
Theoretical Study of Hydroxylation of α- and β-Pinene by a Cytochrome P450 Monooxygenase Model
 ,
,  ,
,  ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Alpha- and Beta-Pinene Bare Systems
2.2. CYP Monooxygenase Model
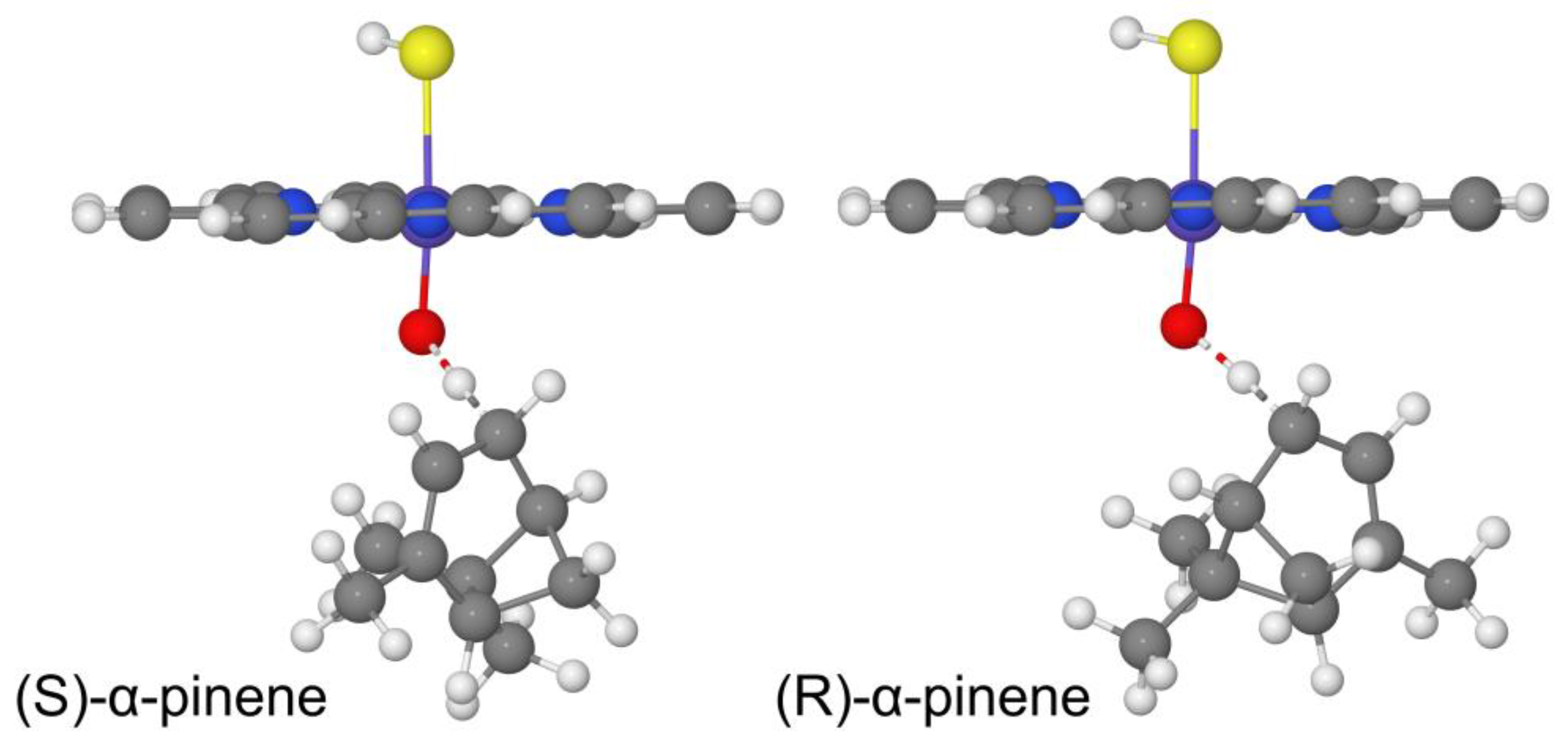
2.3. R and S Enantiomers Cis/Trans Reaction
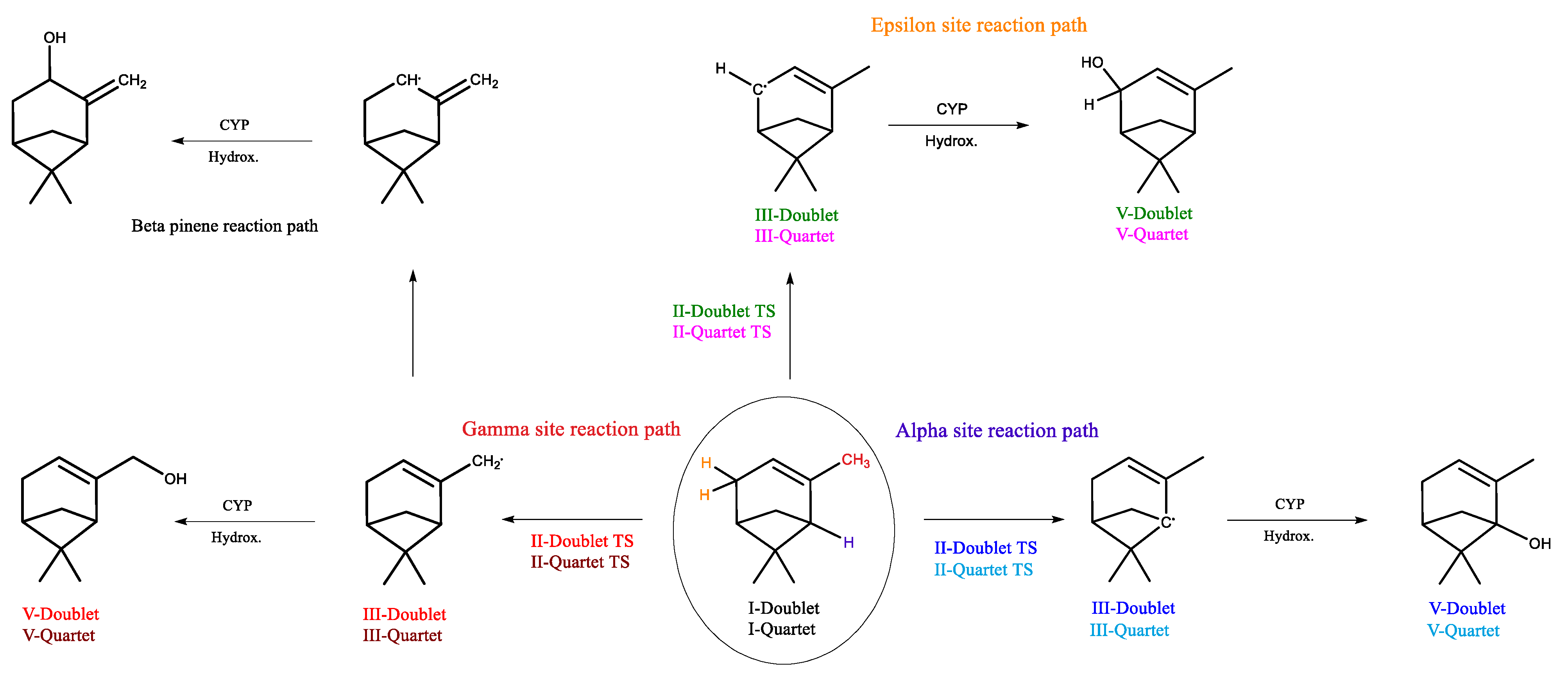
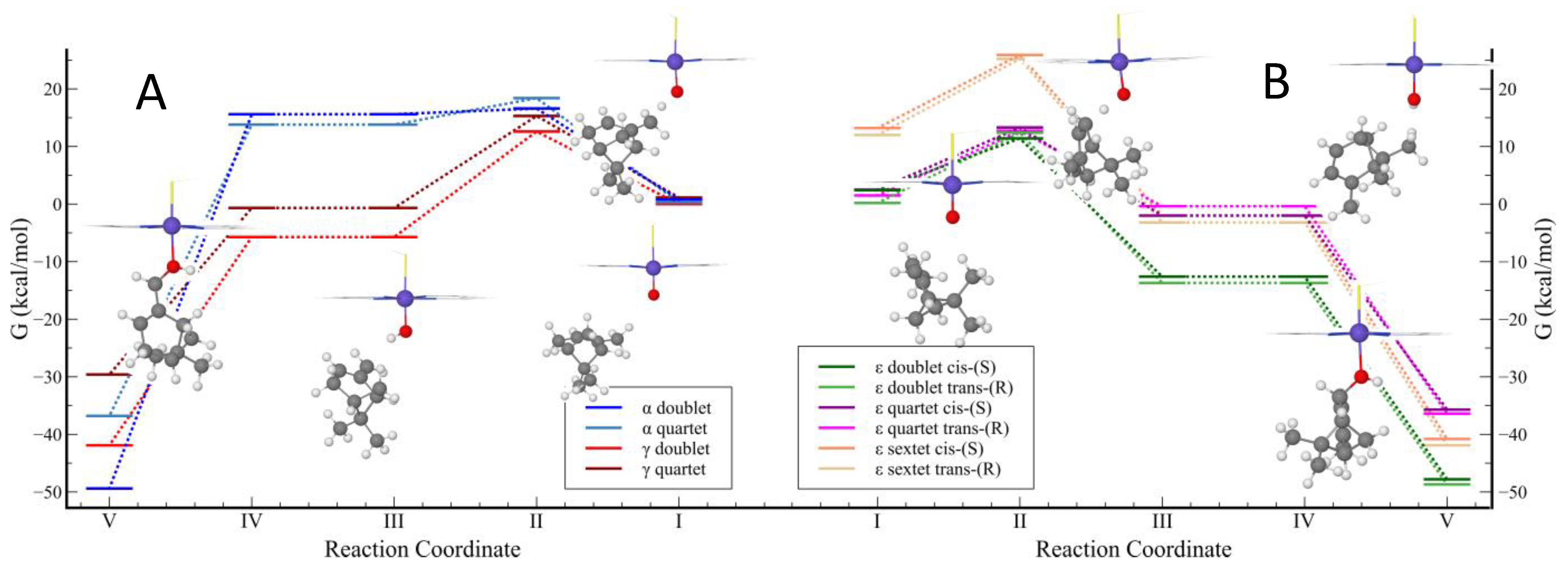
2.4. CYP-Catalyzed Hydroxylation of β-Pinene
2.5. CYP-Catalyzed Hydroxylation of α-Pinene
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Munro, A.W.; McLean, K.J.; Grant, J.L.; Makris, T.M. Structure and Function of the Cytochrome P450 Peroxygenase Enzymes. Biochem. Soc. Trans. 2018, 46, 183–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaik, S.; Cohen, S.; Wang, Y.; Chen, H.; Kumar, D.; Thiel, W. P450 Enzymes: Their Structure, Reactivity, and Selectivity—Modeled by QM/MM Calculations. Chem. Rev. 2010, 110, 949–1017. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, H.; Wang, Y.; Yang, C.; Yang, L.; Han, K. Theoretical Study of the Mechanism of Acetaldehyde Hydroxylation by Compound I of CYP2E1. J. Phys. Chem. B 2006, 110, 6154–6159. [Google Scholar] [CrossRef]
- Krámos, B.; Oláh, J. The Mechanism of Human Aromatase (CYP 19A1) Revisited: DFT and QM/MM Calculations Support a Compound I-Mediated Pathway for the Aromatization Process. Struct. Chem. 2015, 26, 279–300. [Google Scholar] [CrossRef] [Green Version]
- Guengerich, F.P. Mechanisms of Cytochrome P450-Catalyzed Oxidations. ACS Catal. 2018, 8, 10964–10976. [Google Scholar] [CrossRef]
- Guengerich, F.P. Common and Uncommon Cytochrome P450 Reactions Related to Metabolism and Chemical Toxicity. Chem. Res. Toxicol. 2001, 14, 611–650. [Google Scholar] [CrossRef]
- Sánchez-Velandia, J.E.; Mejía, S.M.; Villa, A.L. Reaction Mechanism of the Isomerization of Monoterpene Epoxides with Fe3+ as Active Catalytic Specie: A Computational Approach. J. Phys. Chem. 2020, 124, 3761–3769. [Google Scholar] [CrossRef]
- Shaya, J.; Correia, G.; Heinrich, B.; Ribierre, J.-C.; Polychronopoulou, K.; Mager, L.; Méry, S. Functionalization of Biphenylcarbazole (CBP) with Siloxane-Hybrid Chains for Solvent-Free Liquid Materials. Molecules 2021, 27, 89. [Google Scholar] [CrossRef]
- Zhang, X.; Peng, Y.; Zhao, J.; Li, Q.; Yu, X.; Acevedo-Rocha, C.G.; Li, A. Bacterial Cytochrome P450-Catalyzed Regio- and Stereoselective Steroid Hydroxylation Enabled by Directed Evolution and Rational Design. Bioresour. Bioprocess. 2020, 7, 2. [Google Scholar] [CrossRef]
- Yang, S.; DeMars, M.D.; Grandner, J.M.; Olson, N.M.; Anzai, Y.; Sherman, D.H.; Houk, K.N. Computational-Based Mechanistic Study and Engineering of Cytochrome P450 MycG for Selective Oxidation of 16-Membered Macrolide Antibiotics. J. Am. Chem. Soc. 2020, 142, 17981–17988. [Google Scholar] [CrossRef]
