
Light, Water, and Melatonin: The Synergistic Regulation of Phase Separation in Dementia



Abstract
:1. Introduction
2. Aberrant Phase Separation Is the Fundamental Molecular Driver behind Dementia
2.1. Phase Separation of α-Synuclein into Amyloid Fibrils in Dementia
2.2. The Underappreciated Role of Hydrogen Bonds and Protein Hydration in Phase Separation in Dementia
2.3. Hydration Water Activates Amyloid Aggregation and Regulates Oligomer Toxicity
2.4. The Synergistic Regulation of Hydrogen Bonds by Light, Water, and Melatonin
3. Light, Water, and Melatonin: Ancient Synergies in a Modern World
3.1. Light, Water, and Melatonin: Viscous Relationships with Hydrogen Bonds
3.1.1. PAW Modulates Melatonin Hydrogen Bonding and Conformation
3.1.2. Reactive Oxygen Species Increase Viscosity
3.1.3. Reduction in Viscosity and Hydrogen Bonds Enhance Melatonin ROS Scavenging
3.2. Light, Melatonin, and Viscosity in the Elevation of ATP Synthesis
3.2.1. Efficiency of ATP Synthase Is Modulated by Viscosity
3.2.2. The 670 nm Wavelength Elevates ATP Production in Mitochondria
3.2.3. Viscosity Modulates COX Activities in Mitochondria
3.3. Melatonin Prevents and Disaggregates Aberrant Protein Aggregation in Dementia via Association with ATP
3.3.1. Melatonin Elevates ATP Production via Modulation of COX and Viscosity
3.3.2. Fibril Disaggregation by Melatonin Is Dose-Dependent
3.3.3. Melatonin Hydrogen Bonding May Modulate Salt Bridge Formation in Aggregates
3.3.4. Hyperphosphorylation Reduces Water Hydration during Fibril Aggregation
3.4. Light, Water, and Melatonin: The Adenosine Moiety Effect of ATP

4. Of Mice and Men: Perfecting the Human Equivalent Dose for Melatonin in the Regulation of Phase Separation in Dementia
4.1. Aiming at Moving Targets in Allometric Scaling of Melatonin Interspecies Conversion
4.2. The Many Faces of Bioavailability in the Interspecies Conversion of Melatonin
4.2.1. Administration Routes Modulate Melatonin Bioavailability
4.2.2. Melatonin Bioavailability Is Inversely Correlated to 6-Sulfatoxymelatonin
4.2.3. Animals Show Large Variations in Melatonin Bioavailability
4.2.4. Solubility and Formulation Modulate Melatonin Bioavailability
4.3. Timing Is Everything in the Dosing of Melatonin for the Regulation of Phase Separation in Dementia
4.3.1. The Rationale for Frequent Division of Melatonin Doses
4.3.2. The Calculation of HED Estimates Adjusted for Differences in Metabolic Rates, Bioavailability, and Formulation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 6-OHMS | 6-sulfatoxymelatonin |
| Aβ | amyloid-β |
| AD | Alzhemer’s disease |
| AMP | adenosine monophosphate |
| α-syn | alpha-synuclein |
| ATP | adenosine triphosphate |
| CP | centipoise |
| COX | cytochrome C oxidase |
| EMF | electromagnetic fields |
| FTD | frontotemporal dementia |
| FUS | fused in sarcoma |
| H2O2 | hydrogen peroxide |
| IDP | intrinsically disordered protein |
| IP | intraperitoneal |
| IV | intravenous |
| LCD | low-complexity domain |
| MD | molecular dynamics |
| MLO | membraneless organelle |
| •OH | hydroxyl radical |
| PD | Parkinson’s disease |
| pI | isoelectric point |
| ROS | reactive oxygen species |
| SD | Sprague Dawley |
| SG | stress granule |
| VaD | Vascular dementia |
References
- Qiu, S.; Miller, M.I.; Joshi, P.S.; Lee, J.C.; Xue, C.; Ni, Y.; Wang, Y.; De Anda-Duran, I.; Hwang, P.H.; Cramer, J.A.; et al. Multimodal Deep Learning for Alzheimer’s Disease Dementia Assessment. Nat. Commun. 2022, 13, 3404. [Google Scholar] [CrossRef]
- Anstey, K.J.; Peters, R.; Clare, L.; Lautenschlager, N.T.; Dodge, H.H.; Barnes, D.E.; Shahar, S.; Brodaty, H.; Rees, G. Joining Forces to Prevent Dementia: The International Research Network on Dementia Prevention (IRNDP). Int. Psychogeriatr. 2017, 29, 1757–1760. [Google Scholar] [CrossRef] [Green Version]
- Qiu, C.; Kivipelto, M.; von Strauss, E. Epidemiology of Alzheimer’s Disease: Occurrence, Determinants, and Strategies toward Intervention. Dialogues Clin. Neurosci. 2009, 11, 111–128. [Google Scholar] [CrossRef]
- Piscopo, P.; Crestini, A.; Carbone, E.; Rivabene, R.; Ancidoni, A.; Lo Giudice, M.; Corbo, M.; Vanacore, N.; Lacorte, E. A Systematic Review on Drugs for Synaptic Plasticity in the Treatment of Dementia. Ageing Res. Rev. 2022, 81, 101726. [Google Scholar] [CrossRef] [PubMed]
- Velandia, P.P.; Miller-Petrie, M.K.; Chen, C.; Chakrabarti, S.; Chapin, A.; Hay, S.; Tsakalos, G.; Wimo, A.; Dieleman, J.L. Global and Regional Spending on Dementia Care from 2000–2019 and Expected Future Health Spending Scenarios from 2020–2050: An Economic Modelling Exercise. EClinicalMedicine 2022, 45, 101337. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The Diagnosis of Dementia due to Alzheimer’s Disease: Recommendations from the National Institute on Aging-Alzheimer’s Association Workgroups on Diagnostic Guidelines for Alzheimer’s Disease. Alzheimer’s Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendrie, H.C. Epidemiology of Dementia and Alzheimer’s Disease. Am. J. Geriatr. Psychiatry 1998, 6 (Suppl. 1), S3–S18. [Google Scholar] [CrossRef]
- O’Brien, J.T.; Thomas, A. Vascular Dementia. Lancet 2015, 386, 1698–1706. [Google Scholar] [CrossRef] [Green Version]
- Iadecola, C. The Pathobiology of Vascular Dementia. Neuron 2013, 80, 844–866. [Google Scholar] [CrossRef] [Green Version]
- Walker, Z.; Possin, K.L.; Boeve, B.F.; Aarsland, D. Lewy Body Dementias. Lancet 2015, 386, 1683–1697. [Google Scholar] [CrossRef] [Green Version]
- Bang, J.; Spina, S.; Miller, B.L. Frontotemporal Dementia. Lancet 2015, 386, 1672–1682. [Google Scholar] [CrossRef] [Green Version]
- Bolla, L.R.; Filley, C.M.; Palmer, R.M. Dementia DDx. Office Diagnosis of the Four Major Types of Dementia. Geriatrics 2000, 55, 34–37, 41, 42, 45, 46. [Google Scholar]
- Li, M.; Fan, Y.; Li, Q.; Wang, X.; Zhao, L.; Zhu, M. Liquid-Liquid Phase Separation Promotes Protein Aggregation and Its Implications in Ferroptosis in Parkinson’s Disease Dementia. Oxidative Med. Cell. Longev. 2022, 2022, 7165387. [Google Scholar] [CrossRef]
- Alberti, S.; Hyman, A.A. Biomolecular Condensates at the Nexus of Cellular Stress, Protein Aggregation Disease and Ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 196–213. [Google Scholar] [CrossRef]
- Mackenzie, I.R.; Nicholson, A.M.; Sarkar, M.; Messing, J.; Purice, M.D.; Pottier, C.; Annu, K.; Baker, M.; Perkerson, R.B.; Kurti, A.; et al. TIA1 Mutations in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia Promote Phase Separation and Alter Stress Granule Dynamics. Neuron 2017, 95, 808–816.e9. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Therneau, T.M.; Lundt, E.S.; Wiste, H.J.; Mielke, M.M.; Knopman, D.S.; Graff-Radford, J.; Lowe, V.J.; Vemuri, P.; Schwarz, C.G.; et al. Long-Term Associations between Amyloid Positron Emission Tomography, Sex, Apolipoprotein E and Incident Dementia and Mortality among Individuals without Dementia: Hazard Ratios and Absolute Risk. Brain Commun. 2022, 4, fcac017. [Google Scholar] [CrossRef]
- Gottesman, R.F.; Wu, A.; Coresh, J.; Knopman, D.S.; Jack, C.R., Jr.; Rahmim, A.; Sharrett, A.R.; Spira, A.P.; Wong, D.F.; Wagenknecht, L.E.; et al. Associations of Vascular Risk and Amyloid Burden with Subsequent Dementia. Ann. Neurol. 2022, 92, 607–619. [Google Scholar] [CrossRef]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular Condensates: Organizers of Cellular Biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.; Bard, J.A.M.; Pilipenko, E.; Drummond, D.A. Chaperones Directly and Efficiently Disperse Stress-Triggered Biomolecular Condensates. bioRxiv 2021. [Google Scholar] [CrossRef]
- Wen, Y.; Ma, J. Phase Separation Drives the Formation of Biomolecular Condensates in the Immune System. Front. Immunol. 2022, 13, 986589. [Google Scholar] [CrossRef]
- Ditlev, J.A.; Case, L.B.; Rosen, M.K. Who’s In and Who’s Out-Compositional Control of Biomolecular Condensates. J. Mol. Biol. 2018, 430, 4666–4684. [Google Scholar] [CrossRef] [PubMed]
- Yeong, V.; Werth, E.G.; Brown, L.M.; Obermeyer, A.C. Formation of Biomolecular Condensates in Bacteria by Tuning Protein Electrostatics. ACS Cent. Sci. 2020, 6, 2301–2310. [Google Scholar] [CrossRef]
- Sagan, S.M.; Weber, S.C. Let’s Phase It: Viruses Are Master Architects of Biomolecular Condensates. Trends Biochem. Sci. 2022, 48, 229–243. [Google Scholar] [CrossRef]
- Geiger, F.; Acker, J.; Papa, G.; Wang, X.; Arter, W.E.; Saar, K.L.; Erkamp, N.A.; Qi, R.; Bravo, J.P.; Strauss, S.; et al. Liquid-Liquid Phase Separation Underpins the Formation of Replication Factories in Rotaviruses. EMBO J. 2021, 40, e107711. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.H.; Davis, E.S.; Daugird, T.A.; Zhao, S.; Quiroga, I.Y.; Uryu, H.; Li, J.; Storey, A.J.; Tsai, Y.-H.; Keeley, D.P.; et al. Phase Separation Drives Aberrant Chromatin Looping and Cancer Development. Nature 2021, 595, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Zbinden, A.; Pérez-Berlanga, M.; De Rossi, P.; Polymenidou, M. Phase Separation and Neurodegenerative Diseases: A Disturbance in the Force. Dev. Cell 2020, 55, 45–68. [Google Scholar] [CrossRef]
- Elbaum-Garfinkle, S. Matter over Mind: Liquid Phase Separation and Neurodegeneration. J. Biol. Chem. 2019, 294, 7160–7168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boeynaems, S.; Alberti, S.; Fawzi, N.L.; Mittag, T.; Polymenidou, M.; Rousseau, F.; Schymkowitz, J.; Shorter, J.; Wolozin, B.; Van Den Bosch, L.; et al. Protein Phase Separation: A New Phase in Cell Biology. Trends Cell Biol. 2018, 28, 420–435. [Google Scholar] [CrossRef] [Green Version]
