
Placental Mesenchymal Stem Cells Alleviate Podocyte Injury in Diabetic Kidney Disease by Modulating Mitophagy via the SIRT1-PGC-1alpha-TFAM Pathway
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
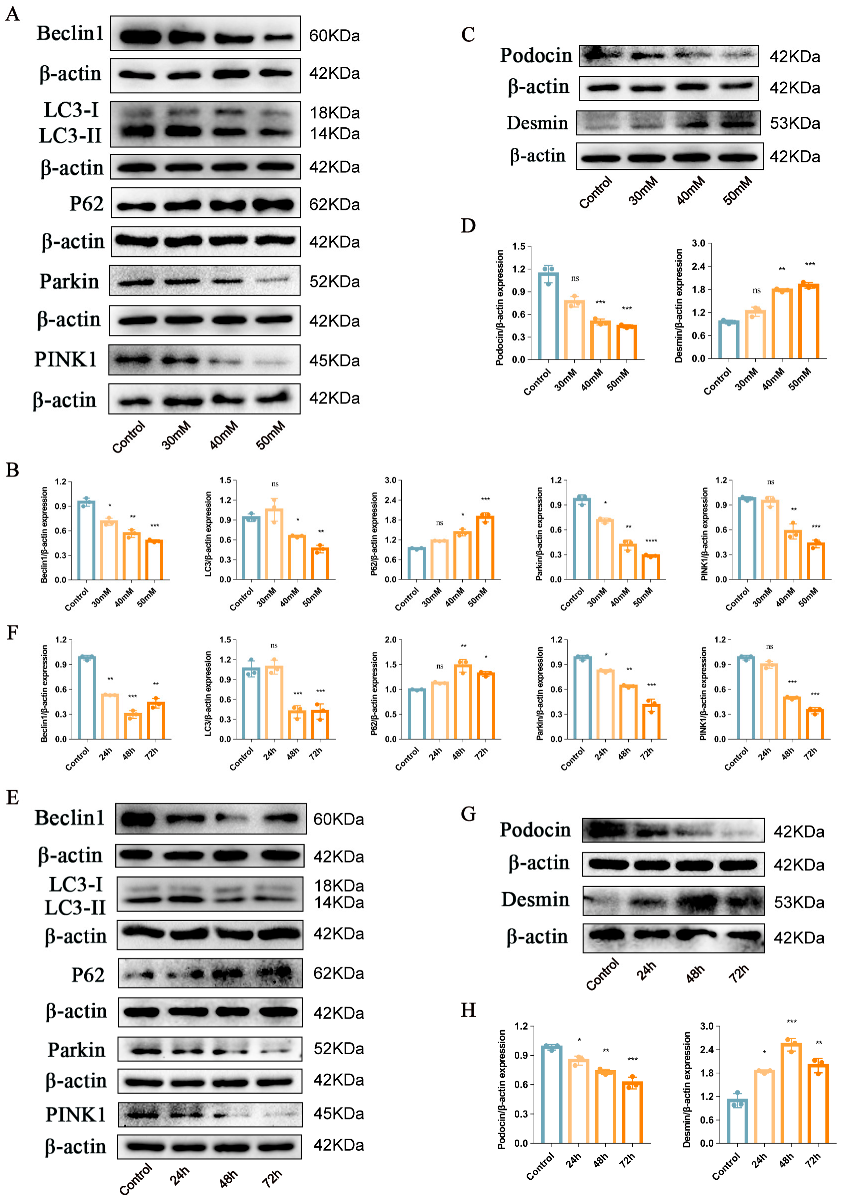
2.1. Podocyte Injury and PINK1/Parkin-Mediated Mitophagy Inhibition Induced by High Glucose in the Mouse Podocyte Cell Line
2.2. P-MSCs Attenuated HG-Induced Podocyte Injury and PINK1/Parkin-Mediated Mitophagy Inhibition
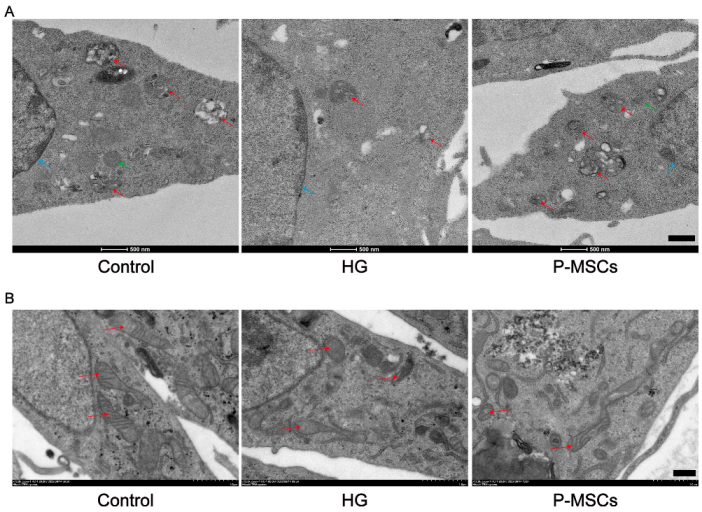
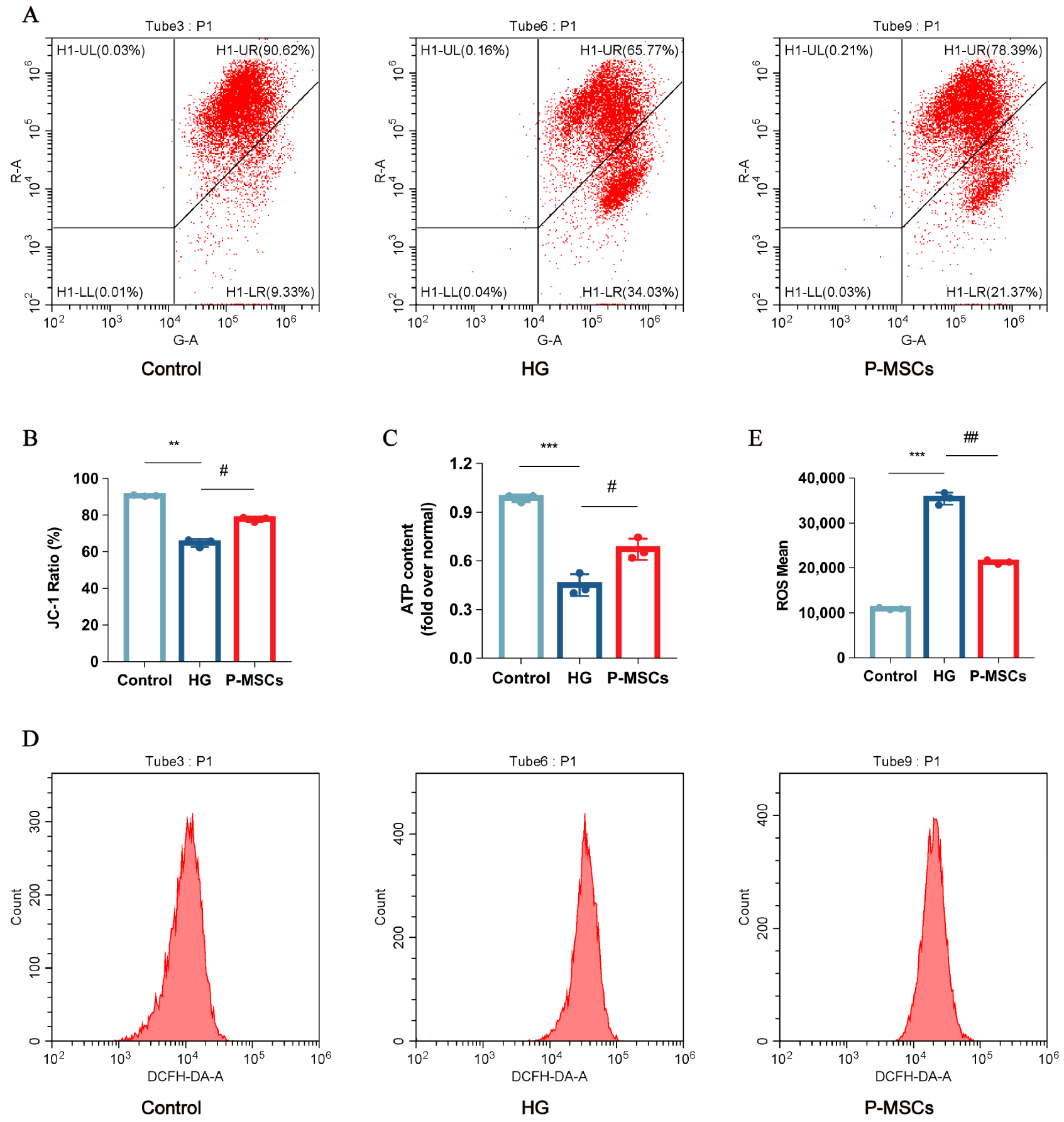
2.3. P-MSCs Extenuated HG-Mediated Mitochondrial Dysfunction and Reactive Oxygen Species Accumulation
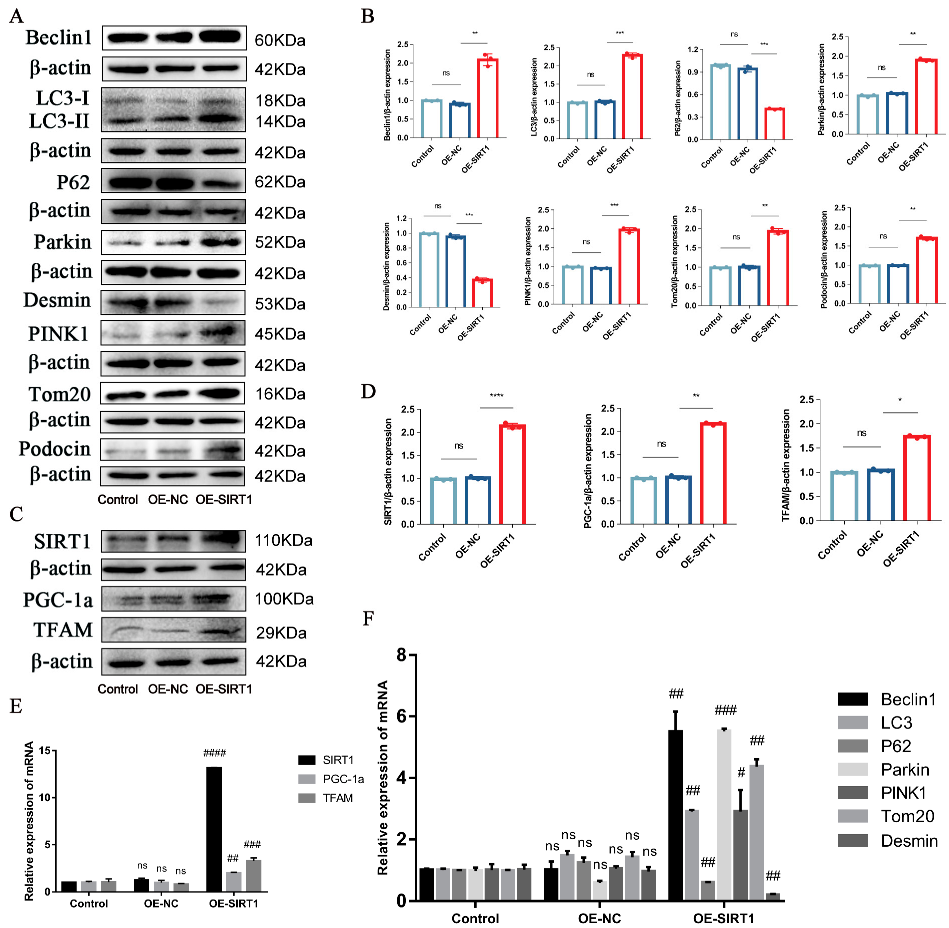
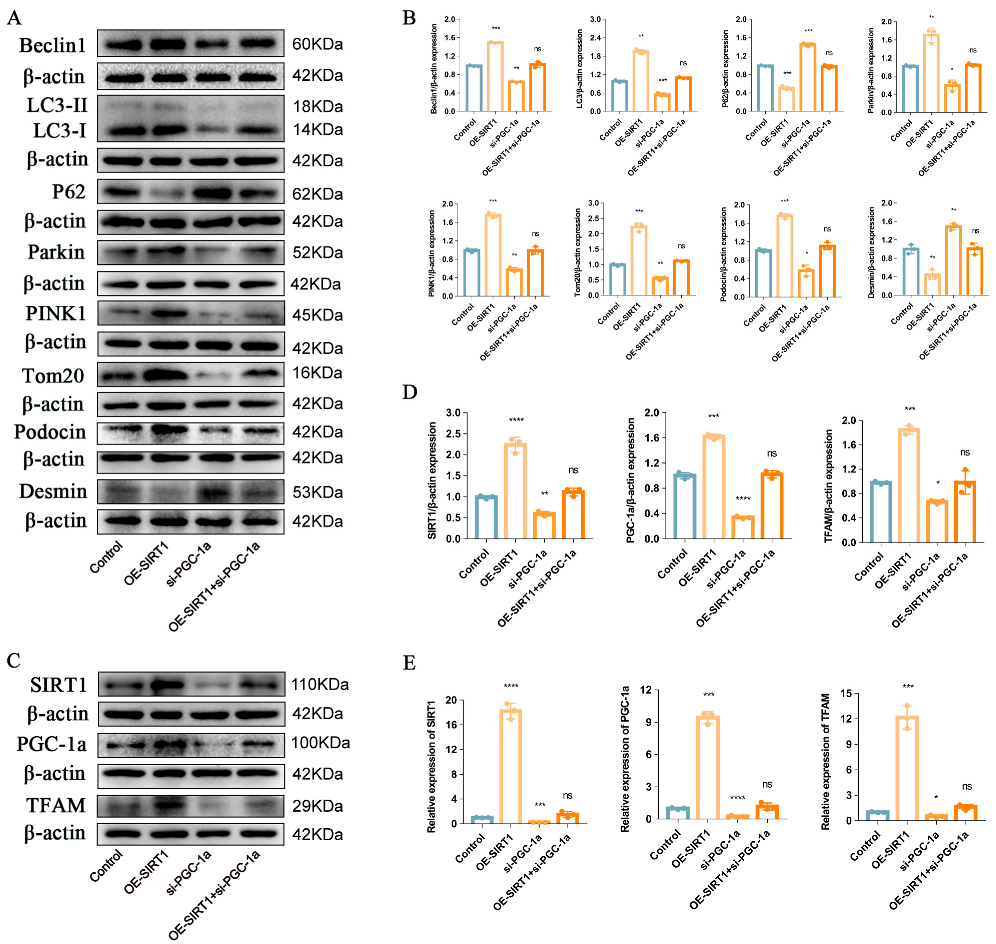
2.4. P-MSCs Alleviated HG-Induced Podocyte Injury and PINK1/Parkin-Mediated Mitophagy Inhibition by Activating the SIRT1-PGC-1α-TFAM Signaling Pathway
2.5. P-MSCs Ameliorated Streptozotocin-Induced Podocyte Injury and PINK1/Parkin-Mediated Mitophagy in DKD Rats
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Culture
4.3. Transfections of Plasmids and Small Interfering RNAs
4.4. WB Assay
4.5. RT-PCR
4.6. Immunofluorescence
4.7. ΔΨm, ATP Content, and ROS Determination
4.8. Transmission Electron Microscopy
4.9. Experimental Animals
4.10. Immunohistochemistry
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DKD | diabetic kidney disease |
| MSCs | mesenchymal stem cells |
| P-MSCs | placenta derived mesenchymal stem cells |
| SIRT1 | silencing information regulator 2 related enzyme 1 |
| PINK1 | phosphatase and tensin homolog–induced kinase |
| PGC-1α | peroxisome proliferator-activated receptor γ coactivator-1alpha |
| TFAM | transcription factor A, mitochondrial |
| FBS | fetal bovine serum |
| β-actin | beta actin |
| HG | high glucose |
| NC | negative control |
| siRNAs | small interfering RNAs |
| PBS | phosphate-buffered saline |
| PBST | phosphate-buffered saline with Tween 20 |
| WB | Western blot |
| RT-PCR | reverse transcription–polymerase chain reaction |
| ΔΨm | mitochondrial membrane potential |
| ROS | reactive oxygen species |
| STZ | streptozotocin |
References
