
Integrated Analysis of DNA Methylation and Gene Expression in Porcine Placental Development
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
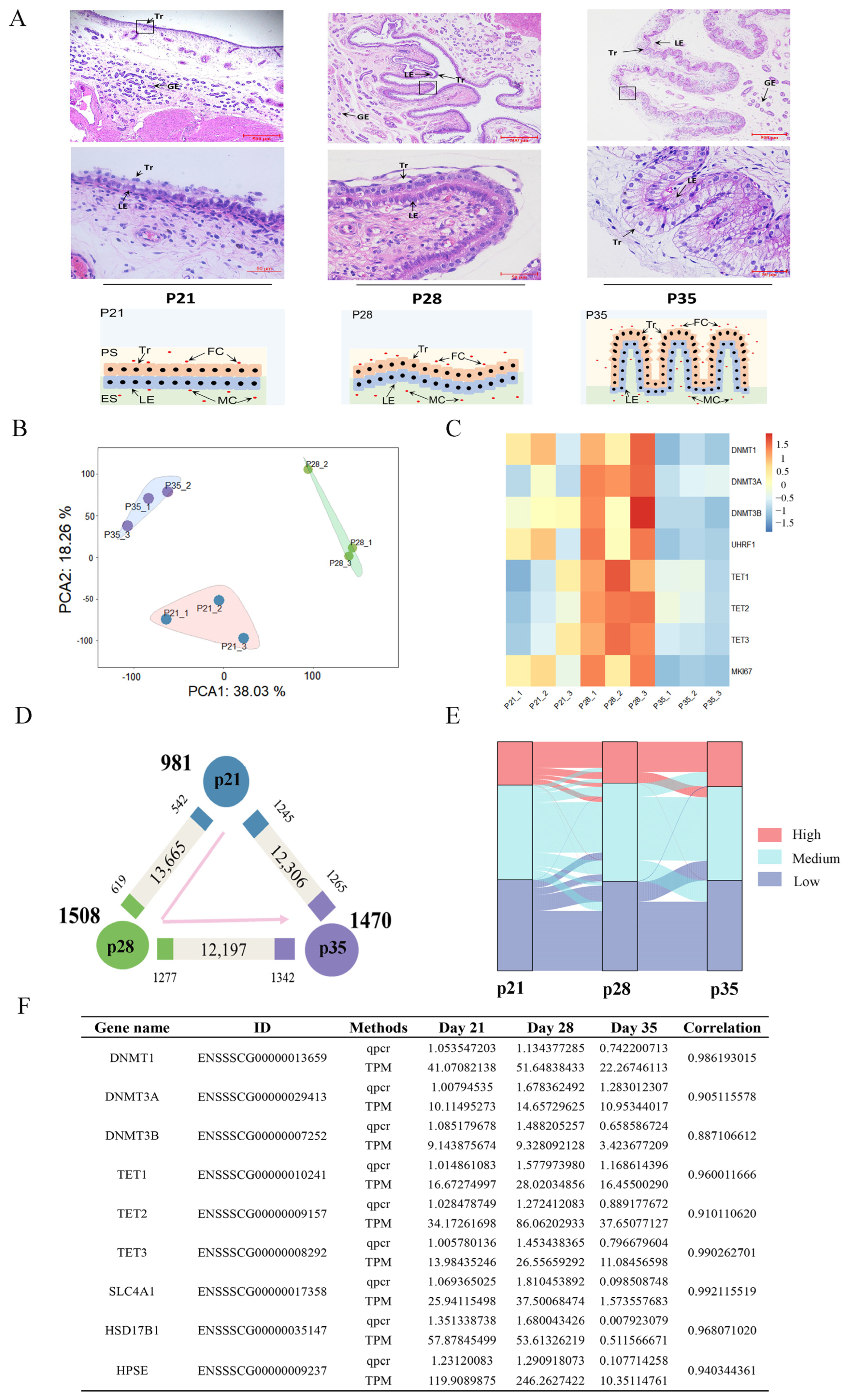
2.1. Morphology and Gene Expression changes during porcine Placental Development
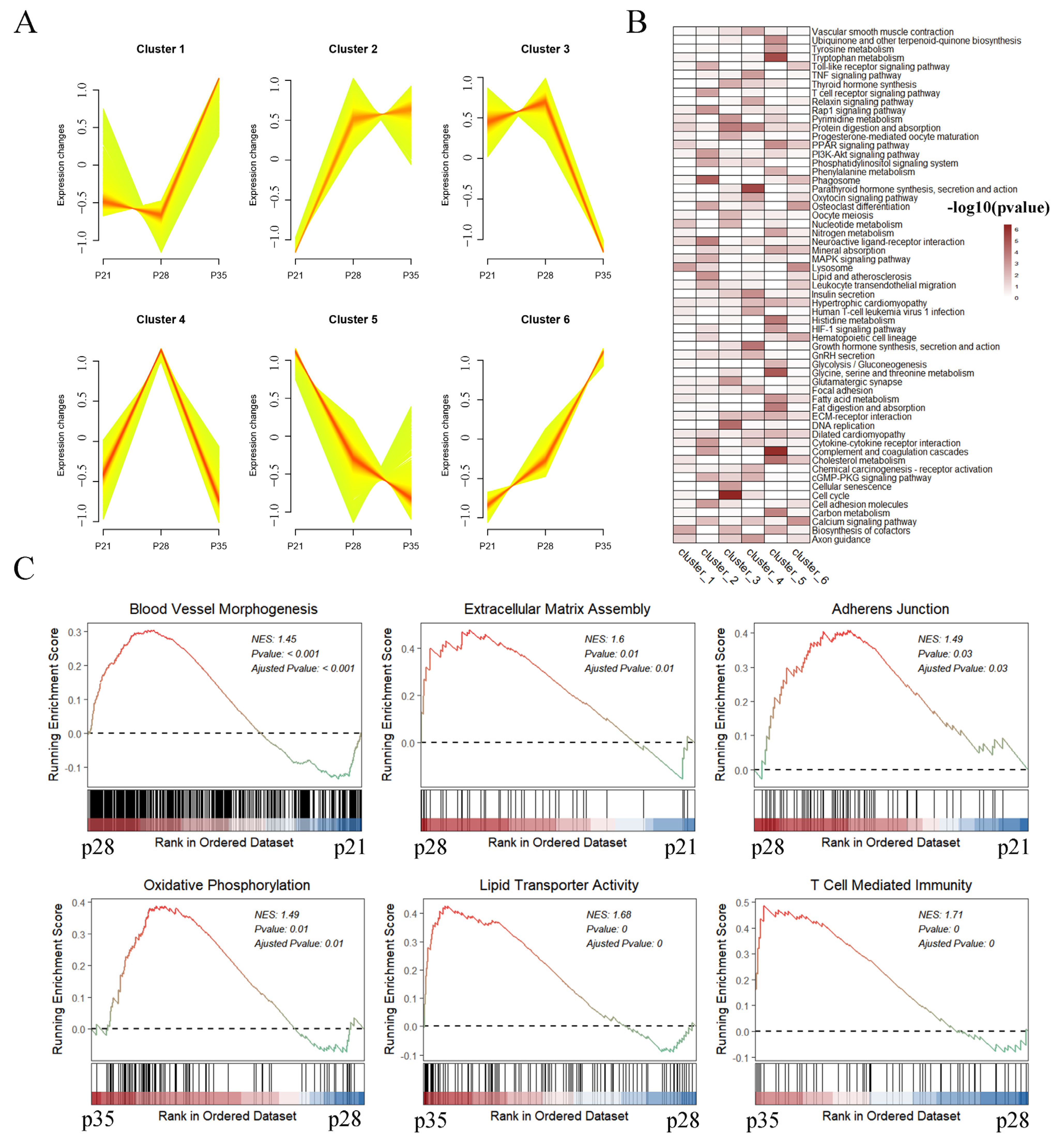
2.2. Functional Enrichment Analysis of Differentially Expressed Genes
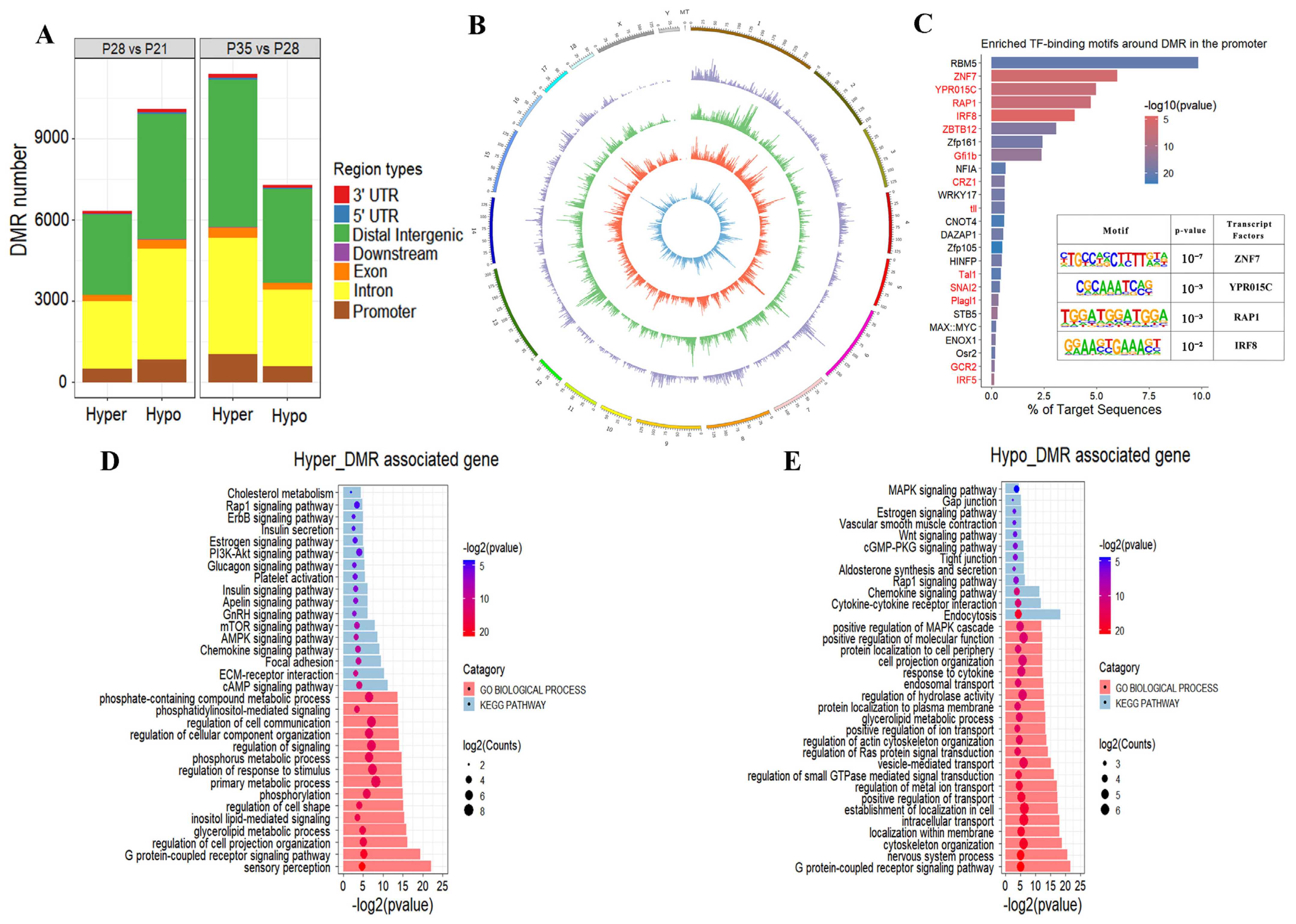
2.3. Characteristics of DNA Methylome during Placental Development
2.4. Dynamic Changes in DNA Methylation Occur in Placental Development
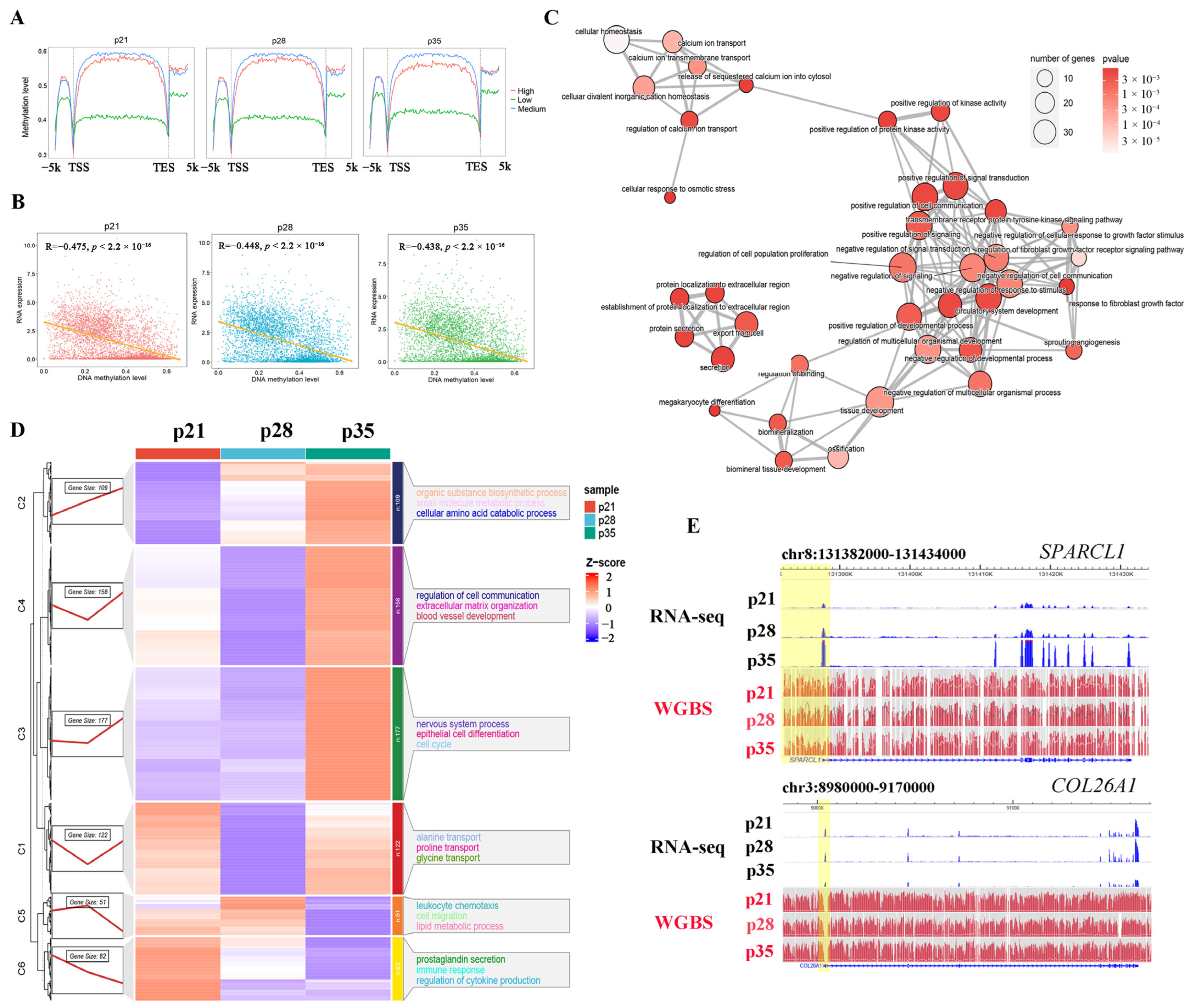
2.5. Association between DNA Methylation and Gene Expression during Placental Development
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Sample Collection
5.2. Hematoxylin–Eosin Staining
5.3. Real-Time Quantitative PCR (qPCR)
5.4. RNA-seq and Data Analysis
5.5. Genome Bisulfite Sequencing and Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kaczynski, P.; Goryszewska-Szczurek, E.; Baryla, M.; Waclawik, A. Novel insights into conceptus–maternal signaling during pregnancy establishment in pigs. Mol. Reprod. Dev. 2022; ahead of print. [Google Scholar]
- Geisert, R.D.; Johnson, G.A.; Burghardt, R.C. Implantation and Establishment of Pregnancy in the Pig. Adv. Anat. Embryol. Cell Biol. 2015, 216, 137–163. [Google Scholar] [PubMed]
- Konwar, C.; Price, E.M.; Wang, L.Q.; Wilson, S.L.; Terry, J.; Robinson, W.P. DNA methylation profiling of acute chorioamnionitis-associated placentas and fetal membranes: Insights into epigenetic variation in spontaneous preterm births. Epigenetics Chromatin 2018, 11, 63. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Xiong, Y.; Qu, B.; Bao, A.; Zhang, Y. DNA Methylation and Recurrent Pregnancy Loss: A Mysterious Compass? Front. Immunol. 2021, 12, 738962. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.; Jauniaux, E. Pathophysiology of placental-derived fetal growth restriction. Am. J. Obstet. Gynecol. 2018, 218 (Suppl. S2), S745–S761. [Google Scholar] [CrossRef] [Green Version]
