kang, S.S.; Wang, Z.; Ahn, E.H.; Xia, Y.; Liu, X.; Sandoval, I.M.; Manfredsson, F.P.; Zhang, Z.; Ye, K. Netrin-1 receptor UNC5C cleavage by active delta-secretase enhances neurodegeneration, promoting Alzheimer’s disease pathologies. Sci. Adv. 2021, 7, eabe4499. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, B.L.; Sun, F.R.; Li, J.Q.; Cao, X.P.; Tan, L. The role of UNC5C in Alzheimer’s disease. Ann. Transl. Med. 2018, 6, 178. [Google Scholar] [CrossRef]
- Nackenoff, A.G.; Hohman, T.J.; Neuner, S.M.; Akers, C.S.; Weitzel, N.C.; Shostak, A.; Ferguson, S.M.; Mobley, B.; Bennett, D.A.; Schneider, J.A.; et al. PLD3 is a neuronal lysosomal phospholipase D associated with beta-amyloid plaques and cognitive function in Alzheimer’s disease. PLoS Genet. 2021, 17, e1009406. [Google Scholar] [CrossRef]
- Wang, J.; Yu, J.T.; Tan, L. PLD3 in Alzheimer’s disease. Mol. Neurobiol. 2015, 51, 480–486. [Google Scholar] [CrossRef]
- Stage, E.; Duran, T.; Risacher, S.L.; Goukasian, N.; Do, T.M.; West, J.D.; Wilhalme, H.; Nho, K.; Phillips, M.; Elashoff, D.; et al. The effect of the top 20 Alzheimer disease risk genes on gray-matter density and FDG PET brain metabolism. Alzheimers Dement. 2016, 5, 53–66. [Google Scholar] [CrossRef]
- Makela, M.; Kaivola, K.; Valori, M.; Paetau, A.; Polvikoski, T.; Singleton, A.B.; Traynor, B.J.; Stone, D.J.; Peuralinna, T.; Tienari, P.J.; et al. Alzheimer risk loci and associated neuropathology in a population-based study (Vantaa 85+). Neurol. Genet. 2018, 4, e211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.G.; Li, Y.; Ng, C.T.; Song, Y.Q. Inflammation in Alzheimer’s Disease and Molecular Genetics: Recent Update. Arch. Immunol. Ther. Exp. 2015, 63, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Yoo, J.; Shin, J.; Chang, Y.; Jung, J.; Jo, D.G.; Kim, J.; Jang, W.; Lengner, C.J.; Kim, B.S.; et al. Modelling APOE varepsilon3/4 allele-associated sporadic Alzheimer’s disease in an induced neuron. Brain 2017, 140, 2193–2209. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Zhou, X.; Yin, W.; Wan, K.; Zhang, W.; Li, C.; Li, M.; Zhu, W.; Zhu, X.; Sun, Z. The Influence of MTHFR Polymorphism on Gray Matter Volume in Patients with Amnestic Mild Cognitive Impairment. Front. Neurosci. 2021, 15, 778123. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Levy, E. Cystatin C in Alzheimer’s disease. Front. Mol. Neurosci. 2012, 5, 79. [Google Scholar] [CrossRef]
- Sun, B.; Zhou, Y.; Halabisky, B.; Lo, I.; Cho, S.H.; Mueller-Steiner, S.; Devidze, N.; Wang, X.; Grubb, A.; Gan, L. Cystatin C-cathepsin B axis regulates amyloid beta levels and associated neuronal deficits in an animal model of Alzheimer’s disease. Neuron 2008, 60, 247–257. [Google Scholar] [CrossRef]
- Mi, W.; Pawlik, M.; Sastre, M.; Jung, S.S.; Radvinsky, D.S.; Klein, A.M.; Sommer, J.; Schmidt, S.D.; Nixon, R.A.; Mathews, P.M.; et al. Cystatin C inhibits amyloid-beta deposition in Alzheimer’s disease mouse models. Nat. Genet. 2007, 39, 1440–1442. [Google Scholar] [CrossRef]