- Bathelt, C.M.; Ridder, L.; Mulholland, A.J.; Harvey, J.N. Aromatic Hydroxylation by Cytochrome P450: Model Calculations of Mechanism and Substituent Effects. J. Am. Chem. Soc. 2003, 125, 15004–15005. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Groves, J.T. Beyond Ferryl-Mediated Hydroxylation: 40 Years of the Rebound Mechanism and C–H Activation. J. Biol. Inorg. Chem. 2017, 22, 185–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groves, J.T. Models and Mechanisms of Cytochrome P450 Action. In Cytochrome P450, Structure, Mechanism, and Biochemistry; Springer: New York, NY, USA, 2005; pp. 1–43. [Google Scholar] [CrossRef]
- Shimizu, D.; Osuka, A. Porphyrinoids as a Platform of Stable Radicals. Chem. Sci. 2018, 9, 1408–1423. [Google Scholar] [CrossRef] [Green Version]
- Golets, M.; Ajaikumar, S.; Mikkola, J.-P. Catalytic Upgrading of Extractives to Chemicals: Monoterpenes to “EXICALS”. Chem. Rev. 2015, 115, 3141–3169. [Google Scholar] [CrossRef]
- Shehayeb, S.; Zaher, S.; Ghannam, L.; Srour, H.; Kanj, A.; Shaya, J.; Karamé, I. Handbook of Greener Synthesis of Nanomaterials and Compounds; Elsevier: Amsterdam, The Netherlands, 2021; pp. 807–860. [Google Scholar]
- Matsuo, A.L.; Figueiredo, C.R.; Arruda, D.C.; Pereira, F.V.; Scutti, J.A.B.; Massaoka, M.H.; Travassos, L.R.; Sartorelli, P.; Lago, J.H.G. α-Pinene Isolated from Schinus Terebinthifolius Raddi (Anacardiaceae) Induces Apoptosis and Confers Antimetastatic Protection in a Melanoma Model. Biochem. Biophys. Res. Commun. 2011, 411, 449–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salehi, B.; Upadhyay, S.; Orhan, I.E.; Jugran, A.K.; Jayaweera, S.L.D.; Dias, D.A.; Sharopov, F.; Taheri, Y.; Martins, N.; Baghalpour, N.; et al. Therapeutic Potential of α- and β-Pinene: A Miracle Gift of Nature. Biomolecules 2019, 9, 738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Guo, S.; Liu, X.; Gao, X. Synergistic Antitumor Effect of α-Pinene and β-Pinene with Paclitaxel against Non-Small-Cell Lung Carcinoma (NSCLC). Drug Res. 2014, 65, 214–218. [Google Scholar] [CrossRef] [PubMed]
- da Franca Rodrigues, K.A.; Amorim, L.V.; Dias, C.N.; Moraes, D.F.C.; Carneiro, S.M.P.; de Amorim Carvalho, F.A. Syzygium Cumini (L.) Skeels Essential Oil and Its Major Constituent α-Pinene Exhibit Anti-Leishmania Activity through Immunomodulation in Vitro. J. Ethnopharmacol. 2014, 160, 32–40. [Google Scholar] [CrossRef] [Green Version]
- Zamyad, M.; Abbasnejad, M.; Esmaeili-Mahani, S.; Mostafavi, A.; Sheibani, V. The Anticonvulsant Effects of Ducrosia Anethifolia (Boiss) Essential Oil Are Produced by Its Main Component Alpha-Pinene in Rats. Arq. Neuro-Psiquiatr. 2018, 77, 106–114. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Zhang, R. Ozonolysis of Alpha-Pinene and Beta-Pinene: Kinetics and Mechanism. J. Chem. Phys. 2005, 122, 114308. [Google Scholar] [CrossRef]
- Oliveira, R.C.d.M.; Bauerfeldt, G.F. Ozonolysis Reactions of Monoterpenes: A Variational Transition State Investigation. J. Phys. Chem. 2015, 119, 2802–2812. [Google Scholar] [CrossRef] [PubMed]
- Guenther, A.; Hewitt, C.N.; Erickson, D.; Fall, R.; Geron, C.; Graedel, T.; Harley, P.; Klinger, L.; Lerdau, M.; Mckay, W.A.; et al. A Global Model of Natural Volatile Organic Compound Emissions. J. Geophys. Res. Atmos. 1995, 100, 8873–8892. [Google Scholar] [CrossRef]