- Aguzzi, A.; Altmeyer, M. Phase Separation: Linking Cellular Compartmentalization to Disease. Trends Cell Biol. 2016, 26, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Rowlinson, J.S. Translation of JD van der Waals’ “The Thermodynamik Theory of Capillarity under the Hypothesis of a Continuous Variation of Density”. J. Stat. Phys. 1979, 20, 197–200. [Google Scholar] [CrossRef]
- Ginzburg, V.V.; Peng, G.; Qiu, F.; Jasnow, D.; Balazs, A.C. Kinetic Model of Phase Separation in Binary Mixtures with Hard Mobile Impurities. Phys. Rev. E 1999, 60 Pt B, 4352–4359. [Google Scholar] [CrossRef]
- Alberti, S.; Saha, S.; Woodruff, J.B.; Franzmann, T.M.; Wang, J.; Hyman, A.A. A User’s Guide for Phase Separation Assays with Purified Proteins. J. Mol. Biol. 2018, 430, 4806–4820. [Google Scholar] [CrossRef] [PubMed]
- Himeno, H.; Shimokawa, N.; Komura, S.; Andelman, D.; Hamada, T.; Takagi, M. Charge-Induced Phase Separation in Lipid Membranes. Soft Matter 2014, 10, 7959–7967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laradji, M.; Guo, H.; Grant, M.; Zuckermann, M.J. The Effect of Surfactants on the Dynamics of Phase Separation. J. Phys. Condens. Matter 1992, 4, 6715. [Google Scholar] [CrossRef]
- Choi, J.-M.; Holehouse, A.S.; Pappu, R.V. Physical Principles Underlying the Complex Biology of Intracellular Phase Transitions. Annu. Rev. Biophys. 2020, 49, 107–133. [Google Scholar] [CrossRef] [Green Version]
- De Sancho, D. Phase Separation in Amino Acid Mixtures Is Governed by Composition. Biophys. J. 2022, 121, 4119–4127. [Google Scholar] [CrossRef]
- Choi, J.-M.; Dar, F.; Pappu, R.V. LASSI: A Lattice Model for Simulating Phase Transitions of Multivalent Proteins. PLoS Comput. Biol. 2019, 15, e1007028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gereben, O.; Pusztai, L. Cluster Formation and Percolation in Ethanol-Water Mixtures. Chem. Phys. 2017, 496, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Servis, M.J.; Wu, D.T.; Braley, J.C. Network Analysis and Percolation Transition in Hydrogen Bonded Clusters: Nitric Acid and Water Extracted by Tributyl Phosphate. Phys. Chem. Chem. Phys. 2017, 19, 11326–11339. [Google Scholar] [CrossRef]
- Pothoczki, S.; Pethes, I.; Pusztai, L.; Temleitner, L.; Csókás, D.; Kohara, S.; Ohara, K.; Bakó, I. Hydrogen Bonding and Percolation in Propan-2-Ol—Water Liquid Mixtures: X-Ray Diffraction Experiments and Computer Simulations. J. Mol. Liq. 2021, 329, 115592. [Google Scholar] [CrossRef]
- Alshareedah, I.; Moosa, M.M.; Banerjee, P.R. Programmable Viscoelasticity in Protein-RNA Condensates with Disordered Sticker-Spacer Polypeptides. bioRxiv 2021. [Google Scholar] [CrossRef]
- Joseph, J.A.; Reinhardt, A.; Aguirre, A.; Chew, P.Y.; Russell, K.O.; Espinosa, J.R.; Garaizar, A.; Collepardo-Guevara, R. Physics-Driven Coarse-Grained Model for Biomolecular Phase Separation with near-Quantitative Accuracy. Nat. Comput. Sci. 2021, 1, 732–743. [Google Scholar] [CrossRef] [PubMed]
- Deniz, A.A. Percolation Physics and Density Transition Frameworks Converge in Biomolecular Condensation. Proc. Natl. Acad. Sci. USA 2022, 119, e2210177119. [Google Scholar] [CrossRef]
- Choi, J.-M.; Hyman, A.A.; Pappu, R.V. Generalized Models for Bond Percolation Transitions of Associative Polymers. Phys. Rev. E 2020, 102, 042403. [Google Scholar] [CrossRef]
- Jadrich, R.; Schweizer, K.S. Percolation, Phase Separation, and Gelation in Fluids and Mixtures of Spheres and Rods. J. Chem. Phys. 2011, 135, 234902. [Google Scholar] [CrossRef] [PubMed]
- Binder, K. Percolation Effects in the Kinetics of Phase Separation. Solid State Commun. 1980, 34, 191–194. [Google Scholar] [CrossRef]
- Kar, M.; Dar, F.; Welsh, T.J.; Vogel, L.T.; Kühnemuth, R.; Majumdar, A.; Krainer, G.; Franzmann, T.M.; Alberti, S.; Seidel, C.A.M.; et al. Phase-Separating RNA-Binding Proteins Form Heterogeneous Distributions of Clusters in Subsaturated Solutions. Proc. Natl. Acad. Sci. USA 2022, 119, e2202222119. [Google Scholar] [CrossRef]
- Thomsen, C.; Grundevik, P.; Elias, P.; Ståhlberg, A.; Aman, P. A Conserved N-Terminal Motif Is Required for Complex Formation between FUS, EWSR1, TAF15 and Their Oncogenic Fusion Proteins. FASEB J. 2013, 27, 4965–4974. [Google Scholar] [CrossRef]
- Andersson, M.K.; Ståhlberg, A.; Arvidsson, Y.; Olofsson, A.; Semb, H.; Stenman, G.; Nilsson, O.; Aman, P. The Multifunctional FUS, EWS and TAF15 Proto-Oncoproteins Show Cell Type-Specific Expression Patterns and Involvement in Cell Spreading and Stress Response. BMC Cell Biol. 2008, 9, 37. [Google Scholar] [CrossRef] [Green Version]
- McSwiggen, D.T.; Mir, M.; Darzacq, X.; Tjian, R. Evaluating Phase Separation in Live Cells: Diagnosis, Caveats, and Functional Consequences. Genes Dev. 2019, 33, 1619–1634. [Google Scholar] [CrossRef]
- Takamuku, M.; Sugishita, T.; Tamaki, H.; Dong, L.; So, M.; Fujiwara, T.; Matsuki, Y. Evolution of α-Synuclein Conformation Ensemble toward Amyloid Fibril via Liquid-Liquid Phase Separation (LLPS) as Investigated by Dynamic Nuclear Polarization-Enhanced Solid-State MAS NMR. Neurochem. Int. 2022, 157, 105345. [Google Scholar] [CrossRef] [PubMed]
- Murray, D.T.; Kato, M.; Lin, Y.; Thurber, K.R.; Hung, I.; McKnight, S.L.; Tycko, R. Structure of FUS Protein Fibrils and Its Relevance to Self-Assembly and Phase Separation of Low-Complexity Domains. Cell 2017, 171, 615–627.e16. [Google Scholar] [CrossRef] [Green Version]
- Mittag, T.; Pappu, R.V. A Conceptual Framework for Understanding Phase Separation and Addressing Open Questions and Challenges. Mol. Cell 2022, 82, 2201–2214. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s Disease: The Amyloid Cascade Hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The Amyloid Hypothesis of Alzheimer’s Disease at 25 Years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Miller, R.L.; Dhavale, D.D.; O’Shea, J.Y.; Andruska, K.M.; Liu, J.; Franklin, E.E.; Buddhala, C.; Loftin, S.K.; Cirrito, J.R.; Perrin, R.J.; et al. Quantifying Regional α -Synuclein, Amyloid β, and Tau Accumulation in Lewy Body Dementia. Ann. Clin. Transl. Neurol. 2022, 9, 106–121. [Google Scholar] [CrossRef]
- Meade, R.M.; Fairlie, D.P.; Mason, J.M. Alpha-Synuclein Structure and Parkinson’s Disease-Lessons and Emerging Principles. Mol. Neurodegener. 2019, 14, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, M.D.; Buxbaum, J.N.; Eisenberg, D.S.; Merlini, G.; Saraiva, M.J.M.; Sekijima, Y.; Sipe, J.D.; Westermark, P. Amyloid Nomenclature 2018: Recommendations by the International Society of Amyloidosis (ISA) Nomenclature Committee. Amyloid 2018, 25, 215–219. [Google Scholar] [CrossRef] [Green Version]
- Benson, M.D.; Buxbaum, J.N.; Eisenberg, D.S.; Merlini, G.; Saraiva, M.J.M.; Sekijima, Y.; Sipe, J.D.; Westermark, P. Amyloid Nomenclature 2020: Update and Recommendations by the International Society of Amyloidosis (ISA) Nomenclature Committee. Amyloid 2020, 27, 217–222. [Google Scholar] [CrossRef]
- Maji, S.K.; Wang, L.; Greenwald, J.; Riek, R. Structure-Activity Relationship of Amyloid Fibrils. FEBS Lett. 2009, 583, 2610–2617. [Google Scholar] [CrossRef] [Green Version]
- Nelson, R.; Eisenberg, D. Recent Atomic Models of Amyloid Fibril Structure. Curr. Opin. Struct. Biol. 2006, 16, 260–265. [Google Scholar] [CrossRef]
- Glabe, C.C. Amyloid Accumulation and Pathogensis of Alzheimer’s Disease: Significance of Monomeric, Oligomeric and Fibrillar Aβ. In Alzheimer’s Disease: Cellular and Molecular Aspects of Amyloid β; Harris, J.R., Fahrenholz, F., Eds.; Springer: Boston, MA, USA, 2005; pp. 167–177. [Google Scholar] [CrossRef]
- Roeters, S.J.; Iyer, A.; Pletikapić, G.; Kogan, V.; Subramaniam, V.; Woutersen, S. Evidence for Intramolecular Antiparallel Beta-Sheet Structure in Alpha-Synuclein Fibrils from a Combination of Two-Dimensional Infrared Spectroscopy and Atomic Force Microscopy. Sci. Rep. 2017, 7, 41051. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-B.; Yoon, J.-S.; Jang, S.-M.; Lee, K.-H.; Shin, S.-M. Computational Study on Oligomer Formation of Fibril-Forming Peptide of α-Synuclein. Bull. Korean Chem. Soc. 2012, 33, 848–854. [Google Scholar] [CrossRef] [Green Version]
- Serpell, L.C.; Berriman, J.; Jakes, R.; Goedert, M.; Crowther, R.A. Fiber Diffraction of Synthetic α-Synuclein Filaments Shows Amyloid-like Cross-β Conformation. Proc. Natl. Acad. Sci. USA 2000, 97, 4897–4902. [Google Scholar] [CrossRef] [Green Version]
- Passarella, D.; Goedert, M. Beta-Sheet Assembly of Tau and Neurodegeneration in Drosophila Melanogaster. Neurobiol. Aging 2018, 72, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Daebel, V.; Chinnathambi, S.; Biernat, J.; Schwalbe, M.; Habenstein, B.; Loquet, A.; Akoury, E.; Tepper, K.; Müller, H.; Baldus, M.; et al. β-Sheet Core of Tau Paired Helical Filaments Revealed by Solid-State NMR. J. Am. Chem. Soc. 2012, 134, 13982–13989. [Google Scholar] [CrossRef] [Green Version]
- von Bergen, M.; Barghorn, S.; Biernat, J.; Mandelkow, E.-M.; Mandelkow, E. Tau Aggregation Is Driven by a Transition from Random Coil to Beta Sheet Structure. Biochim. Biophys. Acta 2005, 1739, 158–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spillantini, M.G.; Divane, A.; Goedert, M. Assignment of Human Alpha-Synuclein (SNCA) and Beta-Synuclein (SNCB) Genes to Chromosomes 4q21 and 5q35. Genomics 1995, 27, 379–381. [Google Scholar] [CrossRef] [PubMed]