- Greig, N.H.; Utsuki, T.; Ingram, D.K.; Wang, Y.; Pepeu, G.; Scali, C.; Yu, Q.S.; Mamczarz, J.; Holloway, H.W.; Giordano, T.; et al. Selective butyrylcholinesterase inhibition elevates brain acetylcholine, augments learning and lowers Alzheimer beta-amyloid peptide in rodent. Proc. Natl. Acad. Sci. USA 2005, 102, 17213–17218. [Google Scholar] [CrossRef]
- Guillozet, A.L.; Smiley, J.F.; Mash, D.C.; Mesulam, M.M. Butyrylcholinesterase in the life cycle of amyloid plaques. Ann. Neurol. 1997, 42, 909–918. [Google Scholar] [CrossRef]
- Letronne, F.; Laumet, G.; Ayral, A.M.; Chapuis, J.; Demiautte, F.; Laga, M.; Vandenberghe, M.E.; Malmanche, N.; Leroux, F.; Eysert, F.; et al. ADAM30 Downregulates APP-Linked Defects Through Cathepsin D Activation in Alzheimer’s Disease. EBioMedicine 2016, 9, 278–292. [Google Scholar] [CrossRef]
- Mariani, E.; Seripa, D.; Ingegni, T.; Nocentini, G.; Mangialasche, F.; Ercolani, S.; Cherubini, A.; Metastasio, A.; Pilotto, A.; Senin, U.; et al. Interaction of CTSD and A2M polymorphisms in the risk for Alzheimer’s disease. J. Neurol. Sci. 2006, 247, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Zhang, S.; Wang, X.; Deng, Y.; Zhao, Y.; Xiao, Y.; Liu, J.; Chu, L.; Qi, X. MEF2C ameliorates learning, memory, and molecular pathological changes in Alzheimer’s disease in vivo and in vitro. Acta Biochim. Biophys. Sin. 2022, 54, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhao, Y. Progress on the roles of MEF2C in neuropsychiatric diseases. Mol. Brain 2022, 15, 8. [Google Scholar] [CrossRef] [PubMed]
- Xue, F.; Tian, J.; Yu, C.; Du, H.; Guo, L. Type I interferon response-related microglial Mef2c deregulation at the onset of Alzheimer’s pathology in 5xFAD mice. Neurobiol. Dis. 2021, 152, 105272. [Google Scholar] [CrossRef]
- Karahan, H.; Smith, D.C.; Kim, B.; Dabin, L.C.; Al-Amin, M.M.; Wijeratne, H.R.S.; Pennington, T.; Viana di Prisco, G.; McCord, B.; Lin, P.B.; et al. Deletion of Abi3 gene locus exacerbates neuropathological features of Alzheimer’s disease in a mouse model of Abeta amyloidosis. Sci. Adv. 2021, 7, eabe3954. [Google Scholar] [CrossRef]
- Satoh, J.I.; Kino, Y.; Yanaizu, M.; Tosaki, Y.; Sakai, K.; Ishida, T.; Saito, Y. Microglia express ABI3 in the brains of Alzheimer’s disease and Nasu-Hakola disease. Intractable Rare Dis. Res. 2017, 6, 262–268. [Google Scholar] [CrossRef]
- Li, K.; Ran, B.; Wang, Y.; Liu, L.; Li, W. PLCgamma2 impacts microglia-related effectors revealing variants and pathways important in Alzheimer’s disease. Front. Cell Dev. Biol. 2022, 10, 999061. [Google Scholar] [CrossRef]
- Claes, C.; England, W.E.; Danhash, E.P.; Kiani Shabestari, S.; Jairaman, A.; Chadarevian, J.P.; Hasselmann, J.; Tsai, A.P.; Coburn, M.A.; Sanchez, J.; et al. The P522R protective variant of PLCG2 promotes the expression of antigen presentation genes by human microglia in an Alzheimer’s disease mouse model. Alzheimers Dement. 2022, 18, 1765–1778. [Google Scholar] [CrossRef]
- Kleineidam, L.; Chouraki, V.; Prochnicki, T.; van der Lee, S.J.; Madrid-Marquez, L.; Wagner-Thelen, H.; Karaca, I.; Weinhold, L.; Wolfsgruber, S.; Boland, A.; et al. PLCG2 protective variant p.P522R modulates tau pathology and disease progression in patients with mild cognitive impairment. Acta Neuropathol. 2020, 139, 1025–1044. [Google Scholar] [CrossRef]