- Negoi, A.; Parvulescu, V.I.; Tudorache, M. Peroxidase-Based Biocatalysis in a Two-Phase System for Allylic Oxidation of α-Pinene. Catal. Today 2018, 306, 199–206. [Google Scholar] [CrossRef]
- Ishida, T.; Asakawa, Y.; Takemoto, T.; Aratani, T. Terpenoids Biotransformation in Mammals III: Biotransformation of A-pinene, Β-pinene, Pinane, 3-carene, Carane, Myrcene, and P-cymene in Rabbits. J. Pharm. Sci. 1981, 70, 406–415. [Google Scholar] [CrossRef] [PubMed]
- Southwell, I.A.; Flynn, T.M.; Degabriele, R. Metabolism of α- and β-Pinene, β-Cymene and 1,8-Cineole in the Brushtail Possum, Trichosurus Vulpecula. Xenobiotica 1980, 10, 17–23. [Google Scholar] [CrossRef]
- Renwick, J.A.A.; Hughes, P.R.; Ty, T.D. Oxidation Products of Pinene in the Bark Beetle, Dendroctonus Frontalis. J. Insect Physiol. 1973, 19, 1735–1740. [Google Scholar] [CrossRef]
- Renwick, J.A.; Hughes, P.R.; Krull, I.S. Selective Production of Cis- and Trans-Verbenol from (-)-and (+)-Alpha by a Bark Beetle. Science 1976, 191, 199–201. [Google Scholar] [CrossRef]
- Chiu, C.C.; Keeling, C.I.; Bohlmann, J. The Cytochrome P450 CYP6DE1 Catalyzes the Conversion of α-Pinene into the Mountain Pine Beetle Aggregation Pheromone Trans-Verbenol. Sci. Rep. 2019, 9, 1477. [Google Scholar] [CrossRef]
- Nadeau, J.A.; Petereit, J.; Tillett, R.L.; Jung, K.; Fotoohi, M.; MacLean, M.; Young, S.; Schlauch, K.; Blomquist, G.J.; Tittiger, C. Comparative Transcriptomics of Mountain Pine Beetle Pheromone-Biosynthetic Tissues and Functional Analysis of CYP6DE3. Bmc Genom. 2017, 18, 311. [Google Scholar] [CrossRef] [Green Version]
- Keeling, C.I.; Henderson, H.; Li, M.; Dullat, H.K.; Ohnishi, T.; Bohlmann, J. CYP345E2, an Antenna-Specific Cytochrome P450 from the Mountain Pine Beetle, Dendroctonus Ponderosae Hopkins, Catalyses the Oxidation of Pine Host Monoterpene Volatiles. Insect Biochem. Mol. Biol. 2013, 43, 1142–1151. [Google Scholar] [CrossRef]
- Schmidt, L.; Göen, T. Human Metabolism of α-Pinene and Metabolite Kinetics after Oral Administration. Arch. Toxicol. 2017, 91, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Aloum, L.; Alefishat, E.; Adem, A.; Petroianu, G. Ionone Is More than a Violet’s Fragrance: A Review. Molecules 2020, 25, 5822. [Google Scholar] [CrossRef] [PubMed]
- Aloum, L.; Semreen, M.H.; Al-Tel, T.H.; Al-Hroub, H.; Mousa, M.; Jayaraj, R.L.; Alefishat, E.; Adem, A.; Petroianu, G.A. Metabolic Conversion of β-Pinene to β-Ionone in Rats. Xenobiotica Fate Foreign Compd. Biol. Syst. 2021, 51, 1427–1435. [Google Scholar] [CrossRef]
- Al-Tel, T.H.; Tarazi, H.; Aloum, L.O.; Lorke, D.E.; Petroianu, G.A. Possible Metabolic Conversion of Pinene to Ionone. Pharmazie 2020, 75, 360–363. [Google Scholar] [CrossRef] [PubMed]
- Rydberg, P.; Olsen, L.; Ryde, U. Quantum-Mechanical Studies of Reactions Performed by Cytochrome P450 Enzymes. Curr. Inorg. Chem. 2012, 2, 292–315. [Google Scholar] [CrossRef] [Green Version]