- Jakes, R.; Spillantini, M.G.; Goedert, M. Identification of Two Distinct Synucleins from Human Brain. FEBS Lett. 1994, 345, 27–32. [Google Scholar] [CrossRef] [Green Version]
- Coelho-Cerqueira, E.; Carmo-Gonçalves, P.; Pinheiro, A.S.; Cortines, J.; Follmer, C. α-Synuclein as an Intrinsically Disordered Monomer—Fact or Artefact? FEBS J. 2013, 280, 4915–4927. [Google Scholar] [CrossRef]
- Hoppe, S.O.; Uzunoğlu, G.; Nussbaum-Krammer, C. α-Synuclein Strains: Does Amyloid Conformation Explain the Heterogeneity of Synucleinopathies? Biomolecules 2021, 11, 931. [Google Scholar] [CrossRef]
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.A.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S.; et al. In Vivo Demonstration That α-Synuclein Oligomers Are Toxic. Proc. Natl. Acad. Sci. USA 2011, 108, 4194–4199. [Google Scholar] [CrossRef] [Green Version]
- Poudyal, M.; Sakunthala, A.; Mukherjee, S.; Gadhe, L.; Maji, S.K. Phase Separation and Other Forms of α-Synuclein Self-Assemblies. Essays Biochem. 2022, 66, 987–1000. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Sakunthala, A.; Gadhe, L.; Poudyal, M.; Sawner, A.S.; Kadu, P.; Maji, S.K. Liquid-Liquid Phase Separation of α-Synuclein: A New Mechanistic Insight for α-Synuclein Aggregation Associated with Parkinson’s Disease Pathogenesis. J. Mol. Biol. 2022, 435, 167713. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Mo, X.; Wang, J.; Ye, X.; Yu, H.; Liu, Y. α-Synuclein Phase Separation and Amyloid Aggregation Are Modulated by C-Terminal Truncations. FEBS Lett. 2022, 596, 1388–1400. [Google Scholar] [CrossRef]
- Hardenberg, M.C.; Sinnige, T.; Casford, S.; Dada, S.T.; Poudel, C.; Robinson, E.A.; Fuxreiter, M.; Kaminksi, C.F.; Kaminski Schierle, G.S.; Nollen, E.A.A.; et al. Observation of an α-Synuclein Liquid Droplet State and Its Maturation into Lewy Body-like Assemblies. J. Mol. Cell Biol. 2021, 13, 282–294. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Singh, N.; Kumar, R.; Patel, K.; Pandey, S.; Datta, D.; Mahato, J.; Panigrahi, R.; Navalkar, A.; Mehra, S.; et al. α-Synuclein Aggregation Nucleates through Liquid-Liquid Phase Separation. Nat. Chem. 2020, 12, 705–716. [Google Scholar] [CrossRef]
- Ray, S.; Singh, N.; Patel, K.; Krishnamoorthy, G.; Maji, S.K. FRAP and FRET Investigation of α-Synuclein Fibrillization via Liquid-Liquid Phase Separation In Vitro and in HeLa Cells. In Protein Aggregation: Methods and Protocols; Cieplak, A.S., Ed.; Springer: New York, NY, USA, 2023; pp. 395–423. [Google Scholar] [CrossRef]
- Xu, B.; Mo, X.; Chen, J.; Yu, H.; Liu, Y. Myricetin Inhibits α-Synuclein Amyloid Aggregation by Delaying the Liquid-to-Solid Phase Transition. Chembiochem 2022, 23, e202200216. [Google Scholar] [CrossRef]
- Davies, H.A.; Rigden, D.J.; Phelan, M.M.; Madine, J. Probing Medin Monomer Structure and Its Amyloid Nucleation Using 13C-Direct Detection NMR in Combination with Structural Bioinformatics. Sci. Rep. 2017, 7, 45224. [Google Scholar] [CrossRef] [Green Version]
- Larsson, A.; Söderberg, L.; Westermark, G.T.; Sletten, K.; Engström, U.; Tjernberg, L.O.; Näslund, J.; Westermark, P. Unwinding Fibril Formation of Medin, the Peptide of the Most Common Form of Human Amyloid. Biochem. Biophys. Res. Commun. 2007, 361, 822–828. [Google Scholar] [CrossRef]
- Peng, S.; Glennert, J.; Westermark, P. Medin-Amyloid: A Recently Characterized Age-Associated Arterial Amyloid Form Affects Mainly Arteries in the Upper Part of the Body. Amyloid 2005, 12, 96–102. [Google Scholar] [CrossRef]
- Marazuela, P.; Solé, M.; Bonaterra-Pastra, A.; Pizarro, J.; Camacho, J.; Martínez-Sáez, E.; Kuiperij, H.B.; Verbeek, M.M.; de Kort, A.M.; Schreuder, F.H.B.M.; et al. MFG-E8 (LACTADHERIN): A Novel Marker Associated with Cerebral Amyloid Angiopathy. Acta Neuropathol. Commun. 2021, 9, 154. [Google Scholar] [CrossRef]
- Larsson, A.; Malmström, S.; Westermark, P. Signs of Cross-Seeding: Aortic Medin Amyloid as a Trigger for Protein AA Deposition. Amyloid 2011, 18, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Larsson, A.; Peng, S.; Persson, H.; Rosenbloom, J.; Abrams, W.R.; Wassberg, E.; Thelin, S.; Sletten, K.; Gerwins, P.; Westermark, P. Lactadherin Binds to Elastin—A Starting Point for Medin Amyloid Formation? Amyloid 2006, 13, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Degenhardt, K.; Veit, M.; Louros, N.; Konstantoulea, K.; Skodras, A.; Wild, K.; Liu, P.; Obermüller, U.; Bansal, V.; et al. Medin Co-Aggregates with Vascular Amyloid-β in Alzheimer’s Disease. Nature 2022, 612, 123–131. [Google Scholar] [CrossRef]
- Degenhardt, K.; Wagner, J.; Skodras, A.; Candlish, M.; Koppelmann, A.J.; Wild, K.; Maxwell, R.; Rotermund, C.; von Zweydorf, F.; Gloeckner, C.J.; et al. Medin Aggregation Causes Cerebrovascular Dysfunction in Aging Wild-Type Mice. Proc. Natl. Acad. Sci. USA 2020, 117, 23925–23931. [Google Scholar] [CrossRef] [PubMed]
- Karamanova, N.; Truran, S.; Serrano, G.E.; Beach, T.G.; Madine, J.; Weissig, V.; Davies, H.A.; Veldhuizen, J.; Nikkhah, M.; Hansen, M.; et al. Endothelial Immune Activation by Medin: Potential Role in Cerebrovascular Disease and Reversal by Monosialoganglioside-Containing Nanoliposomes. J. Am. Heart Assoc. 2020, 9, e014810. [Google Scholar] [CrossRef] [PubMed]
- Migrino, R.Q.; Karamanova, N.; Truran, S.; Serrano, G.E.; Davies, H.A.; Madine, J.; Beach, T.G. Cerebrovascular Medin Is Associated with Alzheimer’s Disease and Vascular Dementia. Alzheimer’s Dement. 2020, 12, e12072. [Google Scholar] [CrossRef]
- Sabha, B.H.; Alzahrani, F.; Almehdar, H.A.; Uversky, V.N.; Redwan, E.M. Disorder in Milk Proteins: Lactadherin Multifunctionality and Structure. Curr. Protein Pept. Sci. 2018, 19, 983–997. [Google Scholar] [CrossRef]
- Fens, M.H.A.M.; Mastrobattista, E.; de Graaff, A.M.; Flesch, F.M.; Ultee, A.; Rasmussen, J.T.; Molema, G.; Storm, G.; Schiffelers, R.M. Angiogenic Endothelium Shows Lactadherin-Dependent Phagocytosis of Aged Erythrocytes and Apoptotic Cells. Blood 2008, 111, 4542–4550. [Google Scholar] [CrossRef]
- Silvestre, J.-S.; Théry, C.; Hamard, G.; Boddaert, J.; Aguilar, B.; Delcayre, A.; Houbron, C.; Tamarat, R.; Blanc-Brude, O.; Heeneman, S.; et al. Lactadherin Promotes VEGF-Dependent Neovascularization. Nat. Med. 2005, 11, 499–506. [Google Scholar] [CrossRef]
- Bu, H.-F.; Zuo, X.-L.; Wang, X.; Ensslin, M.A.; Koti, V.; Hsueh, W.; Raymond, A.S.; Shur, B.D.; Tan, X.-D. Milk Fat Globule-EGF Factor 8/lactadherin Plays a Crucial Role in Maintenance and Repair of Murine Intestinal Epithelium. J. Clin. Investig. 2007, 117, 3673–3683. [Google Scholar] [CrossRef] [Green Version]
- Younger, S.; Jang, H.; Davies, H.A.; Niemiec, M.J.; Garcia, J.G.N.; Nussinov, R.; Migrino, R.Q.; Madine, J.; Arce, F.T. Medin Oligomer Membrane Pore Formation: A Potential Mechanism of Vascular Dysfunction. Biophys. J. 2020, 118, 2769–2782. [Google Scholar] [CrossRef] [PubMed]
- Migrino, R.Q.; Davies, H.A.; Truran, S.; Karamanova, N.; Franco, D.A.; Beach, T.G.; Serrano, G.E.; Truong, D.; Nikkhah, M.; Madine, J. Amyloidogenic Medin Induces Endothelial Dysfunction and Vascular Inflammation through the Receptor for Advanced Glycation Endproducts. Cardiovasc. Res. 2017, 113, 1389–1402. [Google Scholar] [CrossRef] [Green Version]
- Arispe, N.; Rojas, E.; Pollard, H.B. Alzheimer Disease Amyloid Beta Protein Forms Calcium Channels in Bilayer Membranes: Blockade by Tromethamine and Aluminum. Proc. Natl. Acad. Sci. USA 1993, 90, 567–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arispe, N.; Pollard, H.B.; Rojas, E. Zn2+ Interaction with Alzheimer Amyloid Beta Protein Calcium Channels. Proc. Natl. Acad. Sci. USA 1996, 93, 1710–1715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konstantoulea, K.; Louros, N.; Rousseau, F.; Schymkowitz, J. Heterotypic Interactions in Amyloid Function and Disease. FEBS J. 2022, 289, 2025–2046. [Google Scholar] [CrossRef]
- Nag, N.; Tripathi, T. Cross-Seeding with Homologous Sequences Alters Amyloid Aggregation Kinetics and Fibril Structure. ACS Chem. Neurosci. 2022, 13, 537–539. [Google Scholar] [CrossRef]
- Mathieu, C.; Pappu, R.V.; Taylor, J.P. Beyond Aggregation: Pathological Phase Transitions in Neurodegenerative Disease. Science 2020, 370, 56–60. [Google Scholar] [CrossRef]
- St George-Hyslop, P.; Lin, J.Q.; Miyashita, A.; Phillips, E.C.; Qamar, S.; Randle, S.J.; Wang, G. The Physiological and Pathological Biophysics of Phase Separation and Gelation of RNA Binding Proteins in Amyotrophic Lateral Sclerosis and Fronto-Temporal Lobar Degeneration. Brain Res. 2018, 1693 Pt A, 11–23. [Google Scholar] [CrossRef]
- Vanderweyde, T.; Apicco, D.J.; Youmans-Kidder, K.; Ash, P.E.A.; Cook, C.; da Rocha, E.L.; Jansen-West, K.; Frame, A.A.; Citro, A.; Leszyk, J.D.; et al. Interaction of Tau with the RNA-Binding Protein TIA1 Regulates Tau Pathophysiology and Toxicity. Cell Rep. 2016, 15, 1455–1466. [Google Scholar] [CrossRef] [Green Version]
- Gui, X.; Luo, F.; Li, Y.; Zhou, H.; Qin, Z.; Liu, Z.; Gu, J.; Xie, M.; Zhao, K.; Dai, B.; et al. Structural Basis for Reversible Amyloids of hnRNPA1 Elucidates Their Role in Stress Granule Assembly. Nat. Commun. 2019, 10, 2006. [Google Scholar] [CrossRef] [Green Version]
- Ayyadevara, S.; Balasubramaniam, M.; Parcon, P.A.; Barger, S.W.; Griffin, W.S.T.; Alla, R.; Tackett, A.J.; Mackintosh, S.G.; Petricoin, E.; Zhou, W.; et al. Proteins That Mediate Protein Aggregation and Cytotoxicity Distinguish Alzheimer’s Hippocampus from Normal Controls. Aging Cell 2016, 15, 924–939. [Google Scholar] [CrossRef] [PubMed]