- Sims, R.; van der Lee, S.J.; Naj, A.C.; Bellenguez, C.; Badarinarayan, N.; Jakobsdottir, J.; Kunkle, B.W.; Boland, A.; Raybould, R.; Bis, J.C.; et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat. Genet. 2017, 49, 1373–1384. [Google Scholar] [CrossRef]
- Li, Y.; Laws, S.M.; Miles, L.A.; Wiley, J.S.; Huang, X.; Masters, C.L.; Gu, B.J. Genomics of Alzheimer’s disease implicates the innate and adaptive immune systems. Cell. Mol. Life Sci. 2021, 78, 7397–7426. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Wang, F.; Wang, Y. Advances in the Structural and Physiological Functions of SHARPIN. Front. Immunol. 2022, 13, 858505. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Lee, D.; Lee, J.J.; Gim, J.; Gunasekaran, T.I.; Choi, K.Y.; Kang, S.; Do, A.R.; Jo, J.; Park, J.; et al. Alzheimer’s Disease Neuroimaging, I. A missense variant in SHARPIN mediates Alzheimer’s disease-specific brain damages. Transl. Psychiatry 2021, 11, 590. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, D.; Menon, R.N.; Gopala, S. SHARPIN: Role in Finding NEMO and in Amyloid-Beta Clearance and Degradation (ABCD) Pathway in Alzheimer’s Disease? Cell. Mol. Neurobiol. 2022, 42, 1267–1281. [Google Scholar] [CrossRef]
- Krishnan, D.; Menon, R.N.; Mathuranath, P.S.; Gopala, S. A novel role for SHARPIN in amyloid-beta phagocytosis and inflammation by peripheral blood-derived macrophages in Alzheimer’s disease. Neurobiol. Aging 2020, 93, 131–141. [Google Scholar] [CrossRef]
- Asanomi, Y.; Shigemizu, D.; Miyashita, A.; Mitsumori, R.; Mori, T.; Hara, N.; Ito, K.; Niida, S.; Ikeuchi, T.; Ozaki, K. A rare functional variant of SHARPIN attenuates the inflammatory response and associates with increased risk of late-onset Alzheimer’s disease. Mol. Med. 2019, 25, 20. [Google Scholar] [CrossRef]
- Zhu, K.; Jin, X.; Chi, Z.; Chen, S.; Wu, S.; Sloan, R.D.; Lin, X.; Neculai, D.; Wang, D.; Hu, H.; et al. Priming of NLRP3 inflammasome activation by Msn kinase MINK1 in macrophages. Cell. Mol. Immunol. 2021, 18, 2372–2382. [Google Scholar] [CrossRef]
- Lawingco, T.; Chaudhury, S.; Brookes, K.J.; Guetta-Baranes, T.; Guerreiro, R.; Bras, J.; Hardy, J.; Francis, P.; Thomas, A.; Belbin, O.; et al. Genetic variants in glutamate-, Abeta-, and tau-related pathways determine polygenic risk for Alzheimer’s disease. Neurobiol. Aging 2021, 101, 299.e13–299.e21. [Google Scholar] [CrossRef]
- Larhammar, M.; Huntwork-Rodriguez, S.; Rudhard, Y.; Sengupta-Ghosh, A.; Lewcock, J.W. The Ste20 Family Kinases MAP4K4, MINK1, and TNIK Converge to Regulate Stress-Induced JNK Signaling in Neurons. J. Neurosci. 2017, 37, 11074–11084. [Google Scholar] [CrossRef]
- Park, Y.H.; Pyun, J.M.; Hodges, A.; Jang, J.W.; Bice, P.J.; Kim, S.; Saykin, A.J.; Nho, K.; AddNeuroMed consortium and the Alzheimer’s Disease Neuroimaging Initiative. Dysregulated expression levels of APH1B in peripheral blood are associated with brain atrophy and amyloid-beta deposition in Alzheimer’s disease. Alzheimers Res. Ther. 2021, 13, 183. [Google Scholar] [CrossRef]
- Barao, S.; Gartner, A.; Leyva-Diaz, E.; Demyanenko, G.; Munck, S.; Vanhoutvin, T.; Zhou, L.; Schachner, M.; Lopez-Bendito, G.; Maness, P.F.; et al. Antagonistic Effects of BACE1 and APH1B-gamma-Secretase Control Axonal Guidance by Regulating Growth Cone Collapse. Cell Rep. 2015, 12, 1367–1376. [Google Scholar] [CrossRef] [Green Version]