- Shaik, S.; Kumar, D.; de Visser, S.P.; Altun, A.; Thiel, W. Theoretical Perspective on the Structure and Mechanism of Cytochrome P450 Enzymes. Chem. Rev. 2005, 105, 2279–2328. [Google Scholar] [CrossRef]
- Sivaramakrishnan, S.; Ouellet, H.; Matsumura, H.; Guan, S.; Moënne-Loccoz, P.; Burlingame, A.L.; de Montellano, P.R.O. Proximal Ligand Electron Donation and Reactivity of the Cytochrome P450 Ferric–Peroxo Anion. J. Am. Chem. Soc. 2012, 134, 6673–6684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Visser, S.P.; Kumar, D.; Shaik, S. How Do Aldehyde Side Products Occur during Alkene Epoxidation by Cytochrome P450? Theory Reveals a State-Specific Multi-State Scenario Where the High-Spin Component Leads to All Side Products. J. Inorg. Biochem. 2004, 98, 1183–1193. [Google Scholar] [CrossRef]
- Szori, M.; Abou-Abdo, T.; Fittschen, C.; Csizmadia, I.G.; Viskolcz, B. Allylic Hydrogen Abstraction II. H-Abstraction from 1,4 Type Polyalkenes as a Model for Free Radical Trapping by Polyunsaturated Fatty Acids (PUFAs). Phys. Chem. Chem. Phys. 2007, 9, 1931–1940. [Google Scholar] [CrossRef]
- Hoomissen, D.J.V.; Vyas, S. Impact of Conjugation and Hyperconjugation on the Radical Stability of Allylic and Benzylic Systems: A Theoretical Study. J. Org. Chem. 2017, 82, 5731–5742. [Google Scholar] [CrossRef]
- Cummins, D.C.; Alvarado, J.G.; Zaragoza, J.P.T.; Mubarak, M.Q.E.; Lin, Y.-T.; de Visser, S.P.; Goldberg, D.P. Hydroxyl Transfer to Carbon Radicals by Mn(OH) vs Fe(OH) Corrole Complexes. Inorg. Chem. 2020, 59, 16053–16064. [Google Scholar] [CrossRef] [PubMed]
- Zaragoza, J.P.T.; Yosca, T.H.; Siegler, M.A.; Moeënne-Loccoz, P.; Green, M.T.; Goldberg, D.P. Direct Observation of Oxygen Rebound with an Iron-Hydroxide Complex. J. Am. Chem. Soc. 2017, 139, 13640–13643. [Google Scholar] [CrossRef] [PubMed]
- Ogliaro, F.; Cohen, S.; Filatov, M.; Harris, N.; Shaik, S. The High-Valent Compound of Cytochrome P450: The Nature of the Fe−S Bond and the Role of the Thiolate Ligand as an Internal Electron Donor. Angew. Chem. Int. Ed. 2001, 40, 647. [Google Scholar] [CrossRef]
- Li, Y.; Wu, J.; Zhao, Q.; Zhang, Y.; Huang, Z. Theoretical Study of an Undisclosed Reaction Class: Direct H-Atom Abstraction from Allylic Radicals by Molecular Oxygen. Energies 2021, 14, 2916. [Google Scholar] [CrossRef]
- He, K.; Falick, A.M.; Chen, B.; Nilsson, F.; Correia, M.A. Identification of the Heme Adduct and an Active Site Peptide Modified during Mechanism-Based Inactivation of Rat Liver Cytochrome P450 2B1 by Secobarbital. Chem. Res. Toxicol. 1996, 9, 614–622. [Google Scholar] [CrossRef]
- Gheidi, M.; Safari, N.; Zahedi, M. Density Functional Theory Studies on the Conversion of Hydroxyheme to Iron-Verdoheme in the Presence of Dioxygen. Dalton Trans. 2017, 46, 2146–2158. [Google Scholar] [CrossRef]
- Luchini, G.; Alegre-Requena, J.V.; Funes-Ardoiz, I.; Paton, R.S. GoodVibes: Automated Thermochemistry for Heterogeneous Computational Chemistry Data. F1000Research 2020, 9, 291. [Google Scholar] [CrossRef]