- Vallet, J.L.; Freking, B. Differences in placental structure during gestation associated with large and small pig fetuses1,2. J. Anim. Sci. 2007, 85, 3267–3275. [Google Scholar] [CrossRef] [PubMed]
- Bidarimath, M.; Tayade, C. Pregnancy and spontaneous fetal loss: A pig perspective. Mol. Reprod. Dev. 2017, 84, 856–869. [Google Scholar] [CrossRef] [Green Version]
- Vallet, J.L.; Miles, J.R.; Freking, B.A. Development of the pig placenta. Soc. Reprod. Fertil. Suppl. 2009, 66, 265–279. [Google Scholar] [CrossRef]
- Johnson, G.A.; Bazer, F.W.; Seo, H. The Early Stages of Implantation and Placentation in the Pig. Adv. Anat. Embryol. Cell Biol. 2021, 234, 61–89. [Google Scholar]
- Wacławik, A. Novel insights into the mechanisms of pregnancy establishment: Regulation of prostaglandin synthesis and signaling in the pig. Reproduction 2011, 142, 389–399. [Google Scholar] [CrossRef] [Green Version]
- Han, K.; Ren, R.; Cao, J.; Zhao, S.; Yu, M. Genome-Wide Identification of Histone Modifications Involved in Placental Development in Pigs. Front. Genet. 2019, 10, 277. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Wang, M.; Su, L.; Li, X.; Zhao, S.; Yu, M. The Expression Pattern of MicroRNAs and the Associated Pathways Involved in the Development of Porcine Placental Folds That Contribute to the Expansion of the Exchange Surface Area1. Biol. Reprod. 2015, 93, 62. [Google Scholar] [CrossRef]
- Hong, L.; Hou, C.; Li, X.; Li, C.; Zhao, S.; Yu, M. Expression of Heparanase Is Associated with Breed-Specific Morphological Characters of Placental Folded Bilayer Between Yorkshire and Meishan Pigs1. Biol. Reprod. 2014, 90, 56. [Google Scholar] [CrossRef] [PubMed]