- Fazzari, P.; Snellinx, A.; Sabanov, V.; Ahmed, T.; Serneels, L.; Gartner, A.; Shariati, S.A.; Balschun, D.; De Strooper, B. Cell autonomous regulation of hippocampal circuitry via Aph1b-gamma-secretase/neuregulin 1 signalling. eLife 2014, 3, e02196. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhu, Y.; Song, X.; Xiao, Y.; Su, G.; Liu, X.; Wang, Z.; Xu, Y.; Liu, J.; Eliezer, D.; et al. 3-O-Sulfation of Heparan Sulfate Enhances Tau Interaction and Cellular Uptake. Angew. Chem. Int. Ed. Engl. 2020, 59, 1818–1827. [Google Scholar] [CrossRef] [PubMed]
- Holmes, B.B.; DeVos, S.L.; Kfoury, N.; Li, M.; Jacks, R.; Yanamandra, K.; Ouidja, M.O.; Brodsky, F.M.; Marasa, J.; Bagchi, D.P.; et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. USA 2013, 110, E3138–E3147. [Google Scholar] [CrossRef] [PubMed]
- Tesi, N.; van der Lee, S.J.; Hulsman, M.; Jansen, I.E.; Stringa, N.; van Schoor, N.; Meijers-Heijboer, H.; Huisman, M.; Scheltens, P.; Reinders, M.J.T.; et al. Centenarian controls increase variant effect sizes by an average twofold in an extreme case-extreme control analysis of Alzheimer’s disease. Eur. J. Hum. Genet. 2019, 27, 244–253. [Google Scholar] [CrossRef]
- Jun, G.R.; Chung, J.; Mez, J.; Barber, R.; Beecham, G.W.; Bennett, D.A.; Buxbaum, J.D.; Byrd, G.S.; Carrasquillo, M.M.; Crane, P.K.; et al. Transethnic genome-wide scan identifies novel Alzheimer’s disease loci. Alzheimers Dement. 2017, 13, 727–738. [Google Scholar] [CrossRef]
- MacLachlan, R.; Kehoe, P.G.; Miners, J.S. Dysregulation of ACE-1 in Normal Aging and the Early Stages of Alzheimer’s Disease. J. Gerontol. A Biol. Sci. Med. Sci. 2022, 77, 1775–1783. [Google Scholar] [CrossRef] [PubMed]
- Braae, A.; Medway, C.; Carrasquillo, M.; Younkin, S.; Alzheimer’s Research, U.K.C.; Kehoe, P.G.; Morgan, K. Blood type gene locus has no influence on ACE association with Alzheimer’s disease. Neurobiol. Aging 2015, 36, 1767.e1–1767.e2. [Google Scholar] [CrossRef]
- Narain, Y.; Yip, A.; Murphy, T.; Brayne, C.; Easton, D.; Evans, J.G.; Xuereb, J.; Cairns, N.; Esiri, M.M.; Furlong, R.A.; et al. The ACE gene and Alzheimer’s disease susceptibility. J. Med. Genet. 2000, 37, 695–697. [Google Scholar] [CrossRef] [PubMed]
- Westwood, A.J.; Beiser, A.; Decarli, C.; Harris, T.B.; Chen, T.C.; He, X.M.; Roubenoff, R.; Pikula, A.; Au, R.; Braverman, L.E.; et al. Insulin-like growth factor-1 and risk of Alzheimer dementia and brain atrophy. Neurology 2014, 82, 1613–1619. [Google Scholar] [CrossRef]
- Zoidis, E.; Ghirlanda-Keller, C.; Schmid, C. Stimulation of glucose transport in osteoblastic cells by parathyroid hormone and insulin-like growth factor I. Mol. Cell. Biochem. 2011, 348, 33–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spangenberg, E.E.; Lee, R.J.; Najafi, A.R.; Rice, R.A.; Elmore, M.R.; Blurton-Jones, M.; West, B.L.; Green, K.N. Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-beta pathology. Brain 2016, 139 Pt 4, 1265–1281. [Google Scholar] [CrossRef] [PubMed]