- Sure, R.; Brandenburg, J.G.; Grimme, S. Small Atomic Orbital Basis Set First-Principles Quantum Chemical Methods for Large Molecular and Periodic Systems: A Critical Analysis of Error Sources. ChemistryOpen 2016, 5, 94–109. [Google Scholar] [CrossRef] [Green Version]
- Lonsdale, R.; Harvey, J.N.; Mulholland, A.J. Effects of Dispersion in Density Functional Based Quantum Mechanical/Molecular Mechanical Calculations on Cytochrome P450 Catalyzed Reactions. J. Chem. Theory Comput. 2012, 8, 4637–4645. [Google Scholar] [CrossRef]
- Siegbahn, P.E.M.; Borowski, T. Modeling Enzymatic Reactions Involving Transition Metals. Acc. Chem. Res. 2006, 39, 729–738. [Google Scholar] [CrossRef]
- Saito, K.; Watabe, Y.; Fujihara, T.; Takayanagi, T.; Hasegawa, J. Spin-inversion Mechanisms in O2 Binding to a Model Heme Complex Revisited by Density Function Theory Calculations. J. Comput. Chem. 2020, 41, 1130–1138. [Google Scholar] [CrossRef] [PubMed]
- de Visser, S.P. What External Perturbations Influence the Electronic Properties of Catalase Compound I? Inorg. Chem. 2006, 45, 9551–9557. [Google Scholar] [CrossRef] [PubMed]
- Reinhard, F.G.C.; de Visser, S.P. Biodegradation of Cosmetics Products: A Computational Study of Cytochrome P450 Metabolism of Phthalates. Inorganics 2017, 5, 77. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional Thermochemistry. I. The Effect of the Exchange-only Gradient Correction. J. Chem. Phys. 1992, 96, 2155–2160. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results Obtained with the Correlation Energy Density Functionals of Becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Bonomo, S.; Jørgensen, F.S.; Olsen, L. Mechanism of Cytochrome P450 17A1-Catalyzed Hydroxylase and Lyase Reactions. J. Chem. Inf. Model. 2017, 57, 1123–1133. [Google Scholar] [CrossRef]
- Strickland, N.; Harvey, J.N. Spin-Forbidden Ligand Binding to the Ferrous−Heme Group: Ab Initio and DFT Studies. J. Phys. Chem. B 2007, 111, 841–852. [Google Scholar] [CrossRef]
- Rydberg, P.; Olsen, L. The Accuracy of Geometries for Iron Porphyrin Complexes from Density Functional Theory. J. Phys. Chem. 2009, 113, 11949–11953. [Google Scholar] [CrossRef]
- Radoń, M. Spin-State Energetics of Heme-Related Models from DFT and Coupled Cluster Calculations. J. Chem. Theory Comput. 2014, 10, 2306–2321. [Google Scholar] [CrossRef]
- Rassolov, V.A.; Pople, J.A.; Ratner, M.A.; Windus, T.L. 6-31G * Basis Set for Atoms K through Zn. J. Chem. Phys. 1998, 109, 1223–1229. [Google Scholar] [CrossRef]
- Rassolov, V.A.; Ratner, M.A.; Pople, J.A.; Redfern, P.C.; Curtiss, L.A. 6-31G* Basis Set for Third-row Atoms. J. Comput. Chem. 2001, 22, 976–984. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for K to Au Including the Outermost Core Orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Wadt, W.R.; Hay, P.J. Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for Main Group Elements Na to Bi. J. Chem. Phys. 1985, 82, 284–298. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for the Transition Metal Atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Grimme, S. Supramolecular Binding Thermodynamics by Dispersion-Corrected Density Functional Theory. Chem. Eur. J. 2012, 18, 9955–9964. [Google Scholar] [CrossRef]