- Brunello, C.A.; Yan, X.; Huttunen, H.J. Internalized Tau Sensitizes Cells to Stress by Promoting Formation and Stability of Stress Granules. Sci. Rep. 2016, 6, 30498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maziuk, B.; Ballance, H.I.; Wolozin, B. Dysregulation of RNA Binding Protein Aggregation in Neurodegenerative Disorders. Front. Mol. Neurosci. 2017, 10, 89. [Google Scholar] [CrossRef] [Green Version]
- Bishof, I.; Dammer, E.B.; Duong, D.M.; Kundinger, S.R.; Gearing, M.; Lah, J.J.; Levey, A.I.; Seyfried, N.T. RNA-Binding Proteins with Basic-Acidic Dipeptide (BAD) Domains Self-Assemble and Aggregate in Alzheimer’s Disease. J. Biol. Chem. 2018, 293, 11047–11066. [Google Scholar] [CrossRef] [Green Version]
- Kapeli, K.; Martinez, F.J.; Yeo, G.W. Genetic Mutations in RNA-Binding Proteins and Their Roles in ALS. Hum. Genet. 2017, 136, 1193–1214. [Google Scholar] [CrossRef] [Green Version]
- Cookson, M.R. RNA-Binding Proteins Implicated in Neurodegenerative Diseases. Wiley Interdiscip. Rev. RNA 2017, 8, e1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cascarina, S.M.; Elder, M.R.; Ross, E.D. Atypical Structural Tendencies among Low-Complexity Domains in the Protein Data Bank Proteome. PLoS Comput. Biol. 2020, 16, e1007487. [Google Scholar] [CrossRef] [Green Version]
- Posey, A.E.; Holehouse, A.S.; Pappu, R.V. Phase Separation of Intrinsically Disordered Proteins. Methods Enzymol. 2018, 611, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsically Disordered Proteins in Overcrowded Milieu: Membrane-Less Organelles, Phase Separation, and Intrinsic Disorder. Curr. Opin. Struct. Biol. 2017, 44, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Hyman, A.A.; Weber, C.A.; Jülicher, F. Liquid-Liquid Phase Separation in Biology. Annu. Rev. Cell Dev. Biol. 2014, 30, 39–58. [Google Scholar] [CrossRef] [Green Version]
- Kragelj, J.; Orand, T.; Delaforge, E.; Tengo, L.; Blackledge, M.; Palencia, A.; Jensen, M.R. Enthalpy-Entropy Compensation in the Promiscuous Interaction of an Intrinsically Disordered Protein with Homologous Protein Partners. Biomolecules 2021, 11, 1204. [Google Scholar] [CrossRef] [PubMed]
- Arbesú, M.; Iruela, G.; Fuentes, H.; Teixeira, J.M.C.; Pons, M. Intramolecular Fuzzy Interactions Involving Intrinsically Disordered Domains. Front. Mol. Biosci. 2018, 5, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flock, T.; Weatheritt, R.J.; Latysheva, N.S.; Babu, M.M. Controlling Entropy to Tune the Functions of Intrinsically Disordered Regions. Curr. Opin. Struct. Biol. 2014, 26, 62–72. [Google Scholar] [CrossRef]
- Workman, R.J.; Drake, J.A.; Pettitt, B.M. Chapter 4—Thermodynamic Perspective of Protein Disorder and Phase Separation: Model Systems. In Structure and Intrinsic Disorder in Enzymology; Gupta, M.N., Uversky, V.N., Eds.; Academic Press: Cambridge, MA, USA, 2023; pp. 97–126. [Google Scholar] [CrossRef]
- Flory, P.J. Thermodynamics of High Polymer Solutions. J. Chem. Phys. 1942, 10, 51–61. [Google Scholar] [CrossRef]
- Huggins, M.L. Some Properties of Solutions of Long-Chain Compounds. J. Phys. Chem. 1942, 46, 151–158. [Google Scholar] [CrossRef]
- Dignon, G.L.; Best, R.B.; Mittal, J. Biomolecular Phase Separation: From Molecular Driving Forces to Macroscopic Properties. Annu. Rev. Phys. Chem. 2020, 71, 53–75. [Google Scholar] [CrossRef] [Green Version]
- Qian, H.; Hopfield, J.J. Entropy-enthalpy Compensation: Perturbation and Relaxation in Thermodynamic Systems. J. Chem. Phys. 1996, 105, 9292–9298. [Google Scholar] [CrossRef]
- Jacobson, K.; Papahadjopoulos, D. Phase Transitions and Phase Separations in Phospholipid Membranes Induced by Changes in Temperature, pH, and Concentration of Bivalent Cations. Biochemistry 1975, 14, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Nott, T.J.; Petsalaki, E.; Farber, P.; Jervis, D.; Fussner, E.; Plochowietz, A.; Craggs, T.D.; Bazett-Jones, D.P.; Pawson, T.; Forman-Kay, J.D.; et al. Phase Transition of a Disordered Nuage Protein Generates Environmentally Responsive Membraneless Organelles. Mol. Cell 2015, 57, 936–947. [Google Scholar] [CrossRef] [Green Version]
- Molliex, A.; Temirov, J.; Lee, J.; Coughlin, M.; Kanagaraj, A.P.; Kim, H.J.; Mittag, T.; Taylor, J.P. Phase Separation by Low Complexity Domains Promotes Stress Granule Assembly and Drives Pathological Fibrillization. Cell 2015, 163, 123–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riback, J.A.; Katanski, C.D.; Kear-Scott, J.L.; Pilipenko, E.V.; Rojek, A.E.; Sosnick, T.R.; Drummond, D.A. Stress-Triggered Phase Separation Is an Adaptive, Evolutionarily Tuned Response. Cell 2017, 168, 1028–1040.e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cinar, H.; Fetahaj, Z.; Cinar, S.; Vernon, R.M.; Chan, H.S.; Winter, R.H.A. Temperature, Hydrostatic Pressure, and Osmolyte Effects on Liquid-Liquid Phase Separation in Protein Condensates: Physical Chemistry and Biological Implications. Chemistry 2019, 25, 13049–13069. [Google Scholar] [CrossRef]
- Protter, D.S.W.; Parker, R. Principles and Properties of Stress Granules. Trends Cell Biol. 2016, 26, 668–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamaki, M.; Kojima, C. pH-Switchable LCST/UCST-Type Thermosensitive Behaviors of Phenylalanine-Modified Zwitterionic Dendrimers. RSC Adv. 2020, 10, 10452–10460. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Zhang, J.; Dai, W.; Zhao, Y. Facile Synthesis of Thermo-, pH-, CO2- and Oxidation-Responsive Poly(amido Thioether)s with Tunable LCST and UCST Behaviors. Polym. Chem. 2017, 8, 5749–5760. [Google Scholar] [CrossRef]
- Jin, X.; Zhou, M.; Chen, S.; Li, D.; Cao, X.; Liu, B. Effects of pH Alterations on Stress- and Aging-Induced Protein Phase Separation. Cell. Mol. Life Sci. 2022, 79, 380. [Google Scholar] [CrossRef]
- Orij, R.; Brul, S.; Smits, G.J. Intracellular pH Is a Tightly Controlled Signal in Yeast. Biochim. Biophys. Acta 2011, 1810, 933–944. [Google Scholar] [CrossRef]
- Kroschwald, S.; Munder, M.C.; Maharana, S.; Franzmann, T.M.; Richter, D.; Ruer, M.; Hyman, A.A.; Alberti, S. Different Material States of Pub1 Condensates Define Distinct Modes of Stress Adaptation and Recovery. Cell Rep. 2018, 23, 3327–3339. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Fichou, Y.; Longhini, A.P.; Llanes, L.C.; Yin, P.; Bazan, G.C.; Kosik, K.S.; Han, S. Liquid-Liquid Phase Separation of Tau Driven by Hydrophobic Interaction Facilitates Fibrillization of Tau. J. Mol. Biol. 2021, 433, 166731. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Barnes, R.; Lin, Y.; Jeon, B.-J.; Najafi, S.; Delaney, K.T.; Fredrickson, G.H.; Shea, J.-E.; Hwang, D.S.; Han, S. Dehydration Entropy Drives Liquid-Liquid Phase Separation by Molecular Crowding. Commun. Chem. 2020, 3, 83. [Google Scholar] [CrossRef]
- Elbaum-Garfinkle, S.; Kim, Y.; Szczepaniak, K.; Chen, C.C.-H.; Eckmann, C.R.; Myong, S.; Brangwynne, C.P. The Disordered P Granule Protein LAF-1 Drives Phase Separation into Droplets with Tunable Viscosity and Dynamics. Proc. Natl. Acad. Sci. USA 2015, 112, 7189–7194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salis, A.; Ninham, B.W. Models and Mechanisms of Hofmeister Effects in Electrolyte Solutions, and Colloid and Protein Systems Revisited. Chem. Soc. Rev. 2014, 43, 7358–7377. [Google Scholar] [CrossRef] [Green Version]
- Kalra, A.; Tugcu, N.; Cramer, S.M.; Garde, S. Salting-in and Salting-out of Hydrophobic Solutes in Aqueous Salt Solutions. J. Phys. Chem. B 2001, 105, 6380–6386. [Google Scholar] [CrossRef]
- Melander, W.; Horváth, C. Salt Effect on Hydrophobic Interactions in Precipitation and Chromatography of Proteins: An Interpretation of the Lyotropic Series. Arch. Biochem. Biophys. 1977, 183, 200–215. [Google Scholar] [CrossRef]
- Collins, K.D.; Washabaugh, M.W. The Hofmeister Effect and the Behaviour of Water at Interfaces. Q. Rev. Biophys. 1985, 18, 323–422. [Google Scholar] [CrossRef]
- Friberg, S.E.; Chiu, M. Hydrotropes. J. Dispers. Sci. Technol. 1988, 9, 443–457. [Google Scholar] [CrossRef]
- Schwierz, N.; Horinek, D.; Netz, R.R. Anionic and Cationic Hofmeister Effects on Hydrophobic and Hydrophilic Surfaces. Langmuir 2013, 29, 2602–2614. [Google Scholar] [CrossRef]
- Furukawa, K.; Aguirre, C.; So, M.; Sasahara, K.; Miyanoiri, Y.; Sakurai, K.; Yamaguchi, K.; Ikenaka, K.; Mochizuki, H.; Kardos, J.; et al. Isoelectric Point-Amyloid Formation of α-Synuclein Extends the Generality of the Solubility and Supersaturation-Limited Mechanism. Curr. Res. Struct. Biol. 2020, 2, 35–44. [Google Scholar] [CrossRef]
- Vugmeyster, L.; Clark, M.A.; Falconer, I.B.; Ostrovsky, D.; Gantz, D.; Qiang, W.; Hoatson, G.L. Flexibility and Solvation of Amyloid-β Hydrophobic Core. J. Biol. Chem. 2016, 291, 18484–18495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, R.; Chiba, S.; Okuwaki, K.; Kanada, R.; Doi, H.; Ono, M.; Mochizuki, Y.; Okuno, Y. Stabilization Mechanism for a Nonfibrillar Amyloid β Oligomer Based on Formation of a Hydrophobic Core Determined by Dissipative Particle Dynamics. ACS Chem. Neurosci. 2020, 11, 385–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majumdar, A.; Das, D.; Madhu, P.; Avni, A.; Mukhopadhyay, S. Excitation Energy Migration Unveils Fuzzy Interfaces within the Amyloid Architecture. Biophys. J. 2020, 118, 2621–2626. [Google Scholar] [CrossRef]
- Fernández, A. Time-Resolved Backbone Desolvation and Mutational Hot Spots in Folding Proteins. Proteins 2002, 47, 447–457. [Google Scholar] [CrossRef]