- Li, C.M.; Hou, L.; Zhang, H.; Zhang, W.Y. CCL17 Induces Trophoblast Migration and Invasion by Regulating Matrix Metalloproteinase and Integrin Expression in Human First-Trimester Placenta. Reprod. Sci. 2014. [Google Scholar] [CrossRef] [PubMed]
- Renaud, S.J.; Kubota, K.; Rumi, M.A.K.; Soares, M.J. The FOS Transcription Factor Family Differentially Controls Trophoblast Migration and Invasion. J. Biol. Chem. 2014, 289, 5025–5039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Cheng, B.; Liu, Q.; Jiang, P.; Yang, J. CRY2 suppresses trophoblast migration and invasion in recurrent spontaneous abortion. J. Biochem. 2020, 167, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, J.; Fan, X.; Zhang, Y.; Zhoua, M.; Li, X.; Wang, L. LASP2 inhibits trophoblast cell migration and invasion in preeclampsia through inactivation of the Wnt/β-catenin signaling pathway. J. Recept. Signal Transduct. 2020, 41, 67–73. [Google Scholar] [CrossRef]
- Henikoff, S.; Greally, J.M. Epigenetics, cellular memory and gene regulation. Curr. Biol. 2016, 26, R644–R648. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Pradhan, S. Mammalian epigenetic mechanisms. IUBMB Life 2014, 66, 240–256. [Google Scholar] [CrossRef]
- Deng, D.; Tan, X.; Han, K.; Ren, R.; Cao, J.; Yu, M. Transcriptomic and ChIP-seq Integrative Analysis Reveals Important Roles of Epigenetically Regulated lncRNAs in Placental Development in Meishan Pigs. Genes 2020, 11, 397. [Google Scholar] [CrossRef] [Green Version]
- Su, L.; Zhao, S.; Zhu, M.; Yu, M. Differential expression of microRNAs in porcine placentas on Days 30 and 90 of gestation. Reprod. Fertil. Dev. 2010, 22, 1175–1182. [Google Scholar] [CrossRef]