- Grimme, S.; Brandenburg, J.G.; Bannwarth, C.; Hansen, A. Consistent Structures and Interactions by Density Functional Theory with Small Atomic Orbital Basis Sets. J. Chem. Phys. 2015, 143, 054107. [Google Scholar] [CrossRef]
- Brandenburg, J.G.; Alessio, M.; Civalleri, B.; Peintinger, M.F.; Bredow, T.; Grimme, S. Geometrical Correction for the Inter- and Intramolecular Basis Set Superposition Error in Periodic Density Functional Theory Calculations. J. Phys. Chem. 2013, 117, 9282–9292. [Google Scholar] [CrossRef]
- Lonsdale, R.; Harvey, J.N.; Mulholland, A.J. Inclusion of Dispersion Effects Significantly Improves Accuracy of Calculated Reaction Barriers for Cytochrome P450 Catalyzed Reactions. J. Phys. Chem. Lett. 2010, 1, 3232–3237. [Google Scholar] [CrossRef]
- Rydberg, P.; Lonsdale, R.; Harvey, J.N.; Mulholland, A.J.; Olsen, L. Trends in Predicted Chemoselectivity of Cytochrome P450 Oxidation: B3LYP Barrier Heights for Epoxidation and Hydroxylation Reactions. J. Mol. Graph. Model. 2014, 52, 30–35. [Google Scholar] [CrossRef]
- Caldeweyher, E.; Ehlert, S.; Hansen, A.; Neugebauer, H.; Spicher, S.; Bannwarth, C.; Grimme, S. A Generally Applicable Atomic-Charge Dependent London Dispersion Correction. J. Chem. Phys. 2019, 150, 154122. [Google Scholar] [CrossRef]
- Caldeweyher, E.; Bannwarth, C.; Grimme, S. Extension of the D3 Dispersion Coefficient Model. J. Chem. Phys. 2017, 147, 034112. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cancès, E. The IEF Version of the PCM Solvation Method: An Overview of a New Method Addressed to Study Molecular Solutes at the QM Ab Initio Level. J. Mol. Struct. Theochem. 1999, 464, 211–226. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Radical | Gcor(Eh) | Gcor(ref) (kcal/mol) | Hydroxylated Product | Gcor(Eh) | Gcor(ref) (kcal/mol) | |
|---|---|---|---|---|---|---|
 | Alpha | −389.788789 | 21.7 | Alpha | −465.658506 | 1.4 |
| Gamma | −389.819706 | 2.27 | Gamma | −465.645667 | 9.45 | |
| Delta | −389.777607 | 28.7 | Delta | −465.660286 | 0.28 | |
| Epsilon | −389.823321 | 0 | Epsilon cis | −465.651306 | 5.91 | |
| Epsilon trans | −465.652273 | 5.31 | ||||
| Zeta | −389.790994 | 20.3 | Zeta | −465.660730 | 0.00 | |
| Theta | −389.794206 | 18.3 | Theta | −465.654240 | 4.07 | |
| Iota | −389.795916 | 17.2 | Iota | −465.646394 | 9 | |
| Kappa | −389.796285 | 17 | Kappa | −465.646120 | 9.17 |
| Radical | Gcor (Eh) | Gcor(ref) (kcal/mol) | Hydroxylated Product | Gcor (Eh) | Gcor(ref) (kcal/mol) | |
|---|---|---|---|---|---|---|
 | Alpha | −389.784833 | 21.9 | Alpha | −465.653831 | 0.57 |
| Gamma | −389.770224 | 31.1 | Gamma | −465.649621 | 3.21 | |
| Delta | −389.819707 | 0 | Delta cis | −465.645132 | 6.03 | |
| Delta trans | −465.644727 | 6.28 | ||||
| Epsilon | −389.797584 | 13.9 | Epsilon cis | −465.6451796 | 6.00 | |
| Epsilon trans | −465.6473512 | 4.63 | ||||
| Zeta | −389.786313 | 20.9 | Zeta | −465.6547380 | 0 | |
| Theta | −389.788711 | 19.4 | Theta | −465.6488230 | 3.71 | |