- Nakagawa, H.; Tamada, T. Hydration and Its Hydrogen Bonding State on a Protein Surface in the Crystalline State as Revealed by Molecular Dynamics Simulation. Front. Chem. 2021, 9, 738077. [Google Scholar] [CrossRef]
- Bellissent-Funel, M.-C.; Hassanali, A.; Havenith, M.; Henchman, R.; Pohl, P.; Sterpone, F.; van der Spoel, D.; Xu, Y.; Garcia, A.E. Water Determines the Structure and Dynamics of Proteins. Chem. Rev. 2016, 116, 7673–7697. [Google Scholar] [CrossRef] [PubMed]
- Levy, Y.; Onuchic, J.N. Water Mediation in Protein Folding and Molecular Recognition. Annu. Rev. Biophys. Biomol. Struct. 2006, 35, 389–415. [Google Scholar] [CrossRef] [Green Version]
- Chaplin, M. Do We Underestimate the Importance of Water in Cell Biology? Nat. Rev. Mol. Cell Biol. 2006, 7, 861–866. [Google Scholar] [CrossRef]
- Lum, K.; Chandler, D.; Weeks, J.D. Hydrophobicity at Small and Large Length Scales. J. Phys. Chem. B 1999, 103, 4570–4577. [Google Scholar] [CrossRef]
- Ahlers, J.; Adams, E.M.; Bader, V.; Pezzotti, S.; Winklhofer, K.F.; Tatzelt, J.; Havenith, M. The Key Role of Solvent in Condensation: Mapping Water in Liquid-Liquid Phase-Separated FUS. Biophys. J. 2021, 120, 1266–1275. [Google Scholar] [CrossRef]
- Conti Nibali, V.; Pezzotti, S.; Sebastiani, F.; Galimberti, D.R.; Schwaab, G.; Heyden, M.; Gaigeot, M.-P.; Havenith, M. Wrapping Up Hydrophobic Hydration: Locality Matters. J. Phys. Chem. Lett. 2020, 11, 4809–4816. [Google Scholar] [CrossRef]
- Laage, D.; Elsaesser, T.; Hynes, J.T. Water Dynamics in the Hydration Shells of Biomolecules. Chem. Rev. 2017, 117, 10694–10725. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, A.C.; Laage, D. Water Dynamics in Protein Hydration Shells: The Molecular Origins of the Dynamical Perturbation. J. Phys. Chem. B 2014, 118, 7715–7729. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chan, H.S. Desolvation Is a Likely Origin of Robust Enthalpic Barriers to Protein Folding. J. Mol. Biol. 2005, 349, 872–889. [Google Scholar] [CrossRef]
- Camino, J.D.; Gracia, P.; Cremades, N. The Role of Water in the Primary Nucleation of Protein Amyloid Aggregation. Biophys. Chem. 2021, 269, 106520. [Google Scholar] [CrossRef] [PubMed]
- Castellano, B.M.; Eggers, D.K. Experimental Support for a Desolvation Energy Term in Governing Equations for Binding Equilibria. J. Phys. Chem. B 2013, 117, 8180–8188. [Google Scholar] [CrossRef] [Green Version]
- Kauzmann, W. Some Factors in the Interpretation of Protein Denaturation. Adv. Protein Chem. 1959, 14, 1–63. [Google Scholar] [CrossRef]
- Blokzijl, W.; Engberts, J.B.F.N. Hydrophobic Effects. Opinions and Facts. Angew. Chem. Int. Ed. Engl. 1993, 32, 1545–1579. [Google Scholar] [CrossRef]
- Muller, N. Search for a Realistic View of Hydrophobic Effects. Acc. Chem. Res. 1990, 23, 23–28. [Google Scholar] [CrossRef]
- Hamaker, H.C. The London—Van Der Waals Attraction between Spherical Particles. Physica 1937, 4, 1058–1072. [Google Scholar] [CrossRef]
- Davis, J.G.; Gierszal, K.P.; Wang, P.; Ben-Amotz, D. Water Structural Transformation at Molecular Hydrophobic Interfaces. Nature 2012, 491, 582–585. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, R.L. Dynamic Hydration Shell Restores Kauzmann’s 1959 Explanation of How the Hydrophobic Factor Drives Protein Folding. Proc. Natl. Acad. Sci. USA 2014, 111, 13052–13056. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Hou, C.; Ma, X.; Guo, S.; Zhang, H.; Shi, L.; Liao, C.; Zheng, B.; Ye, L.; Yang, L.; et al. Entropy-Enthalpy Compensations Fold Proteins in Precise Ways. Int. J. Mol. Sci. 2021, 22, 9653. [Google Scholar] [CrossRef]
- Mancera, R.L. Computer Simulation of the Effect of Salt on the Hydrophobic Effect. J. Chem. Soc. Faraday Trans. 1998, 94, 3549–3559. [Google Scholar] [CrossRef]
- Tuladhar, A.; Dewan, S.; Pezzotti, S.; Brigiano, F.S.; Creazzo, F.; Gaigeot, M.-P.; Borguet, E. Ions Tune Interfacial Water Structure and Modulate Hydrophobic Interactions at Silica Surfaces. J. Am. Chem. Soc. 2020, 142, 6991–7000. [Google Scholar] [CrossRef] [PubMed]
- Arya, S.; Singh, A.K.; Bhasne, K.; Dogra, P.; Datta, A.; Das, P.; Mukhopadhyay, S. Femtosecond Hydration Map of Intrinsically Disordered α-Synuclein. Biophys. J. 2018, 114, 2540–2551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, J.A.; Ivanova, M.I.; Sawaya, M.R.; Cascio, D.; Reyes, F.E.; Shi, D.; Sangwan, S.; Guenther, E.L.; Johnson, L.M.; Zhang, M.; et al. Structure of the Toxic Core of α-Synuclein from Invisible Crystals. Nature 2015, 525, 486–490. [Google Scholar] [CrossRef] [Green Version]
- Thirumalai, D.; Reddy, G.; Straub, J.E. Role of Water in Protein Aggregation and Amyloid Polymorphism. Acc. Chem. Res. 2012, 45, 83–92. [Google Scholar] [CrossRef]
- Krone, M.G.; Hua, L.; Soto, P.; Zhou, R.; Berne, B.J.; Shea, J.-E. Role of Water in Mediating the Assembly of Alzheimer Amyloid-Beta Abeta16-22 Protofilaments. J. Am. Chem. Soc. 2008, 130, 11066–11072. [Google Scholar] [CrossRef] [Green Version]
- Klement, K.; Wieligmann, K.; Meinhardt, J.; Hortschansky, P.; Richter, W.; Fändrich, M. Effect of Different Salt Ions on the Propensity of Aggregation and on the Structure of Alzheimer’s abeta(1-40) Amyloid Fibrils. J. Mol. Biol. 2007, 373, 1321–1333. [Google Scholar] [CrossRef] [PubMed]
- van der Lubbe, S.C.C.; Fonseca Guerra, C. The Nature of Hydrogen Bonds: A Delineation of the Role of Different Energy Components on Hydrogen Bond Strengths and Lengths. Chem. Asian J. 2019, 14, 2760–2769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muthachikavil, A.V.; Peng, B.; Kontogeorgis, G.M.; Liang, X. Distinguishing Weak and Strong Hydrogen Bonds in Liquid Water-A Potential of Mean Force-Based Approach. J. Phys. Chem. B 2021, 125, 7187–7198. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Fichou, Y.; Zeng, Z.; Hu, N.Y.; Han, S. Electrostatically Driven Complex Coacervation and Amyloid Aggregation of Tau Are Independent Processes with Overlapping Conditions. ACS Chem. Neurosci. 2020, 11, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Camino, J.D.; Gracia, P.; Chen, S.W.; Sot, J.; de la Arada, I.; Sebastián, V.; Arrondo, J.L.R.; Goñi, F.M.; Dobson, C.M.; Cremades, N. The Extent of Protein Hydration Dictates the Preference for Heterogeneous or Homogeneous Nucleation Generating Either Parallel or Antiparallel β-Sheet α-Synuclein Aggregates. Chem. Sci. 2020, 11, 11902–11914. [Google Scholar] [CrossRef] [PubMed]
- Tarus, B.; Straub, J.E.; Thirumalai, D. Probing the Initial Stage of Aggregation of the Abeta(10-35)-Protein: Assessing the Propensity for Peptide Dimerization. J. Mol. Biol. 2005, 345, 1141–1156. [Google Scholar] [CrossRef]
- Esler, W.P.; Felix, A.M.; Stimson, E.R.; Lachenmann, M.J.; Ghilardi, J.R.; Lu, Y.A.; Vinters, H.V.; Mantyh, P.W.; Lee, J.P.; Maggio, J.E. Activation Barriers to Structural Transition Determine Deposition Rates of Alzheimer’s Disease a Beta Amyloid. J. Struct. Biol. 2000, 130, 174–183. [Google Scholar] [CrossRef]
- Chang, L.; Ernst, T.; Poland, R.E.; Jenden, D.J. In Vivo Proton Magnetic Resonance Spectroscopy of the Normal Aging Human Brain. Life Sci. 1996, 58, 2049–2056. [Google Scholar] [CrossRef]
- Naber, D.; Korte, U.; Krack, K. Content of Water-Soluble and Total Proteins in the Aging Human Brain. Exp. Gerontol. 1979, 14, 59–63. [Google Scholar] [CrossRef]
- Chong, S.-H.; Ham, S. Dynamics of Hydration Water Plays a Key Role in Determining the Binding Thermodynamics of Protein Complexes. Sci. Rep. 2017, 7, 8744. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, S.S.; Samanta, N.; Ebbinghaus, S.; Marcos, J.C. The Synergic Effect of Water and Biomolecules in Intracellular Phase Separation. Nat. Rev. Chem. 2019, 3, 552–561. [Google Scholar] [CrossRef]
- Wang, T.; Jo, H.; DeGrado, W.F.; Hong, M. Water Distribution, Dynamics, and Interactions with Alzheimer’s β-Amyloid Fibrils Investigated by Solid-State NMR. J. Am. Chem. Soc. 2017, 139, 6242–6252. [Google Scholar] [CrossRef] [PubMed]
- Schwierz, N.; Frost, C.V.; Geissler, P.L.; Zacharias, M. Dynamics of Seeded Aβ40-Fibril Growth from Atomistic Molecular Dynamics Simulations: Kinetic Trapping and Reduced Water Mobility in the Locking Step. J. Am. Chem. Soc. 2016, 138, 527–539. [Google Scholar] [CrossRef] [PubMed]
- van der Spoel, D.; van Maaren, P.J.; Larsson, P.; Tîmneanu, N. Thermodynamics of Hydrogen Bonding in Hydrophilic and Hydrophobic Media. J. Phys. Chem. B 2006, 110, 4393–4398. [Google Scholar] [CrossRef] [PubMed]
- Arunan, E.; Desiraju, G.R.; Klein, R.A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D.C.; Crabtree, R.H.; Dannenberg, J.J.; Hobza, P.; et al. Definition of the hydrogen bond (IUPAC Recommendations 2011). J. Macromol. Sci. Part A Pure Appl. Chem. 2011, 83, 1637–1641. [Google Scholar] [CrossRef]
- Dong, K.; Zhang, S.; Wang, J. Understanding the Hydrogen Bonds in Ionic Liquids and Their Roles in Properties and Reactions. Chem. Commun. 2016, 52, 6744–6764. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.; Liu, L.; Liu, X.; Zhang, X.; Zhang, S. Insight into the Relationship between Viscosity and Hydrogen Bond of a Series of Imidazolium Ionic Liquids: A Molecular Dynamics and Density Functional Theory Study. Ind. Eng. Chem. Res. 2019, 58, 18848–18854. [Google Scholar] [CrossRef]
- Ma, Y.; Liu, Y.; Su, H.; Wang, L.; Zhang, J. Relationship between Hydrogen Bond and Viscosity for a Series of Pyridinium Ionic Liquids: Molecular Dynamics and Quantum Chemistry. J. Mol. Liq. 2018, 255, 176–184. [Google Scholar] [CrossRef]