- Hwang, J.H.; An, S.M.; Kwon, S.; Park, D.H.; Kim, T.W.; Kang, D.G.; Yu, G.E.; Kim, I.-S.; Park, H.C.; Ha, J.; et al. DNA methylation patterns and gene expression associated with litter size in Berkshire pig placenta. PLoS ONE 2017, 12, e0184539. [Google Scholar] [CrossRef] [Green Version]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, C.-C.; Fondell, J.D. Nuclear Receptor Recruitment of Histone-Modifying Enzymes to Target Gene Promoters. Vitam. Horm. 2004, 68, 93–122. [Google Scholar] [PubMed]
- Zocher, S.; Overall, R.W.; Berdugo-Vega, G.; Rund, N.; Karasinsky, A.; Adusumilli, V.S.; Steinhauer, C.; Scheibenstock, S.; Händler, K.; Schultze, J.L.; et al. De novo DNA methylation controls neuronal maturation during adult hippocampal neurogenesis. EMBO J. 2021, 40, e107100. [Google Scholar] [CrossRef]
- Liu, H.; Song, Y.; Qiu, H.; Liu, Y.; Luo, K.; Yi, Y.; Jiang, G.; Lu, M.; Zhang, Z.; Yin, J.; et al. Downregulation of FOXO3a by DNMT1 promotes breast cancer stem cell properties and tumorigenesis. Cell Death Differ. 2020, 27, 966–983. [Google Scholar] [CrossRef] [Green Version]
- Jung, M.; Pfeifer, G.P. Aging and DNA methylation. BMC Biol. 2015, 13, 7. [Google Scholar] [CrossRef] [Green Version]
- Kaczynski, P.; van der Weijden, V.; Goryszewska-Szczurek, E.; Baryla, M.; Ulbrich, S.E.; Waclawik, A. Novel role for conceptus signals in mRNA expression regulation by DNA methylation in porcine endometrium during early pregnancy. Biol. Reprod. 2023, 108, 150–168. [Google Scholar] [CrossRef] [PubMed]
- Serman, L.; Vlahović, M.; Sijan, M.; Bulić-Jakus, F.; Serman, A.; Sincić, N.; Matijević, R.; Jurić-Lekić, G.; Katusić, A. The impact of 5-azacytidine on placental weight, glycoprotein pattern and proliferating cell nuclear antigen expression in rat placenta. Placenta 2007, 28, 803–811. [Google Scholar] [CrossRef] [Green Version]
- Luo, N.; Cheng, W.; Zhou, Y.; Gu, B.; Zhao, Z.; Zhao, Y. Screening Candidate Genes Regulating Placental Development from Trophoblast Transcriptome at Early Pregnancy in Dazu Black Goats (Capra hircus). Animals 2021, 11, 2132. [Google Scholar] [CrossRef]