| Iota | −389.789920 | 18.7 | Iota | −465.6375598 | 10.78 | |
| Kappa | −389.790510 | 18.3 | Kappa | −465.6399752 | 9.26 |
| RC | Site | Spin | Gref (kcal/mol) | RC | Site | Spin | Gref (kcal/mol) |
|---|---|---|---|---|---|---|---|
| 1 | Epsilon cis-(S) | S = 1/2 | 2.41 | 1 | Epsilon cis-(R) | S = 1/2 | 2.40 |
| 2 | 11.4 | 2 | 11.4 | ||||
| 3 | −12.6 | 3 | −12.6 | ||||
| 1 | Epsilon trans-(S) | S = 1/2 | 0.184 | 1 | Epsilon trans-(R) | S = 1/2 | 0.184 |
| 2 | 12.4 | 2 | 12.4 | ||||
| 3 | −13.7 | 3 | −13.7 |
| Site | Spin | ΔrE | ΔE‡ | ΔrH | ΔH‡ | ΔrG | ΔG‡ | ΔrGcor | ΔG‡cor | HydΔrGcor |
|---|---|---|---|---|---|---|---|---|---|---|
| Delta cis-(R) | S = 1/2 | −4.11 | 17.4 | −6.13 | 12.8 | −3.82 | 17.6 | −7.05 | 12.4 | −47.4 |
| Delta trans-(S) | S = 1/2 | −4.64 | 15.4 | −5.56 | 10.9 | −5.10 | 15.4 | −8.30 | 10.8 | −48.4 |
| Delta cis-(R) | S = 3/2 | −0.183 | 18.7 | −1.59 | 13.8 | −0.548 | 18.8 | −2.64 | 13.6 | −45.6 |
| Delta trans-(S) | S = 3/2 | −1.03 | 16.5 | −1.16 | 13.0 | −3.67 | 13.4 | −5.08 | 9.87 | −47.3 |
| Delta cis-(R) | S = 5/2 | −6.52 | 19.6 | −7.19 | 15.9 | −13.3 | 15.8 | −9.18 | 12.8 | −54.0 |
| Delta trans-(S) | S = 5/2 | −7.36 | 17.3 | −7.35 | 14.4 | −15.1 | 12.5 | −11.5 | 10.1 | −54.2 |
| Alpha | S = 1/2 | +17.4 | 21.5 | +15.4 | 16.8 | +18.9 | 22.2 | +17.4 | 15.3 | −50.0 |
| Alpha | S = 3/2 | +13.0 | 18.8 | +13.6 | 15.3 | +14.3 | 18.5 | +10.8 | 12.8 | −43.4 |
| Reactant | G (hartree) | Mole Fraction | Population % | ΔrG | ΔG‡ |
|---|---|---|---|---|---|
| alpha-doublet | −1976.037587 | 0.49 | 48.77 | 28.1 | 15.3 |
| alpha-quartet | −1976.030510 | 0 | 0.03 | 10.8 | 12.8 |
| delta-trans-doublet | −1976.035861 | 0.08 | 7.83 | −8.3 | 10.8 |
| delta-cis-doublet | −1976.036419 | 0.14 | 14.14 | −7.05 | 12.4 |
| delta-cis-quartet | −1976.037104 | 0.29 | 29.23 | −2.64 | 13.6 |
| delta-trans-quartet | −1976.014863 | 0 | 0 | −5.08 | 9.87 |
| delta-cis-sextet | −1976.015837 | 0 | 0 | −9.18 | 12.8 |
| delta-trans-sextet | −1976.014863 | 0 | 0 | −11.5 | 10.1 |
| Reactant | G (hartree) | Mole Fraction | Population % | ΔrG | ΔG‡ |
|---|---|---|---|---|---|
| alpha-doublet | −1975.973971 | 0 | 0 | 28.1 | 15.3 |
| alpha-quartet | −1975.973075 | 0 | 0 | 10.8 | 12.8 |
| delta-trans-doublet | −1976.009716 | 0.534 | 53.4 | −8.3 | 10.8 |
| delta-cis-doublet | −1976.009578 | 0.461 | 46.1 | −7.05 | 12.4 |
| delta-cis-quartet | −1976.004151 | 0.00147 | 0.147 | −2.64 | 13.6 |
| delta-trans-quartet | −1976.004891 | 0.00321 | 0.321 | −5.08 | 9.87 |
| delta-cis-sextet | −1976.000132 | 0 | 0 | −9.18 | 12.8 |
| delta-trans-sextet | −1976.001255 | 0 | 0 | −11.5 | 10.1 |
| Site | Spin | ΔrE | ΔE‡ | ΔrH | ΔH‡ | ΔrG | ΔG‡ | ΔrGcor | ΔG‡cor | HydΔrGcor |
|---|---|---|---|---|---|---|---|---|---|---|
| Epsilon cis-(S) | S = 1/2 | −13.6 | 15.9 | −12.9 | 12.9 | −13.4 | 12.5 | −15.0 | 9.00 | −50.2 |
| Epsilon trans-(R) | S = 1/2 | −14.8 | 14.6 | −14.8 | 11.0 | −10.5 | 16.6 | −13.8 | 12.2 | −48.9 |
| Epsilon cis-(S) | S = 3/2 | −0.1 | 18.0 | −0.215 | 14.1 | −0.981 | 16.4 | −3.06 | 12.3 | −36.8 |