- Lin, Y.C.; Chang, Y.H.; Yang, S.Y.; Wu, K.D.; Chu, T.S. Update of pathophysiology and management of diabetic kidney disease. J. Formos. Med. Assoc. 2018, 117, 662–675. [Google Scholar] [CrossRef] [PubMed]
- Gregg, E.W.; Li, Y.; Wang, J.; Burrows, N.R.; Ali, M.K.; Rolka, D.; Williams, D.E.; Geiss, L. Changes in diabetes-related complications in the United States, 1990–2010. N. Engl. J. Med. 2014, 370, 1514–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alicic, R.Z.; Rooney, M.T.; Tuttle, K.R. Diabetic Kidney Disease: Challenges, Progress, and Possibilities. Clin. J. Am. Soc. Nephrol. 2017, 12, 2032–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gnudi, L.; Coward, R.J.M.; Long, D.A. Diabetic Nephropathy: Perspective on Novel Molecular Mechanisms. Trends Endocrinol. Metab. 2016, 27, 820–830. [Google Scholar] [CrossRef] [Green Version]
- Lennon, R.; Randles, M.J.; Humphries, M.J. The importance of podocyte adhesion for a healthy glomerulus. Front. Endocrinol. 2014, 5, 160. [Google Scholar] [CrossRef] [Green Version]
- Maier, J.I.; Rogg, M.; Helmstädter, M.; Sammarco, A.; Schilling, O.; Sabass, B.; Miner, J.H.; Dengjel, J.; Walz, G.; Werner, M.; et al. EPB41L5 controls podocyte extracellular matrix assembly by adhesome-dependent force transmission. Cell Rep. 2021, 34, 108883. [Google Scholar] [CrossRef]
- Susztak, K.; Raff, A.C.; Schiffer, M.; Böttinger, E.P. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes 2006, 55, 225–233. [Google Scholar] [CrossRef]
- Chen, C.; Zhou, M.; Ge, Y.; Wang, X. SIRT1 and aging related signaling pathways. Mech. Ageing Dev. 2020, 187, 111215. [Google Scholar] [CrossRef]
- Zhong, Y.; Lee, K.; He, J.C. SIRT1 Is a Potential Drug Target for Treatment of Diabetic Kidney Disease. Front. Endocrinol. 2018, 9, 624. [Google Scholar] [CrossRef]
- Hasegawa, K.; Wakino, S.; Simic, P.; Sakamaki, Y.; Minakuchi, H.; Fujimura, K.; Hosoya, K.; Komatsu, M.; Kaneko, Y.; Kanda, T.; et al. Renal tubular Sirt1 attenuates diabetic albuminuria by epigenetically suppressing Claudin-1 overexpression in podocytes. Nat. Med. 2013, 19, 1496–1504. [Google Scholar] [CrossRef] [Green Version]