- Gong, J.; Nv, X.; Li, Z.; Zeng, K.; He, Z.; Chen, X. Investigation On Reproductive Capacity in Tibetan Pig. Southwest China J. Agric. Sci. 2009, 22, 807–810. [Google Scholar]
- Kaczmarek, M.M.; Najmula, J.; Guzewska, M.M.; Przygrodzka, E. MiRNAs in the Peri-Implantation Period: Contribution to Embryo–Maternal Communication in Pigs. Int. J. Mol. Sci. 2020, 21, 2229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Luan, Y.; Liu, T.; Lee, H.J.; Fang, L.; Wang, Y.; Wang, X.; Zhang, B.; Jin, Q.; Ang, K.C.; et al. A map of cis-regulatory elements and 3D genome structures in zebrafish. Nature 2020, 588, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Apicella, C.; Ruano, C.S.M.; Méhats, C.; Miralles, F.; Vaiman, D. The Role of Epigenetics in Placental Development and the Etiology of Preeclampsia. Int. J. Mol. Sci. 2019, 20, 2837. [Google Scholar] [CrossRef] [Green Version]
- Burton, G.J.; Fowden, A.L. The placenta: A multifaceted, transient organ. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2015, 370, 20140066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrilla, I.; Gil, M.A.; Cuello, C.; Cambra, J.M.; Plaza, A.G.; Lucas, X.; Vazquez, J.L.; Vazquez, J.M.; Rodriguez-Martinez, H.; Martinez, E.A. Immunological uterine response to pig embryos before and during implantation. Reprod. Domest. Anim. 2022, 57 (Suppl. S5), 4–13. [Google Scholar] [CrossRef] [PubMed]
- Lessey, B.A. Adhesion molecules and implantation. J. Reprod. Immunol. 2002, 55, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Albelda, S.M.; Buck, C.A. Integrins and other cell adhesion molecules. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1990, 4, 2868–2880. [Google Scholar] [CrossRef]
- McCarty, J.H. αvβ8 integrin adhesion and signaling pathways in development, physiology and disease. J. Cell Sci. 2020, 133, jcs239434. [Google Scholar] [CrossRef]