| Epsilon trans-(R) | S = 3/2 | +0.814 | 16.7 | +0.413 | 12.7 | −1.10 | 15.0 | −1.87 | 11.2 | −37.9 |
| Epsilon cis-(S) | S = 5/2 | −16.0 | 18.7 | −15.8 | 14.7 | −18.6 | 15.5 | −18.7 | 12.7 | −53.9 |
| Epsilon trans-(R) | S = 5/2 | −15.7 | 17.5 | −16.8 | 13.0 | −14.1 | 17.3 | −15.2 | 13.2 | −53.9 |
| Alpha | S = 1/2 | +18.4 | 22.8 | +18.0 | 18.7 | +18.3 | 21.4 | +14.8 | 15.9 | −50.2 |
| Alpha | S = 3/2 | +16.7 | 24.9 | +15.9 | 20.6 | +15.7 | 23.1 | +13.5 | 18.1 | −37.1 |
| Gamma | S = 1/2 | −3.18 | 16.1 | −4.05 | 12.5 | −0.516 | 16.9 | −5.73 | 12.6 | −41.9 |
| Gamma | S = 3/2 | +1.09 | 18.0 | +0.612 | 14.0 | +0.437 | 15.8 | −1.80 | 14.2 | −30.8 |
| Reactant | G (hartree) | Mole Fraction | Population % | ΔrG | ΔG‡ |
|---|---|---|---|---|---|
| alpha-doublet | −1976.041937 | 0.0871 | 8.71 | 14.8 | 17.6 |
| alpha-quartet | −1976.024096 | 0.209 | 20.9 | 13.5 | 18.1 |
| epsilon-trans-doublet | −1976.042897 | 0.241 | 24.1 | −13.8 | 12.2 |
| epsilon-cis-doublet | −1976.039343 | 0.00557 | 0.557 | −15.0 | 8.99 |
| epsilon-trans-quartet | −1976.040800 | 0.0261 | 2.61 | −1.87 | 11.2 |
| epsilon-cis-quartet | −1976.041490 | 0.0542 | 5.42 | −3.06 | 12.3 |
| epsilon-trans-sextet | −1976.024055 | 0 | 0 | −15.2 | 13.2 |
| epsilon-cis-sextet | −1976.022164 | 0 | 0 | −18.7 | 12.7 |
| gamma-doublet | −1976.043191 | 0.329 | 32.9 | −5.73 | 12.6 |
| gamma-quartet | −1976.041393 | 0.0489 | 4.89 | −1.80 | 14.2 |
| Reactant | G (hartree) | Mole Fraction | Population % | ΔrG | ΔG‡ |
|---|---|---|---|---|---|
| alpha-doublet | −1976.018408 | 0 | 0 | 14.8 | 17.6 |
| alpha-quartet | −1976.021248 | 0 | 0 | 13.5 | 18.1 |
| epsilon-trans-doublet | −1976.064967 | 0.864 | 86.4 | −13.8 | 12.2 |
| epsilon-cis-doublet | −1976.063223 | 0.136 | 13.6 | −15.0 | 8.99 |
| epsilon-trans-quartet | −1976.043787 | 0 | 0 | −1.87 | 11.2 |
| epsilon-cis-quartet | −1976.046371 | 0 | 0 | −3.06 | 12.3 |
| epsilon-trans-sextet | −1976.048297 | 0 | 0 | −15.2 | 13.2 |
| epsilon-cis-sextet | −1976.051903 | 0 | 0 | −18.7 | 12.7 |
| gamma-doublet | −1976.052329 | 0 | 0 | −5.73 | 12.6 |
| gamma-quartet | −1976.044266 | 0 | 0 | −1.80 | 14.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shaya, J.; Aloum, L.; Lu, C.-S.; Corridon, P.R.; Aoudi, A.; Shunnar, A.; Alefishat, E.; Petroianu, G. Theoretical Study of Hydroxylation of α- and β-Pinene by a Cytochrome P450 Monooxygenase Model. Int. J. Mol. Sci. 2023, 24, 5150. https://doi.org/10.3390/ijms24065150
Shaya J, Aloum L, Lu C-S, Corridon PR, Aoudi A, Shunnar A, Alefishat E, Petroianu G. Theoretical Study of Hydroxylation of α- and β-Pinene by a Cytochrome P450 Monooxygenase Model. International Journal of Molecular Sciences. 2023; 24(6):5150. https://doi.org/10.3390/ijms24065150
Chicago/Turabian StyleShaya, Janah, Lujain Aloum, Chung-Shin Lu, Peter R. Corridon, Abdulrahman Aoudi, Abeer Shunnar, Eman Alefishat, and Georg Petroianu. 2023. "Theoretical Study of Hydroxylation of α- and β-Pinene by a Cytochrome P450 Monooxygenase Model" International Journal of Molecular Sciences 24, no. 6: 5150. https://doi.org/10.3390/ijms24065150