- Bekdash, R.A. Early Life Nutrition and Mental Health: The Role of DNA Methylation. Nutrients 2021, 13, 3111. [Google Scholar] [CrossRef] [PubMed]
- Iraola-Guzman, S.; Estivill, X.; Rabionet, R. DNA methylation in neurodegenerative disorders: A missing link between genome and environment? Clin. Genet. 2011, 80, 1–14. [Google Scholar] [CrossRef]
- Schwartzentruber, J.; Cooper, S.; Liu, J.Z.; Barrio-Hernandez, I.; Bello, E.; Kumasaka, N.; Young, A.M.H.; Franklin, R.J.M.; Johnson, T.; Estrada, K.; et al. Genome-wide meta-analysis, fine-mapping and integrative prioritization implicate new Alzheimer’s disease risk genes. Nat. Genet. 2021, 53, 392–402. [Google Scholar] [CrossRef]
- Jia, L.; Li, F.; Wei, C.; Zhu, M.; Qu, Q.; Qin, W.; Tang, Y.; Shen, L.; Wang, Y.; Shen, L.; et al. Prediction of Alzheimer’s disease using multi-variants from a Chinese genome-wide association study. Brain 2021, 144, 924–937. [Google Scholar] [CrossRef]
- Huang, T.; Shu, Y.; Cai, Y.D. Genetic differences among ethnic groups. BMC Genom. 2015, 16, 1093. [Google Scholar] [CrossRef]
- Nazarian, A.; Yashin, A.I.; Kulminski, A.M. Genome-wide analysis of genetic predisposition to Alzheimer’s disease and related sex disparities. Alzheimers Res. Ther. 2019, 11, 5. [Google Scholar] [CrossRef]
- Vila-Castelar, C.; Tariot, P.N.; Sink, K.M.; Clayton, D.; Langbaum, J.B.; Thomas, R.G.; Chen, Y.; Su, Y.; Chen, K.; Hu, N.; et al. Sex differences in cognitive resilience in preclinical autosomal-dominant Alzheimer’s disease carriers and non-carriers: Baseline findings from the API ADAD Colombia Trial. Alzheimers Dement. 2022, 18, 2272–2282. [Google Scholar] [CrossRef]
- Stern, Y. Cognitive reserve in ageing and Alzheimer’s disease. Lancet Neurol. 2012, 11, 1006–1012. [Google Scholar] [CrossRef]
- Escott-Price, V.; Hardy, J. Genome-wide association studies for Alzheimer’s disease: Bigger is not always better. Brain Commun. 2022, 4, fcac125. [Google Scholar] [CrossRef]
- Marioni, R.E.; Harris, S.E.; Zhang, Q.; McRae, A.F.; Hagenaars, S.P.; Hill, W.D.; Davies, G.; Ritchie, C.W.; Gale, C.R.; Starr, J.M.; et al. GWAS on family history of Alzheimer’s disease. Transl. Psychiatry 2018, 8, 99. [Google Scholar] [CrossRef]
- Gallagher, M.D.; Chen-Plotkin, A.S. The Post-GWAS Era: From Association to Function. Am. J. Hum. Genet. 2018, 102, 717–730. [Google Scholar] [CrossRef]
- Grupe, A.; Abraham, R.; Li, Y.; Rowland, C.; Hollingworth, P.; Morgan, A.; Jehu, L.; Segurado, R.; Stone, D.; Schadt, E.; et al. Evidence for novel susceptibility genes for late-onset Alzheimer’s disease from a genome-wide association study of putative functional variants. Hum. Mol. Genet. 2007, 16, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Bertram, L.; Lange, C.; Mullin, K.; Parkinson, M.; Hsiao, M.; Hogan, M.F.; Schjeide, B.M.; Hooli, B.; Divito, J.; Ionita, I.; et al. Genome-wide association analysis reveals putative Alzheimer’s disease susceptibility loci in addition to APOE. Am. J. Hum. Genet. 2008, 83, 623–632. [Google Scholar] [CrossRef]
- Lambert, J.C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B.; et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Seshadri, S.; Fitzpatrick, A.L.; Ikram, M.A.; DeStefano, A.L.; Gudnason, V.; Boada, M.; Bis, J.C.; Smith, A.V.; Carassquillo, M.M.; Lambert, J.C.; et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA 2010, 303, 1832–1840. [Google Scholar] [CrossRef] [PubMed]