- Frank, J.W.; Steinhauser, C.B.; Wang, X.; Burghardt, R.C.; Bazer, F.W.; Johnson, G.A. Loss of ITGB3 in ovine conceptuses decreases conceptus expression of NOS3 and SPP1: Implications for the developing placental vasculature†. Biol. Reprod. 2021, 104, 657–668. [Google Scholar] [CrossRef]
- Aran, D.; Toperoff, G.; Rosenberg, M.; Hellman, A. Replication timing-related and gene body-specific methylation of active human genes. Hum. Mol. Genet. 2010, 20, 670–680. [Google Scholar] [CrossRef]
- Xie, W.; Barr, C.L.; Kim, A.; Yue, F.; Lee, A.Y.; Eubanks, J.; Dempster, E.; Ren, B. Base-Resolution Analyses of Sequence and Parent-of-Origin Dependent DNA Methylation in the Mouse Genome. Cell 2012, 148, 816–831. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, M.V.C.; Bourc’His, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef]
- Weber, M.; Hellmann, I.; Stadler, M.B.; Ramos, L.; Pääbo, S.; Rebhan, M.; Schübeler, D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 2007, 39, 457–466. [Google Scholar] [CrossRef]
- Chen, C.-P.; Huang, J.-P.; Chu, T.-Y.; Aplin, J.; Wu, Y.-H. Human placental multipotent mesenchymal stromal cells modulate trophoblast migration via Rap1 activation. Placenta 2013, 34, 913–923. [Google Scholar] [CrossRef]
- Chang, C.W.; Cheong, M.L.; Chang, G.D.; Tsai, M.S.; Chen, H. Involvement of Epac1/Rap1/CaMKI/HDAC5 signaling cascade in the regulation of placental cell fusion. Mol. Hum. Reprod. 2013, 19, 745–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guvakova, M.A.; Lee, W.S.Y.; Furstenau, D.K.; Prabakaran, I.; Li, D.C.; Hung, R.; Kushnir, N. The small GTPase Rap1 promotes cell movement rather than stabilizes adhesion in epithelial cells responding to insulin-like growth factor I. Biochem. J. 2014, 463, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Caruana, I.; Savoldo, B.; Hoyos, V.; Weber, G.; Liu, H.; Kim, E.S.; Ittmann, M.M.; Marchetti, D.; Dotti, G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med. 2015, 21, 524–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connor, B.B.; Pope, B.D.; Peters, M.M.; Ris-Stalpers, C.; Parker, K.K. The role of extracellular matrix in normal and pathological pregnancy: Future applications of microphysiological systems in reproductive medicine. Exp. Biol. Med. 2020, 245, 1163–1174. [Google Scholar] [CrossRef]
- Nasser, N.J. Heparanase involvement in physiology and disease. Cell. Mol. Life Sci. 2008, 65, 1706–1715. [Google Scholar] [CrossRef]
- Liu, X.; Zhao, J.; Luan, X.; Li, S.; Zhai, J.; Liu, J.; Du, Y. SPARCL1 impedes trophoblast migration and invasion by down-regulating ERK phosphorylation and AP-1 production and altering EMT-related molecule expression. Placenta 2019, 89, 33–41. [Google Scholar] [CrossRef]
- Huang, Z.; Huang, S.; Song, T.; Yin, Y.; Tan, C. Placental Angiogenesis in Mammals: A Review of the Regulatory Effects of Signaling Pathways and Functional Nutrients. Adv. Nutr. 2021, 12, 2415–2434. [Google Scholar] [CrossRef] [PubMed]
- Gram, A.; Boos, A.; Kowalewski, M.P. Cellular localization, expression and functional implications of the utero-placental endothelin system during maintenance and termination of canine gestation. J. Reprod. Dev. 2017, 63, 235–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, F.; Allen, J.E.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean Irina, M.; Austine-Orimoloye, O.; Azov Andrey, G.; Barnes, I.; Bennett, R.; et al. Ensembl 2022. Nucleic Acids Res. 2021, 50, D988–D995. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. feature Counts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Mi, H.; Huang, X.; Muruganujan, A.; Tang, H.; Mills, C.; Kang, D.; Thomas, P.D. PANTHER version 11: Expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2016, 45, D183–D189. [Google Scholar] [CrossRef] [Green Version]