- Tagawa, A.; Yasuda, M.; Kume, S.; Yamahara, K.; Nakazawa, J.; Chin-Kanasaki, M.; Araki, H.; Araki, S.; Koya, D.; Asanuma, K.; et al. Impaired Podocyte Autophagy Exacerbates Proteinuria in Diabetic Nephropathy. Diabetes 2016, 65, 755–767. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Feng, J.; Li, X.; Wu, D.; Wang, Q.; Li, S.; Shi, C. Mitophagy in Diabetic Kidney Disease. Front. Cell Dev. Biol. 2021, 9, 778011. [Google Scholar] [CrossRef]
- Eiyama, A.; Okamoto, K. PINK1/Parkin-mediated mitophagy in mammalian cells. Curr. Opin. Cell Biol. 2015, 33, 95–101. [Google Scholar] [CrossRef]
- Ivankovic, D.; Chau, K.Y.; Schapira, A.H.; Gegg, M.E. Mitochondrial and lysosomal biogenesis are activated following PINK1/parkin-mediated mitophagy. J. Neurochem. 2016, 136, 388–402. [Google Scholar] [CrossRef] [Green Version]
- Qian, X.; Li, X.; Shi, Z.; Bai, X.; Xia, Y.; Zheng, Y.; Xu, D.; Chen, F.; You, Y.; Fang, J.; et al. KDM3A Senses Oxygen Availability to Regulate PGC-1α-Mediated Mitochondrial Biogenesis. Mol. Cell 2019, 76, 885–895. [Google Scholar] [CrossRef]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef]
- Saxena, S.; Mathur, A.; Kakkar, P. Critical role of mitochondrial dysfunction and impaired mitophagy in diabetic nephropathy. J. Cell. Physiol. 2019, 234, 19223–19236. [Google Scholar] [CrossRef]
- Yue, Y.; Yeh, J.N.; Chiang, J.Y.; Sung, P.H.; Chen, Y.L.; Liu, F.; Yip, H.K. Intrarenal arterial administration of human umbilical cord-derived mesenchymal stem cells effectively preserved the residual renal function of diabetic kidney disease in rat. Stem Cell Res. Ther. 2022, 13, 186. [Google Scholar] [CrossRef]
- Lin, W.; Li, H.Y.; Yang, Q.; Chen, G.; Lin, S.; Liao, C.; Zhou, T. Administration of mesenchymal stem cells in diabetic kidney disease: A systematic review and meta-analysis. Stem Cell Res. Ther. 2021, 12, 43. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Zhang, Q.; Wang, W.; Liu, Q.; Liu, S.; Song, Y.; Wang, X.; Zhang, Y.; Li, S.; et al. Human umbilical cord mesenchymal stem cells attenuate podocyte injury under high glucose via TLR2 and TLR4 signaling. Diabetes Res. Clin. Pract. 2021, 173, 108702. [Google Scholar] [CrossRef]