- Feulner, T.M.; Laws, S.M.; Friedrich, P.; Wagenpfeil, S.; Wurst, S.H.; Riehle, C.; Kuhn, K.A.; Krawczak, M.; Schreiber, S.; Nikolaus, S.; et al. Examination of the current top candidate genes for AD in a genome-wide association study. Mol. Psychiatry 2010, 15, 756–766. [Google Scholar] [CrossRef]
- Wijsman, E.M.; Pankratz, N.D.; Choi, Y.; Rothstein, J.H.; Faber, K.M.; Cheng, R.; Lee, J.H.; Bird, T.D.; Bennett, D.A.; Diaz-Arrastia, R.; et al. Genome-wide association of familial late-onset Alzheimer’s disease replicates BIN1 and CLU and nominates CUGBP2 in interaction with APOE. PLoS Genet. 2011, 7, e1001308. [Google Scholar] [CrossRef]
- Logue, M.W.; Schu, M.; Vardarajan, B.N.; Buros, J.; Green, R.C.; Go, R.C.; Griffith, P.; Obisesan, T.O.; Shatz, R.; Borenstein, A.; et al. Multi-Institutional Research on Alzheimer Genetic Epidemiology Study, G. A comprehensive genetic association study of Alzheimer disease in African Americans. Arch. Neurol. 2011, 68, 1569–1579. [Google Scholar] [CrossRef]
- Lee, J.H.; Cheng, R.; Barral, S.; Reitz, C.; Medrano, M.; Lantigua, R.; Jimenez-Velazquez, I.Z.; Rogaeva, E.; St George-Hyslop, P.H.; Mayeux, R. Identification of novel loci for Alzheimer disease and replication of CLU, PICALM, and BIN1 in Caribbean Hispanic individuals. Arch. Neurol. 2011, 68, 320–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaj, P.; Paziewska, A.; Bik, W.; Dabrowska, M.; Baranowska-Bik, A.; Styczynska, M.; Chodakowska-Zebrowska, M.; Pfeffer-Baczuk, A.; Barcikowska, M.; Baranowska, B.; et al. Identification of a late onset Alzheimer’s disease candidate risk variant at 9q21.33 in Polish patients. J. Alzheimers Dis. 2012, 32, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Kamboh, M.I.; Demirci, F.Y.; Wang, X.; Minster, R.L.; Carrasquillo, M.M.; Pankratz, V.S.; Younkin, S.G.; Saykin, A.J.; Alzheimer’s Disease Neuroimaging, I.; Jun, G.; et al. Genome-wide association study of Alzheimer’s disease. Transl. Psychiatry 2012, 2, e117. [Google Scholar] [CrossRef]
- Cruchaga, C.; Karch, C.M.; Jin, S.C.; Benitez, B.A.; Cai, Y.; Guerreiro, R.; Harari, O.; Norton, J.; Budde, J.; Bertelsen, S.; et al. Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer’s disease. Nature 2014, 505, 550–554. [Google Scholar] [CrossRef]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Floudas, C.S.; Um, N.; Kamboh, M.I.; Barmada, M.M.; Visweswaran, S. Identifying genetic interactions associated with late-onset Alzheimer’s disease. BioData Min. 2014, 7, 35. [Google Scholar] [CrossRef]
- Jun, G.; Asai, H.; Zeldich, E.; Drapeau, E.; Chen, C.; Chung, J.; Park, J.H.; Kim, S.; Haroutunian, V.; Foroud, T.; et al. PLXNA4 is associated with Alzheimer disease and modulates tau phosphorylation. Ann. Neurol. 2014, 76, 379–392. [Google Scholar] [CrossRef]
- Tosto, G.; Fu, H.; Vardarajan, B.N.; Lee, J.H.; Cheng, R.; Reyes-Dumeyer, D.; Lantigua, R.; Medrano, M.; Jimenez-Velazquez, I.Z.; Elkind, M.S.; et al. F-box/LRR-repeat protein 7 is genetically associated with Alzheimer’s disease. Ann. Clin. Transl. Neurol. 2015, 2, 810–820. [Google Scholar] [CrossRef]