- Goertz, M.P.; Houston, J.E.; Zhu, X.-Y. Hydrophilicity and the Viscosity of Interfacial Water. Langmuir 2007, 23, 5491–5497. [Google Scholar] [CrossRef]
- Luzar, A.; Chandler, D. Hydrogen-Bond Kinetics in Liquid Water. Nature 1996, 379, 55–57. [Google Scholar] [CrossRef]
- Beal, C. The Viscosity of Air, Water, Natural Gas, Crude Oil and Its Associated Gases at Oil Field Temperatures and Pressures. Trans. AIME 1946, 165, 94–115. [Google Scholar] [CrossRef]
- Hinrikus, H.; Bachmann, M.; Lass, J. Understanding Physical Mechanism of Low-Level Microwave Radiation Effect. Int. J. Radiat. Biol. 2018, 94, 877–882. [Google Scholar] [CrossRef]
- Zong, D.; Hu, H.; Duan, Y.; Sun, Y. Viscosity of Water under Electric Field: Anisotropy Induced by Redistribution of Hydrogen Bonds. J. Phys. Chem. B 2016, 120, 4818–4827. [Google Scholar] [CrossRef] [PubMed]
- Tatoń, G. The Influence of Electromagnetic Wave Originating from WiFi Router on Water Viscosity. Prz. Elektrotech. 2018, 1, 280–282. [Google Scholar] [CrossRef] [Green Version]
- Hinrikus, H.; Lass, J.; Karai, D.; Pilt, K.; Bachmann, M. Microwave Effect on Diffusion: A Possible Mechanism for Non-Thermal Effect. Electromagn. Biol. Med. 2015, 34, 327–333. [Google Scholar] [CrossRef]
- Ghauri, S.A.; Ansari, M.S. Increase of Water Viscosity under the Influence of Magnetic Field. J. Appl. Phys. 2006, 100, 066101. [Google Scholar] [CrossRef]
- Read, F.H. Electromagnetic Radiation; John Wiley and Sons: Chichester, UK; New York, NY, USA, 1980. [Google Scholar]
- Sellers, P.J.; Berry, J.A.; Collatz, G.J.; Field, C.B.; Hall, F.G. Canopy Reflectance, Photosynthesis, and Transpiration. III. A Reanalysis Using Improved Leaf Models and a New Canopy Integration Scheme. Remote Sens. Environ. 1992, 42, 187–216. [Google Scholar] [CrossRef]
- Sellers, P.J. Canopy Reflectance, Photosynthesis, and Transpiration, II. The Role of Biophysics in the Linearity of Their Interdependence. Remote Sens. Environ. 1987, 21, 143–183. [Google Scholar] [CrossRef]
- Gates, D.M.; Tantraporn, W. The Reflectivity of Deciduous Trees and Herbaceous Plants in the Infrared to 25 Microns. Science 1952, 115, 613–616. [Google Scholar] [CrossRef] [PubMed]
- Baldocchi, D.D.; Ryu, Y.; Dechant, B.; Eichelmann, E.; Hemes, K.; Ma, S.; Sanchez, C.R.; Shortt, R.; Szutu, D.; Valach, A.; et al. Outgoing Near-infrared Radiation from Vegetation Scales with Canopy Photosynthesis across a Spectrum of Function, Structure, Physiological Capacity, and Weather. J. Geophys. Res. Biogeosci. 2020, 125, e2019JG005534. [Google Scholar] [CrossRef]
- Aitken, W.W.; Lombard, J.; Wang, K.; Toro, M.; Byrne, M.; Nardi, M.I.; Kardys, J.; Parrish, A.; Dong, C.; Szapocznik, J.; et al. Relationship of Neighborhood Greenness to Alzheimer’s Disease and Non-Alzheimer’s Dementia among 249,405 U.S. Medicare Beneficiaries. J. Alzheimer’s Dis. 2021, 81, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Veleva, B.I.; Caljouw, M.A.A.; van der Steen, J.T.; Mertens, B.J.A.; Chel, V.G.M.; Numans, M.E. The Effect of Ultraviolet B Irradiation Compared with Oral Vitamin D Supplementation on the Well-Being of Nursing Home Residents with Dementia: A Randomized Controlled Trial. Int. J. Environ. Res. Public Health 2020, 17, 1684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purushothuman, S.; Johnstone, D.M.; Nandasena, C.; van Eersel, J.; Ittner, L.M.; Mitrofanis, J.; Stone, J. Near Infrared Light Mitigates Cerebellar Pathology in Transgenic Mouse Models of Dementia. Neurosci. Lett. 2015, 591, 155–159. [Google Scholar] [CrossRef]
- Purushothuman, S.; Johnstone, D.M.; Nandasena, C.; Mitrofanis, J.; Stone, J. Photobiomodulation with near Infrared Light Mitigates Alzheimer’s Disease-Related Pathology in Cerebral Cortex—Evidence from Two Transgenic Mouse Models. Alzheimer’s Res. Ther. 2014, 6, 2. [Google Scholar] [CrossRef] [Green Version]
- Comerota, M.M.; Krishnan, B.; Taglialatela, G. Near Infrared Light Decreases Synaptic Vulnerability to Amyloid Beta Oligomers. Sci. Rep. 2017, 7, 15012. [Google Scholar] [CrossRef] [Green Version]
- Comerota, M.M.; Tumurbaatar, B.; Krishnan, B.; Kayed, R.; Taglialatela, G. Near Infrared Light Treatment Reduces Synaptic Levels of Toxic Tau Oligomers in Two Transgenic Mouse Models of Human Tauopathies. Mol. Neurobiol. 2019, 56, 3341–3355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stepanov, Y.V.; Golovynska, I.; Zhang, R.; Golovynskyi, S.; Stepanova, L.I.; Gorbach, O.; Dovbynchuk, T.; Garmanchuk, L.V.; Ohulchanskyy, T.Y.; Qu, J. Near-Infrared Light Reduces β-Amyloid-Stimulated Microglial Toxicity and Enhances Survival of Neurons: Mechanisms of Light Therapy for Alzheimer’s Disease. Alzheimer’s Res. Ther. 2022, 14, 84. [Google Scholar] [CrossRef] [PubMed]
- Chao, L.L. Effects of Home Photobiomodulation Treatments on Cognitive and Behavioral Function, Cerebral Perfusion, and Resting-State Functional Connectivity in Patients with Dementia: A Pilot Trial. Photobiomodulation Photomed. Laser Surg. 2019, 37, 133–141. [Google Scholar] [CrossRef]
- Berman, M.H.; Halper, J.P.; Nichols, T.W.; Jarrett, H.; Lundy, A.; Huang, J.H. Photobiomodulation with Near Infrared Light Helmet in a Pilot, Placebo Controlled Clinical Trial in Dementia Patients Testing Memory and Cognition. J. Neurol. Neurosci. 2017, 8, 176. [Google Scholar] [CrossRef] [Green Version]
- Nizamutdinov, D.; Qi, X.; Berman, M.H.; Dougal, G.; Dayawansa, S.; Wu, E.; Yi, S.S.; Stevens, A.B.; Huang, J.H. Transcranial Near Infrared Light Stimulations Improve Cognition in Patients with Dementia. Aging Dis. 2021, 12, 954–963. [Google Scholar] [CrossRef]
- Dougal, G.; Ennaceur, A.; Chazot, P.L. Effect of Transcranial Near-Infrared Light 1068 Nm Upon Memory Performance in Aging Healthy Individuals: A Pilot Study. Photobiomodulation Photomed. Laser Surg. 2021, 39, 654–660. [Google Scholar] [CrossRef]
- Han, M.; Wang, Q.; Wang, X.; Zeng, Y.; Huang, Y.; Meng, Q.; Zhang, J.; Wei, X. Near Infra-Red Light Treatment of Alzheimer’s Disease. J. Innov. Opt. Health Sci. 2018, 11, 1750012. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Peng, J.; Luo, Y.; Zhou, J.; Li, T.; Cao, L.; Peng, S.; Zuo, Z.; Wang, Z. Far Infrared Light Irradiation Enhances Aβ Clearance via Increased Exocytotic Microglial ATP and Ameliorates Cognitive Deficit in Alzheimer’s Disease-like Mice. J. Neuroinflamm. 2022, 19, 145. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.-X.; Reiter, R.J.; Zimmerman, S.; Hardeland, R. Melatonin: Both a Messenger of Darkness and a Participant in the Cellular Actions of Non-Visible Solar Radiation of Near Infrared Light. Biology 2023, 12, 89. [Google Scholar] [CrossRef]
- Zimmerman, S.; Reiter, R.J. Melatonin and the Optics of the Human Body. Melatonin Res. 2019, 2, 138–160. [Google Scholar] [CrossRef]
- Chang, S.-Y.; Lee, M.Y.; Chung, P.-S.; Kim, S.; Choi, B.; Suh, M.-W.; Rhee, C.-K.; Jung, J.Y. Enhanced Mitochondrial Membrane Potential and ATP Synthesis by Photobiomodulation Increases Viability of the Auditory Cell Line after Gentamicin-Induced Intrinsic Apoptosis. Sci. Rep. 2019, 9, 19248. [Google Scholar] [CrossRef] [Green Version]
- Hamblin, M.R. Mechanisms and Applications of the Anti-Inflammatory Effects of Photobiomodulation. AIMS Biophys. 2017, 4, 337–361. [Google Scholar] [CrossRef]
- Karu, T. Mitochondrial Mechanisms of Photobiomodulation in Context of New Data about Multiple Roles of ATP. Photomed. Laser Surg. 2010, 28, 159–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coon, S.L.; Klein, D.C. Evolution of Arylalkylamine N-Acetyltransferase: Emergence and Divergence. Mol. Cell. Endocrinol. 2006, 252, 2–10. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Choi, G.-H.; Back, K. Functional Characterization of Serotonin N-Acetyltransferase in Archaeon Thermoplasma Volcanium. Antioxidants 2022, 11, 596. [Google Scholar] [CrossRef]
- Kang, K.; Lee, K.; Park, S.; Byeon, Y.; Back, K. Molecular Cloning of Rice Serotonin N-Acetyltransferase, the Penultimate Gene in Plant Melatonin Biosynthesis. J. Pineal Res. 2013, 55, 7–13. [Google Scholar] [CrossRef]
- Yoshizawa, T.; Nozawa, R.-S.; Jia, T.Z.; Saio, T.; Mori, E. Biological Phase Separation: Cell Biology Meets Biophysics. Biophys. Rev. 2020, 12, 519–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, J.T.; Shakya, A. Phase Separation of DNA: From Past to Present. Biophys. J. 2021, 120, 1139–1149. [Google Scholar] [CrossRef]
- Dion, W.; Ballance, H.; Lee, J.; Pan, Y.; Irfan, S.; Edwards, C.; Sun, M.; Zhang, J.; Zhang, X.; Liu, S.; et al. Four-Dimensional Nuclear Speckle Phase Separation Dynamics Regulate Proteostasis. Sci. Adv. 2022, 8, eabl4150. [Google Scholar] [CrossRef]
- Poudyal, R.R.; Pir Cakmak, F.; Keating, C.D.; Bevilacqua, P.C. Physical Principles and Extant Biology Reveal Roles for RNA-Containing Membraneless Compartments in Origins of Life Chemistry. Biochemistry 2018, 57, 2509–2519. [Google Scholar] [CrossRef]
- Loh, D.; Reiter, R.J. Melatonin: Regulation of Biomolecular Condensates in Neurodegenerative Disorders. Antioxidants 2021, 10, 1483. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.-Y.; Xu, Y.-Y.; Tong, X.-Y.; Wang, G.; Zhang, H.-Y. The Legend of ATP: From Origin of Life to Precision Medicine. Metabolites 2022, 12, 461. [Google Scholar] [CrossRef]
- Franzmann, T.M.; Alberti, S. Protein Phase Separation as a Stress Survival Strategy. Cold Spring Harb. Perspect. Biol. 2019, 11, a034058. [Google Scholar] [CrossRef] [Green Version]