- Li, F.X.; Lin, X.; Xu, F.; Shan, S.K.; Guo, B.; Lei, L.M.; Zheng, M.H.; Wang, Y.; Xu, Q.S.; Yuan, L.Q. The Role of Mesenchymal Stromal Cells-Derived Small Extracellular Vesicles in Diabetes and Its Chronic Complications. Front. Endocrinol. 2021, 12, 780974. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, J.S.; Gonçalves, R.G.J.; Vasques, J.F.; Rocha, B.S.; Nascimento-Carlos, B.; Montagnoli, T.L.; Mendez-Otero, R.; de Sá, M.P.L.; Zapata-Sudo, G. Mesenchymal Stem Cell Therapy in Diabetic Cardiomyopathy. Cells 2022, 11, 240. [Google Scholar] [CrossRef] [PubMed]
- Na, K.R.; Jeong, J.Y.; Shin, J.A.; Chang, Y.K.; Suh, K.S.; Lee, K.W.; Choi, D.E. Mitochondrial Dysfunction in Podocytes Caused by CRIF1 Deficiency Leads to Progressive Albuminuria and Glomerular Sclerosis in Mice. Int. J. Mol. Sci. 2021, 22, 4827. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ge, X.; Li, X.; He, J.; Wei, X.; Du, J.; Sun, J.; Li, X.; Xun, Z.; Liu, W.; et al. High-fat diet promotes renal injury by inducing oxidative stress and mitochondrial dysfunction. Cell Death Dis. 2020, 11, 914. [Google Scholar] [CrossRef]
- Yao, J.; Wang, J.; Xu, Y.; Guo, Q.; Sun, Y.; Liu, J.; Li, S.; Guo, Y.; Wei, L. CDK9 inhibition blocks the initiation of PINK1-PRKN-mediated mitophagy by regulating the SIRT1-FOXO3-BNIP3 axis and enhances the therapeutic effects involving mitochondrial dysfunction in hepatocellular carcinoma. Autophagy 2022, 18, 1879–1897. [Google Scholar] [CrossRef]
- Yao, Z.Q.; Zhang, X.; Zhen, Y.; He, X.Y.; Zhao, S.; Li, X.F.; Yang, B.; Gao, F.; Guo, F.Y.; Fu, L.; et al. A novel small-molecule activator of Sirtuin-1 induces autophagic cell death/mitophagy as a potential therapeutic strategy in glioblastoma. Cell Death Dis. 2018, 9, 767. [Google Scholar] [CrossRef] [Green Version]
- Fang, E.F.; Scheibye-Knudsen, M.; Brace, L.E.; Kassahun, H.; SenGupta, T.; Nilsen, H.; Mitchell, J.R.; Croteau, D.L.; Bohr, V.A. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell 2014, 157, 882–896. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekaran, K.; Anjaneyulu, M.; Choi, J.; Kumar, P.; Salimian, M.; Ho, C.Y.; Russell, J.W. Role of mitochondria in diabetic peripheral neuropathy: Influencing the NAD(+)-dependent SIRT1-PGC-1α-TFAM pathway. Int. Rev. Neurobiol. 2019, 145, 177–209. [Google Scholar]