- Hirano, A.; Ohara, T.; Takahashi, A.; Aoki, M.; Fuyuno, Y.; Ashikawa, K.; Morihara, T.; Takeda, M.; Kamino, K.; Oshima, E.; et al. A genome-wide association study of late-onset Alzheimer’s disease in a Japanese population. Psychiatr. Genet. 2015, 25, 139–146. [Google Scholar] [CrossRef]
- Herold, C.; Hooli, B.V.; Mullin, K.; Liu, T.; Roehr, J.T.; Mattheisen, M.; Parrado, A.R.; Bertram, L.; Lange, C.; Tanzi, R.E. Family-based association analyses of imputed genotypes reveal genome-wide significant association of Alzheimer’s disease with OSBPL6, PTPRG, and PDCL3. Mol. Psychiatry 2016, 21, 1608–1612. [Google Scholar] [CrossRef]
- Hao, S.; Wang, R.; Zhang, Y.; Zhan, H. Prediction of Alzheimer’s Disease-Associated Genes by Integration of GWAS Summary Data and Expression Data. Front. Genet. 2018, 9, 653. [Google Scholar] [CrossRef]
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; van der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat. Genet. 2019, 51, 414–430. [Google Scholar] [CrossRef]
- Kunkle, B.W.; Schmidt, M.; Klein, H.U.; Naj, A.C.; Hamilton-Nelson, K.L.; Larson, E.B.; Evans, D.A.; De Jager, P.L.; Crane, P.K.; Buxbaum, J.D.; et al. Novel Alzheimer Disease Risk Loci and Pathways in African American Individuals Using the African Genome Resources Panel: A Meta-analysis. JAMA Neurol. 2021, 78, 102–113. [Google Scholar] [CrossRef]
- Shigemizu, D.; Asanomi, Y.; Akiyama, S.; Mitsumori, R.; Niida, S.; Ozaki, K. Whole-genome sequencing reveals novel ethnicity-specific rare variants associated with Alzheimer’s disease. Mol. Psychiatry 2022, 27, 2554–2562. [Google Scholar] [CrossRef]
- Yong, W.S.; Hsu, F.M.; Chen, P.Y. Profiling genome-wide DNA methylation. Epigenetics Chromatin 2016, 9, 26. [Google Scholar] [CrossRef]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef]
- Sabarwal, A.; Kumar, K.; Singh, R.P. Hazardous effects of chemical pesticides on human health-Cancer and other associated disorders. Environ. Toxicol. Pharmacol. 2018, 63, 103–114. [Google Scholar] [CrossRef]
- Gao, X.; Chen, Q.; Yao, H.; Tan, J.; Liu, Z.; Zhou, Y.; Zou, Z. Epigenetics in Alzheimer’s Disease. Front. Aging Neurosci. 2022, 14, 911635. [Google Scholar] [CrossRef]
- Tam, V.; Patel, N.; Turcotte, M.; Bosse, Y.; Pare, G.; Meyre, D. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [CrossRef]
- Wang, H.; Bennett, D.A.; De Jager, P.L.; Zhang, Q.Y.; Zhang, H.Y. Genome-wide epistasis analysis for Alzheimer’s disease and implications for genetic risk prediction. Alzheimers Res. Ther. 2021, 13, 55. [Google Scholar] [CrossRef]
- Vaz, M.; Silvestre, S. Alzheimer’s disease: Recent treatment strategies. Eur. J. Pharmacol. 2020, 887, 173554. [Google Scholar] [CrossRef]
- Panza, F.; Lozupone, M.; Seripa, D.; Imbimbo, B.P. Amyloid-beta immunotherapy for alzheimer disease: Is it now a long shot? Ann. Neurol. 2019, 85, 303–315. [Google Scholar] [CrossRef]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef]
- U.S. Food & Drug Administration. Available online: https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-alzheimers-disease-treatment (accessed on 10 January 2023).