- Lerner, A.B.; Case, J.D.; Takahashi, Y.; Lee, T.H.; Mori, W. Isolation of melatonin, the pineal gland factor that lightens melanocytes1. J. Am. Chem. Soc. 1958, 80, 2587. [Google Scholar] [CrossRef]
- Loh, D.; Reiter, R.J. Melatonin and Phase Separation: Potential Interactions and Significance. Melatonin Res. 2022, 5, 186–191. [Google Scholar] [CrossRef]
- Loh, D.; Reiter, R.J. Melatonin: Regulation of Prion Protein Phase Separation in Cancer Multidrug Resistance. Molecules 2022, 27, 705. [Google Scholar] [CrossRef]
- Loh, D.; Reiter, R.J. Melatonin: Regulation of Viral Phase Separation and Epitranscriptomics in Post-Acute Sequelae of COVID-19. Int. J. Mol. Sci. 2022, 23, 8122. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Akinc, M.; Wiench, J.; Pruski, M.; Schilling, C.H. Relationship between Water Mobility and Viscosity of Nanometric Alumina Suspensions. J. Am. Ceram. Soc. 2005, 88, 2762–2768. [Google Scholar] [CrossRef]
- Ye, H.; Zhang, H.; Zhang, Z.; Zheng, Y. Size and Temperature Effects on the Viscosity of Water inside Carbon Nanotubes. Nanoscale Res. Lett. 2011, 6, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaat, M.; Zheng, Y. Fluidity and Phase Transitions of Water in Hydrophobic and Hydrophilic Nanotubes. Sci. Rep. 2019, 9, 5689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaban, V.V.; Prezhdo, V.V.; Prezhdo, O.V. Confinement by Carbon Nanotubes Drastically Alters the Boiling and Critical Behavior of Water Droplets. ACS Nano 2012, 6, 2766–2773. [Google Scholar] [CrossRef] [Green Version]
- Striolo, A. Water Self-Diffusion through Narrow Oxygenated Carbon Nanotubes. Nanotechnology 2007, 18, 475704. [Google Scholar] [CrossRef]
- Babu, J.S.; Sathian, S.P. The Role of Activation Energy and Reduced Viscosity on the Enhancement of Water Flow through Carbon Nanotubes. J. Chem. Phys. 2011, 134, 194509. [Google Scholar] [CrossRef]
- Zhang, D.; Yang, X.; Jiang, W.; Jin, L.; Gao, Y.; Wang, Z. Pauli Repulsion Enhances Mobility of Ultraconfined Water. ACS Nano 2021, 15, 2490–2496. [Google Scholar] [CrossRef]
- Najafi, S.; Lin, Y.; Longhini, A.P.; Zhang, X.; Delaney, K.T.; Kosik, K.S.; Fredrickson, G.H.; Shea, J.-E.; Han, S. Liquid-Liquid Phase Separation of Tau by Self and Complex Coacervation. Protein Sci. 2021, 30, 1393–1407. [Google Scholar] [CrossRef]
- Jawerth, L.; Fischer-Friedrich, E.; Saha, S.; Wang, J.; Franzmann, T.; Zhang, X.; Sachweh, J.; Ruer, M.; Ijavi, M.; Saha, S.; et al. Protein Condensates as Aging Maxwell Fluids. Science 2020, 370, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Tejedor, A.R.; Sanchez-Burgos, I.; Estevez-Espinosa, M.; Garaizar, A.; Collepardo-Guevara, R.; Ramirez, J.; Espinosa, J.R. Protein Structural Transitions Critically Transform the Network Connectivity and Viscoelasticity of RNA-Binding Protein Condensates but RNA Can Prevent It. Nat. Commun. 2022, 13, 5717. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-C.; Mai, F.-D.; Yang, K.-H.; Chen, L.-Y.; Yang, C.-P.; Liu, Y.-C. Quantitative Evaluation on Activated Property-Tunable Bulk Liquid Water with Reduced Hydrogen Bonds Using Deconvoluted Raman Spectroscopy. Anal. Chem. 2015, 87, 808–815. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.-H.; Chang, C.-C.; Mai, F.-D.; Yang, C.-P.; Liu, Y.-C. Plasmon-Activated Water Can Form Stronger Intermolecular Hydrogen Bonding with Water-Soluble Alcohols and Dissolve More Hydrophobic Solutes. Chem. Eng. J. 2022, 427, 131949. [Google Scholar] [CrossRef]
- Zhang, J.; Yan, X.; Tian, Y.; Li, W.; Wang, H.; Li, Q.; Li, Y.; Li, Z.; Wu, T. Synthesis of a New Water-Soluble Melatonin Derivative with Low Toxicity and a Strong Effect on Sleep Aid. ACS Omega 2020, 5, 6494–6499. [Google Scholar] [CrossRef] [Green Version]
- Renn, T.-Y.; Yang, C.-P.; Wu, U.-I.; Chen, L.-Y.; Mai, F.-D.; Tikhonova, M.A.; Amstislavskaya, T.G.; Liao, W.-C.; Lin, C.-T.; Liu, Y.-C.; et al. Water Composed of Reduced Hydrogen Bonds Activated by Localized Surface Plasmon Resonance Effectively Enhances Anti-Viral and Anti-Oxidative Activities of Melatonin. Chem. Eng. J. 2022, 427, 131626. [Google Scholar] [CrossRef]
- Rodrigues, A.C.C.; de M. Camargo, L.T.F.; Lopes, Y.F.; Sallum, L.O.; Napolitano, H.B.; Camargo, A.J. Aqueous Solvation Study of Melatonin Using Ab Initio Molecular Dynamics. J. Mol. Liq. 2021, 343, 117451. [Google Scholar] [CrossRef]
- Florio, G.M.; Zwier, T.S. Solvation of a Flexible Biomolecule in the Gas Phase: The Ultraviolet and Infrared Spectroscopy of Melatonin−Water Clusters. J. Phys. Chem. A 2003, 107, 974–983. [Google Scholar] [CrossRef]
- Ritwiset, A.; Khajonrit, J.; Krongsuk, S.; Maensiri, S. Molecular Insight on the Formation Structure and Dynamics of Melatonin in an Aqueous Solution and at the Water-Air Interface: A Molecular Dynamics Study. J. Mol. Graph. Model. 2021, 108, 107983. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.-M.; Wu, U.-I.; Lin, T.-B.; Lan, C.-T.; Chien, W.-C.; Huang, W.-L.; Shieh, J.-Y. Total Sleep Deprivation Inhibits the Neuronal Nitric Oxide Synthase and Cytochrome Oxidase Reactivities in the Nodose Ganglion of Adult Rats. J. Anat. 2006, 209, 239–250. [Google Scholar] [CrossRef]
- Cheng, C.-H.; Lin, K.-J.; Hong, C.-T.; Wu, D.; Chang, H.-M.; Liu, C.-H.; Hsiao, I.-T.; Yang, C.-P.; Liu, Y.-C.; Hu, C.-J. Plasmon-Activated Water Reduces Amyloid Burden and Improves Memory in Animals with Alzheimer’s Disease. Sci. Rep. 2019, 9, 13252. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.-C.; Cheng, C.-Y.; Chen, L.-Y.; Chang, C.-C.; Yang, C.-P.; Mai, F.-D.; Liao, W.-C.; Chang, H.-M.; Liu, Y.-C. Plasmon-Activated Water Effectively Relieves Hepatic Oxidative Damage Resulting from Chronic Sleep Deprivation. RSC Adv. 2018, 8, 9618–9626. [Google Scholar] [CrossRef] [Green Version]
- Mitroka, S.; Zimmeck, S.; Troya, D.; Tanko, J.M. How Solvent Modulates Hydroxyl Radical Reactivity in Hydrogen Atom Abstractions. J. Am. Chem. Soc. 2010, 132, 2907–2913. [Google Scholar] [CrossRef]
- Vassilev, P.; Louwerse, M.J.; Baerends, E.J. Hydroxyl Radical and Hydroxide Ion in Liquid Water: A Comparative Electron Density Functional Theory Study. J. Phys. Chem. B 2005, 109, 23605–23610. [Google Scholar] [CrossRef]
- Wong, H.-S.; Dighe, P.A.; Mezera, V.; Monternier, P.-A.; Brand, M.D. Production of Superoxide and Hydrogen Peroxide from Specific Mitochondrial Sites under Different Bioenergetic Conditions. J. Biol. Chem. 2017, 292, 16804–16809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giulivi, C.; Boveris, A.; Cadenas, E. Hydroxyl Radical Generation during Mitochondrial Electron Transfer and the Formation of 8-Hydroxydesoxyguanosine in Mitochondrial DNA. Arch. Biochem. Biophys. 1995, 316, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Aleardi, A.M.; Benard, G.; Augereau, O.; Malgat, M.; Talbot, J.C.; Mazat, J.P.; Letellier, T.; Dachary-Prigent, J.; Solaini, G.C.; Rossignol, R. Gradual Alteration of Mitochondrial Structure and Function by Beta-Amyloids: Importance of Membrane Viscosity Changes, Energy Deprivation, Reactive Oxygen Species Production, and Cytochrome c Release. J. Bioenerg. Biomembr. 2005, 37, 207–225. [Google Scholar] [CrossRef] [PubMed]
- Gough, D.R.; Cotter, T.G. Hydrogen Peroxide: A Jekyll and Hyde Signalling Molecule. Cell Death Dis. 2011, 2, e213. [Google Scholar] [CrossRef] [Green Version]
- Ren, M.; Deng, B.; Zhou, K.; Kong, X.; Wang, J.-Y.; Lin, W. Single Fluorescent Probe for Dual-Imaging Viscosity and H2O2 in Mitochondria with Different Fluorescence Signals in Living Cells. Anal. Chem. 2017, 89, 552–555. [Google Scholar] [CrossRef]
- Zheng, A.; Liu, H.; Gao, X.; Xu, K.; Tang, B. A Mitochondrial-Targeting Near-Infrared Fluorescent Probe for Revealing the Effects of Hydrogen Peroxide And Heavy Metal Ions on Viscosity. Anal. Chem. 2021, 93, 9244–9249. [Google Scholar] [CrossRef]
- Li, S.; Wang, P.; Feng, W.; Xiang, Y.; Dou, K.; Liu, Z. Simultaneous Imaging of Mitochondrial Viscosity and Hydrogen Peroxide in Alzheimer’s Disease by a Single near-Infrared Fluorescent Probe with a Large Stokes Shift. Chem. Commun. 2020, 56, 1050–1053. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.X.; Manchester, L.C.; Reiter, R.J.; Plummer, B.F. Cyclic 3-Hydroxymelatonin: A Melatonin Metabolite Generated as a Result of Hydroxyl Radical Scavenging. Biol. Signals Recept. 1999, 8, 70–74. [Google Scholar] [CrossRef]
- Galano, A. On the Direct Scavenging Activity of Melatonin towards Hydroxyl and a Series of Peroxyl Radicals. Phys. Chem. Chem. Phys. 2011, 13, 7178–7188. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, D.; Biswas, K.; Bandyopadhyay, U.; Reiter, R.J.; Banerjee, R.K. Melatonin Protects against Stress-Induced Gastric Lesions by Scavenging the Hydroxyl Radical. J. Pineal Res. 2000, 29, 143–151. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.X.; Osuna, C.; Gitto, E. Actions of Melatonin in the Reduction of Oxidative Stress. A Review. J. Biomed. Sci. 2000, 7, 444–458. [Google Scholar] [CrossRef] [PubMed]
- Galano, A.; Reiter, R.J. Melatonin and Its Metabolites vs Oxidative Stress: From Individual Actions to Collective Protection. J. Pineal Res. 2018, 65, e12514. [Google Scholar] [CrossRef] [Green Version]
- Purushothaman, A.; Sheeja, A.A.; Janardanan, D. Hydroxyl Radical Scavenging Activity of Melatonin and Its Related Indolamines. Free Radic. Res. 2020, 54, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Persson, L.B.; Ambati, V.S.; Brandman, O. Cellular Control of Viscosity Counters Changes in Temperature and Energy Availability. Cell 2020, 183, 1572–1585.e16. [Google Scholar] [CrossRef]