- Wang, J. The Repair Effect and Mechanism of Human Placenta Derived Mesenchymal Stem Cells on Immunoinflammatory Injury in Diabetic Kidney Disease. Ph.D. Thesis, Nanchang University, Nanchang, China, 2021. [Google Scholar]
- Nagata, M. Podocyte injury and its consequences. Kidney Int. 2016, 89, 1221–1230. [Google Scholar] [CrossRef]
- Dai, H.; Liu, Q.; Liu, B. Research Progress on Mechanism of Podocyte Depletion in Diabetic Nephropathy. J. Diabetes Res. 2017, 2017, 2615286. [Google Scholar] [CrossRef] [Green Version]
- Funk, J.; Ott, V.; Herrmann, A.; Rapp, W.; Raab, S.; Riboulet, W.; Vandjour, A.; Hainaut, E.; Benardeau, A.; Singer, T.; et al. Semiautomated quantitative image analysis of glomerular immunohistochemistry markers desmin, vimentin, podocin, synaptopodin and WT-1 in acute and chronic rat kidney disease models. Histochem. Cell Biol. 2016, 145, 315–326. [Google Scholar] [CrossRef]
- Matsusaka, T.; Sandgren, E.; Shintani, A.; Kon, V.; Pastan, I.; Fogo, A.B.; Ichikawa, I. Podocyte injury damages other podocytes. J. Am. Soc. Nephrol. 2011, 22, 1275–1285. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Yan, Y.; Hu, Q.; Zhang, X. LncRNA MALAT1/microRNA let-7f/KLF5 axis regulates podocyte injury in diabetic nephropathy. Life Sci. 2021, 266, 118794. [Google Scholar] [CrossRef]
- Higgins, G.C.; Coughlan, M.T. Mitochondrial dysfunction and mitophagy: The beginning and end to diabetic nephropathy? Br. J. Pharmacol. 2014, 171, 1917–1942. [Google Scholar] [CrossRef] [Green Version]
- Yi, X.; Yan, W.; Guo, T.; Liu, N.; Wang, Z.; Shang, J.; Wei, X.; Cui, X.; Sun, Y.; Ren, S.; et al. Erythropoietin Mitigates Diabetic Nephropathy by Restoring PINK1/Parkin-Mediated Mitophagy. Front. Pharmacol. 2022, 13, 883057. [Google Scholar] [CrossRef]
- Liu, X.; Lu, J.; Liu, S.; Huang, D.; Chen, M.; Xiong, G.; Li, S. Huangqi-Danshen decoction alleviates diabetic nephropathy in db/db mice by inhibiting PINK1/Parkin-mediated mitophagy. Am. J. Transl. Res. 2020, 12, 989–998. [Google Scholar]