- Nehls, M. Unified theory of Alzheimer’s disease (UTAD): Implications for prevention and curative therapy. J. Mol. Psychiatry 2016, 4, 3. [Google Scholar] [CrossRef]
- Gauthier, S.; Alam, J.; Fillit, H.; Iwatsubo, T.; Liu-Seifert, H.; Sabbagh, M.; Salloway, S.; Sampaio, C.; Sims, J.R.; Sperling, B.; et al. Combination Therapy for Alzheimer’s Disease: Perspectives of the EU/US CTAD Task Force. J. Prev. Alzheimers Dis. 2019, 6, 164–168. [Google Scholar] [CrossRef]
- Rosenberg, A.; Mangialasche, F.; Ngandu, T.; Solomon, A.; Kivipelto, M. Multidomain Interventions to Prevent Cognitive Impairment, Alzheimer’s Disease, and Dementia: From FINGER to World-Wide FINGERS. J. Prev. Alzheimers Dis. 2020, 7, 29–36. [Google Scholar] [CrossRef]
- Kivipelto, M.; Mangialasche, F.; Ngandu, T. Lifestyle interventions to prevent cognitive impairment, dementia and Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 653–666. [Google Scholar] [CrossRef]
- Ngandu, T.; Lehtisalo, J.; Solomon, A.; Levalahti, E.; Ahtiluoto, S.; Antikainen, R.; Backman, L.; Hanninen, T.; Jula, A.; Laatikainen, T.; et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): A randomised controlled trial. Lancet 2015, 385, 2255–2263. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andrade-Guerrero, J.; Santiago-Balmaseda, A.; Jeronimo-Aguilar, P.; Vargas-Rodríguez, I.; Cadena-Suárez, A.R.; Sánchez-Garibay, C.; Pozo-Molina, G.; Méndez-Catalá, C.F.; Cardenas-Aguayo, M.-d.-C.; Diaz-Cintra, S.; et al. Alzheimer’s Disease: An Updated Overview of Its Genetics. Int. J. Mol. Sci. 2023, 24, 3754. https://doi.org/10.3390/ijms24043754
Andrade-Guerrero J, Santiago-Balmaseda A, Jeronimo-Aguilar P, Vargas-Rodríguez I, Cadena-Suárez AR, Sánchez-Garibay C, Pozo-Molina G, Méndez-Catalá CF, Cardenas-Aguayo M-d-C, Diaz-Cintra S, et al. Alzheimer’s Disease: An Updated Overview of Its Genetics. International Journal of Molecular Sciences. 2023; 24(4):3754. https://doi.org/10.3390/ijms24043754
Chicago/Turabian StyleAndrade-Guerrero, Jesús, Alberto Santiago-Balmaseda, Paola Jeronimo-Aguilar, Isaac Vargas-Rodríguez, Ana Ruth Cadena-Suárez, Carlos Sánchez-Garibay, Glustein Pozo-Molina, Claudia Fabiola Méndez-Catalá, Maria-del-Carmen Cardenas-Aguayo, Sofía Diaz-Cintra, and et al. 2023. "Alzheimer’s Disease: An Updated Overview of Its Genetics" International Journal of Molecular Sciences 24, no. 4: 3754. https://doi.org/10.3390/ijms24043754