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J.; et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Kumar, L.; Futschik, M.E. Mfuzz: A software package for soft clustering of microarray data. Bioinformation. 2007, 2, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Figueroa, M.E.; Melnick, A.; Mason, C.E. methylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012, 13, R87. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.-G.; He, Q.-Y. ChIPseeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015, 31, 2382–2383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Lowdon, R.F.; Li, D.; Lawson, H.; Madden, P.A.; Costello, J.F.; Wang, T. Exploring long-range genome interactions using the WashU Epigenome Browser. Nat. Methods 2013, 10, 375–376. [Google Scholar] [CrossRef] [Green Version]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, B.; Zhou, C.; Zang, X.; Zhao, X.; Xiao, L.; Zeng, J.; Hong, L.; Wu, Z.; Gu, T. Integrated Analysis of DNA Methylation and Gene Expression in Porcine Placental Development. Int. J. Mol. Sci. 2023, 24, 5169. https://doi.org/10.3390/ijms24065169
Tan B, Zhou C, Zang X, Zhao X, Xiao L, Zeng J, Hong L, Wu Z, Gu T. Integrated Analysis of DNA Methylation and Gene Expression in Porcine Placental Development. International Journal of Molecular Sciences. 2023; 24(6):5169. https://doi.org/10.3390/ijms24065169
Chicago/Turabian StyleTan, Baohua, Chen Zhou, Xupeng Zang, Xinming Zhao, Liyao Xiao, Jiekang Zeng, Linjun Hong, Zhenfang Wu, and Ting Gu. 2023. "Integrated Analysis of DNA Methylation and Gene Expression in Porcine Placental Development" International Journal of Molecular Sciences 24, no. 6: 5169. https://doi.org/10.3390/ijms24065169