- Onishi, M.; Yamano, K.; Sato, M.; Matsuda, N.; Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021, 40, e104705. [Google Scholar] [CrossRef]
- Sulkshane, P.; Ram, J.; Thakur, A.; Reis, N.; Kleifeld, O.; Glickman, M.H. Ubiquitination and receptor-mediated mitophagy converge to eliminate oxidation-damaged mitochondria during hypoxia. Redox Biol. 2021, 45, 102047. [Google Scholar] [CrossRef]
- Lv, M.; Zhang, S.; Jiang, B.; Cao, S.; Dong, Y.; Cao, L.; Guo, S. Adipose-derived stem cells regulate metabolic homeostasis and delay aging by promoting mitophagy. FASEB J. 2021, 35, e21709. [Google Scholar] [CrossRef]
- Hu, Z.; Yuan, Y.; Zhang, X.; Lu, Y.; Dong, N.; Jiang, X.; Xu, J.; Zheng, D. Human Umbilical Cord Mesenchymal Stem Cell-Derived Exosomes Attenuate Oxygen-Glucose Deprivation/Reperfusion-Induced Microglial Pyroptosis by Promoting FOXO3a-Dependent Mitophagy. Oxidative Med. Cell. Longev. 2021, 2021, 6219715. [Google Scholar] [CrossRef]
- Lee, S.E.; Jang, J.E.; Kim, H.S.; Jung, M.K.; Ko, M.S.; Kim, M.O.; Park, H.S.; Oh, W.; Choi, S.J.; Jin, H.J.; et al. Mesenchymal stem cells prevent the progression of diabetic nephropathy by improving mitochondrial function in tubular epithelial cells. Exp. Mol. Med. 2019, 51, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Liu, Z.; Shu, M.; Zhang, L.; Xia, W.; Tang, L.; Li, J.; Huang, B.; Li, H. Human placental mesenchymal stem cells ameliorate chemotherapy-induced damage in the testis by reducing apoptosis/oxidative stress and promoting autophagy. Stem Cell Res. Ther. 2021, 12, 199. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xu, M.; Ma, C.; Lyu, Y.; Ma, X.; Ma, X. Exosomes derived from human placental mesenchymal stem cells reduce human lung microvascular endothelial cell injury induced by lipopolysaccharide via enhancing autophagy. Chin. J. Cell. Mol. Immunol. 2021, 37, 225–232. [Google Scholar]
- Seok, J.; Jun, S.; Cho, J.; Park, S.; Lee, J.O.; Kim, G.J. Human placenta-derived mesenchymal stem cells induce trophoblast invasion via dynamic effects on mitochondrial function. J. Cell. Physiol. 2021, 236, 6678–6690. [Google Scholar] [CrossRef]
- Gillmore, T.; Farrell, A.; Alahari, S.; Sallais, J.; Kurt, M.; Park, C.; Ausman, J.; Litvack, M.; Post, M.; Caniggia, I. Dichotomy in hypoxia-induced mitochondrial fission in placental mesenchymal cells during development and preeclampsia: Consequences for trophoblast mitochondrial homeostasis. Cell Death Dis. 2022, 13, 191. [Google Scholar] [CrossRef]
- Wang, W.; Sun, W.; Cheng, Y.; Xu, Z.; Cai, L. Role of sirtuin-1 in diabetic nephropathy. J. Mol. Med. 2019, 97, 291–309. [Google Scholar] [CrossRef] [Green Version]
- Sarmah, D.; Datta, A.; Kaur, H.; Kalia, K.; Borah, A.; Rodriguez, A.M.; Yavagal, D.R.; Bhattacharya, P. Sirtuin-1-Mediated NF-κB Pathway Modulation to Mitigate Inflammasome Signaling and Cellular Apoptosis is One of the Neuroprotective Effects of Intra-arterial Mesenchymal Stem Cell Therapy Following Ischemic Stroke. Stem Cell Rev. Rep. 2022, 18, 821–838. [Google Scholar] [CrossRef]
- Audzeyenka, I.; Rachubik, P.; Typiak, M.; Kulesza, T.; Topolewska, A.; Rogacka, D.; Angielski, S.; Saleem, M.A.; Piwkowska, A. Hyperglycemia alters mitochondrial respiration efficiency and mitophagy in human podocytes. Exp. Cell Res. 2021, 407, 112758. [Google Scholar] [CrossRef]
- Yuan, Y.; Yuan, L.; Li, L.; Liu, F.; Liu, J.; Chen, Y.; Cheng, J.; Lu, Y. Mitochondrial transfer from mesenchymal stem cells to macrophages restricts inflammation and alleviates kidney injury in diabetic nephropathy mice via PGC-1α activation. Stem Cells 2021, 39, 913–928. [Google Scholar] [CrossRef]
- Zhao, M.; Wang, Y.; Li, L.; Liu, S.; Wang, C.; Yuan, Y.; Yang, G.; Chen, Y.; Cheng, J.; Lu, Y.; et al. Mitochondrial ROS promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting TFAM-mediated mtDNA maintenance. Theranostics 2021, 11, 1845–1863. [Google Scholar] [CrossRef]
- Liang, D.; Zhuo, Y.; Guo, Z.; He, L.; Wang, X.; He, Y.; Li, L.; Dai, H. SIRT1/PGC-1 pathway activation triggers autophagy/mitophagy and attenuates oxidative damage in intestinal epithelial cells. Biochimie 2020, 170, 10–20. [Google Scholar] [CrossRef]
- Fierabracci, A.; Lazzari, L.; Muraca, M.; Parolini, O. How far are we from the clinical use of placental-derived mesenchymal stem cells? Expert. Opin. Biol. Ther. 2015, 15, 613–617. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.; Tang, Y.; Hu, K.; Jiao, W.; Ying, L.; Zhu, L.; Liu, J.; Xu, J. Three-week topical treatment with placenta-derived mesenchymal stem cells hydrogel in a patient with diabetic foot ulcer: A case report. Medicine 2017, 96, e9212. [Google Scholar] [CrossRef]
- Liang, L.; Li, Z.; Ma, T.; Han, Z.; Du, W.; Geng, J.; Jia, H.; Zhao, M.; Wang, J.; Zhang, B.; et al. Transplantation of Human Placenta-Derived Mesenchymal Stem Cells Alleviates Critical Limb Ischemia in Diabetic Nude Rats. Cell Transplant. 2017, 26, 45–61. [Google Scholar] [CrossRef] [Green Version]
- Li, J.J.; Ma, F.X.; Wang, Y.W.; Chen, F.; Lu, S.H.; Chi, Y.; Du, W.J.; Song, B.Q.; Hu, L.D.; Chen, H.; et al. Knockdown of IL-8 Provoked Premature Senescence of Placenta-Derived Mesenchymal Stem Cells. Stem Cells Dev. 2017, 26, 912–931. [Google Scholar] [CrossRef]
- Wang, J.; Zeng, X.X.; Cai, W.; Han, Z.B.; Zhu, L.Y.; Liu, J.Y.; Xu, J.X. Safety and Efficacy of Placenta-Derived Mesenchymal Stem Cell Treatment for Diabetic Patients with Critical Limb Ischemia: A Pilot Study. Exp. Clin. Endocrinol. Diabetes 2021, 129, 542–548. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, X.; Wang, J.; Li, R.; Huang, M.; Yue, G.; Guan, L.; Deng, Y.; Cai, W.; Xu, J. Placental Mesenchymal Stem Cells Alleviate Podocyte Injury in Diabetic Kidney Disease by Modulating Mitophagy via the SIRT1-PGC-1alpha-TFAM Pathway. Int. J. Mol. Sci. 2023, 24, 4696. https://doi.org/10.3390/ijms24054696
Han X, Wang J, Li R, Huang M, Yue G, Guan L, Deng Y, Cai W, Xu J. Placental Mesenchymal Stem Cells Alleviate Podocyte Injury in Diabetic Kidney Disease by Modulating Mitophagy via the SIRT1-PGC-1alpha-TFAM Pathway. International Journal of Molecular Sciences. 2023; 24(5):4696. https://doi.org/10.3390/ijms24054696
Chicago/Turabian StyleHan, Xiudan, Jiao Wang, Ruilin Li, Meiling Huang, Guanru Yue, Lulu Guan, Yuanyuan Deng, Wei Cai, and Jixiong Xu. 2023. "Placental Mesenchymal Stem Cells Alleviate Podocyte Injury in Diabetic Kidney Disease by Modulating Mitophagy via the SIRT1-PGC-1alpha-TFAM Pathway" International Journal of Molecular Sciences 24, no. 5: 4696. https://doi.org/10.3390/ijms24054696