- Scalettar, B.A.; Abney, J.R.; Hackenbrock, C.R. Dynamics, Structure, and Function Are Coupled in the Mitochondrial Matrix. Proc. Natl. Acad. Sci. USA 1991, 88, 8057–8061. [Google Scholar] [CrossRef] [Green Version]
- Hao, X.; Zhan, J.; Geng, C.; Lin, W. Discriminating Normal and Inflammatory Mice Models by Viscosity Changes with a Two-Photon Fluorescent Probe. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2023, 284, 121807. [Google Scholar] [CrossRef]
- Guan, X.; Hong, J.; Li, Q.; Feng, G. High-Fidelity Imaging Probe for Lysosomes and Selective Visualization of Cancer Cells and Tissues. Sens. Actuators B Chem. 2022, 369, 132325. [Google Scholar] [CrossRef]
- Song, H.; Zhang, W.; Zhang, Y.; Yin, C.; Huo, F. Viscosity Activated NIR Fluorescent Probe for Visualizing Mitochondrial Viscosity Dynamic and Fatty Liver Mice. Chem. Eng. J. 2022, 445, 136448. [Google Scholar] [CrossRef]
- Li, S.; Huo, F.; Yin, C. NIR Fluorescent Probe for Dual-Response Viscosity and Hydrogen Sulfide and Its Application in Parkinson’s Disease Model. Dyes Pigments 2022, 197, 109825. [Google Scholar] [CrossRef]
- Pan, W.; Han, L.; Cao, X.; Shen, S.; Pang, X.; Zhu, Y. Dual-Response near-Infrared Fluorescent Probe for Detecting Cyanide and Mitochondrial Viscosity and Its Application in Bioimaging. Food Chem. 2022, 407, 135163. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Huo, F.; Wen, Y.; Yin, C. A Dual-Response NIR Probe Reveals Positive Correlation between Biothiols and Viscosity under Cellular Stress Change. Chem. Commun. 2022, 58, 4881–4884. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, Q.; Bu, Y.; Xu, T.; Zhu, X.; Zhang, J.; Yu, Z.; Wang, L.; Zhong, F.; Zhou, H. Real-Time Imaging Mitochondrial Viscosity Dynamic during Mitophagy Mediated by Photodynamic Therapy. Anal. Chim. Acta 2021, 1178, 338847. [Google Scholar] [CrossRef]
- Zhou, Y.; Liu, Z.; Qiao, G.; Tang, B.; Li, P. Visualization of Endoplasmic Reticulum Viscosity in the Liver of Mice with Nonalcoholic Fatty Liver Disease by a near-Infrared Fluorescence Probe. Chin. Chem. Lett. 2021, 32, 3641–3645. [Google Scholar] [CrossRef]
- Liu, Y.; Ma, Y.; Gao, W.; Ma, S.; Lin, W. Construction of a Fluorescent Probe with Large Stokes Shift and Deep Red Emission for Sensing of the Viscosity in Hyperglycemic Mice. Dyes Pigments 2021, 195, 109674. [Google Scholar] [CrossRef]
- Xiao, H.; Li, P.; Tang, B. Small Molecular Fluorescent Probes for Imaging of Viscosity in Living Biosystems. Chemistry 2021, 27, 6880–6898. [Google Scholar] [CrossRef]
- Shen, W.; Wang, P.; Xie, Z.; Zhou, H.; Hu, Y.; Fu, M.; Zhu, Q. A Bifunctional Probe Reveals Increased Viscosity and Hydrogen Sulfide in Zebra Fish Model of Parkinson’s Disease. Talanta 2021, 234, 122621. [Google Scholar] [CrossRef]
- Fang, Z.; Su, Z.; Qin, W.; Li, H.; Fang, B.; Du, W.; Wu, Q.; Peng, B.; Li, P.; Yu, H.; et al. Two-Photon Dual-Channel Fluorogenic Probe for in Situ Imaging the Mitochondrial H2S/viscosity in the Brain of Drosophila Parkinson’s Disease Model. Chin. Chem. Lett. 2020, 31, 2903–2908. [Google Scholar] [CrossRef]
- Park, S.J.; Shin, B.K.; Lee, H.W.; Song, J.M.; Je, J.T.; Kim, H.M. Asymmetric Cyanine as a Far-Red Fluorescence Probe for Mitochondrial Viscosity. Dyes Pigments 2020, 174, 108080. [Google Scholar] [CrossRef]
- Yang, J.; Guo, Y.; Pistolozzi, M.; Yan, J. Research Progress of Multi-Functional Fluorescent Probes for Alzheimer’s Disease Monitoring. Dyes Pigments 2021, 193, 109466. [Google Scholar] [CrossRef]
- Zhu, L.; Fu, M.; Yin, B.; Wang, L.; Chen, Y.; Zhu, Q. A Red-Emitting Fluorescent Probe for Mitochondria-Target Microviscosity in Living Cells and Blood Viscosity Detection in Hyperglycemia Mice. Dyes Pigments 2020, 172, 107859. [Google Scholar] [CrossRef]
- Tan, H.-Y.; Qiu, Y.-T.; Sun, H.; Yan, J.-W.; Zhang, L. A Lysosome-Targeting Dual-Functional Fluorescent Probe for Imaging Intracellular Viscosity and Beta-Amyloid. Chem. Commun. 2019, 55, 2688–2691. [Google Scholar] [CrossRef]
- Li, H.; Xin, C.; Zhang, G.; Han, X.; Qin, W.; Zhang, C.-W.; Yu, C.; Jing, S.; Li, L.; Huang, W. A Mitochondria-Targeted Two-Photon Fluorogenic Probe for the Dual-Imaging of Viscosity and H2O2 Levels in Parkinson’s Disease Models. J. Mater. Chem. B Mater. Biol. Med. 2019, 7, 4243–4251. [Google Scholar] [CrossRef]
- Yin, J.; Peng, M.; Lin, W. Visualization of Mitochondrial Viscosity in Inflammation, Fatty Liver, and Cancer Living Mice by a Robust Fluorescent Probe. Anal. Chem. 2019, 91, 8415–8421. [Google Scholar] [CrossRef] [PubMed]
- Ren, M.; Zhou, K.; Wang, L.; Liu, K.; Lin, W. Construction of a Ratiometric Two-Photon Fluorescent Probe to Monitor the Changes of Mitochondrial Viscosity. Sens. Actuators B Chem. 2018, 262, 452–459. [Google Scholar] [CrossRef]
- Jiang, N.; Fan, J.; Zhang, S.; Wu, T.; Wang, J.; Gao, P.; Qu, J.; Zhou, F.; Peng, X. Dual Mode Monitoring Probe for Mitochondrial Viscosity in Single Cell. Sens. Actuators B Chem. 2014, 190, 685–693. [Google Scholar] [CrossRef]
- Aras, S.; Tek, I.; Varli, M.; Yalcin, A.; Cengiz, O.K.; Atmis, V.; Atli, T. Plasma Viscosity: Is a Biomarker for the Differential Diagnosis of Alzheimer’s Disease and Vascular Dementia? Am. J. Alzheimer’s Dis. Other Dement. 2013, 28, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Peng, J.; Zhang, Q.; Song, S.; Lin, W. A New NIR Emission Mitochondrial Targetable Fluorescent Probe and Its Application in Detecting Viscosity Changes in Mouse Liver and Kidney Injury. Talanta 2022, 249, 123647. [Google Scholar] [CrossRef]
- Yu, F.-T.; Huang, Z.; Yang, J.-X.; Yang, L.-M.; Xu, X.-Y.; Huang, J.-Y.; Kong, L. Two Quinoline-Based Two-Photon Fluorescent Probes for Imaging of Viscosity in Subcellular Organelles of Living HeLa Cells. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2022, 283, 121769. [Google Scholar] [CrossRef]
- Wei, Y.-F.; Weng, X.-F.; Sha, X.-L.; Sun, R.; Xu, Y.-J.; Ge, J.-F. Simultaneous Imaging of Lysosomal and Mitochondrial Viscosity under Different Conditions Using a NIR Probe. Sens. Actuators B Chem. 2021, 326, 128954. [Google Scholar] [CrossRef]
- Yan, F.; Sun, X.; Ma, T.; Zhang, Y.; Jiang, Y.; Wang, R.; Ma, C.; Wei, J.; Chen, L.; Cui, Y. A Viscosity-Dependent Carbon Dots with Anti-VEGF Properties for Monitoring and Promoting Apoptosis in Cancerous Cell. Chem. Eng. J. 2021, 407, 127801. [Google Scholar] [CrossRef]
- Wu, Y.; Shu, W.; Zeng, C.; Guo, B.; Shi, J.; Jing, J.; Zhang, X. A Mitochondria Targetable and Viscosity Sensitive Fluorescent Probe and Its Applications for Distinguishing Cancerous Cells. Dyes Pigments 2019, 168, 134–139. [Google Scholar] [CrossRef]
- Yang, Z.; He, Y.; Lee, J.-H.; Park, N.; Suh, M.; Chae, W.-S.; Cao, J.; Peng, X.; Jung, H.; Kang, C.; et al. A Self-Calibrating Bipartite Viscosity Sensor for Mitochondria. J. Am. Chem. Soc. 2013, 135, 9181–9185. [Google Scholar] [CrossRef]
- Liang, L.-F.; Da, X.; Chen, T.-S.; Pei, Y.-H. Nucleoplasmic viscosity of living cells investigated by fluorescence correlation spectroscopy. Guang Pu Xue Yu Guang Pu Fen Xi 2009, 29, 459–462. [Google Scholar] [PubMed]
- Zhang, S.; Zhang, Y.; Zhao, L.; Xu, L.; Han, H.; Huang, Y.; Fei, Q.; Sun, Y.; Ma, P.; Song, D. A Novel Water-Soluble near-Infrared Fluorescent Probe for Monitoring Mitochondrial Viscosity. Talanta 2021, 233, 122592. [Google Scholar] [CrossRef]
- Chernyak, B.V.; Izyumov, D.S.; Lyamzaev, K.G.; Pashkovskaya, A.A.; Pletjushkina, O.Y.; Antonenko, Y.N.; Sakharov, D.V.; Wirtz, K.W.A.; Skulachev, V.P. Production of Reactive Oxygen Species in Mitochondria of HeLa Cells under Oxidative Stress. Biochim. Biophys. Acta 2006, 1757, 525–534. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Shi, Y.-D.; Ding, A.-X.; Tan, Z.-L.; Chen, H.; Liu, R.; Wang, R.; Lu, Z.-L. Imaging Viscosity and Peroxynitrite by a Mitochondria-Targeting Two-Photon Ratiometric Fluorescent Probe. Sens. Actuators B Chem. 2018, 276, 238–246. [Google Scholar] [CrossRef]
- Xu, T.; Pagadala, V.; Mueller, D.M. Understanding Structure, Function, and Mutations in the Mitochondrial ATP Synthase. Microb. Cell Factories 2015, 2, 105–125. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, R.K.; Scanlon, J.A.B.; Al-Shawi, M.K. The Rotary Mechanism of the ATP Synthase. Arch. Biochem. Biophys. 2008, 476, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Stock, D.; Gibbons, C.; Arechaga, I.; Leslie, A.G.; Walker, J.E. The Rotary Mechanism of ATP Synthase. Curr. Opin. Struct. Biol. 2000, 10, 672–679. [Google Scholar] [CrossRef]
- Usukura, E.; Suzuki, T.; Furuike, S.; Soga, N.; Saita, E.-I.; Hisabori, T.; Kinosita, K., Jr.; Yoshida, M. Torque Generation and Utilization in Motor Enzyme F0F1-ATP Synthase: Half-Torque F1 with Short-Sized Pushrod Helix and Reduced ATP Synthesis by Half-Torque F0F1. J. Biol. Chem. 2012, 287, 1884–1891. [Google Scholar] [CrossRef] [Green Version]
- Novichkova, N.S.; Malyan, A.N. The Effect of the Viscosity of a Trehalose Solution on ATP Hydrolysis by Chloroplast F1-ATPase. Biophysics 2019, 64, 853–857. [Google Scholar] [CrossRef]
- Nakanishi-Matsui, M.; Kashiwagi, S.; Hosokawa, H.; Cipriano, D.J.; Dunn, S.D.; Wada, Y.; Futai, M. Stochastic High-Speed Rotation of Escherichia Coli ATP Synthase F1 Sector: The Epsilon Subunit-Sensitive Rotation. J. Biol. Chem. 2006, 281, 4126–4131. [Google Scholar] [CrossRef] [Green Version]
- Capaldi, R.A.; Aggeler, R. Mechanism of the F(1)F(0)-Type ATP Synthase, a Biological Rotary Motor. Trends Biochem. Sci. 2002, 27, 154–160. [Google Scholar] [CrossRef]
- Martin, J.L.; Ishmukhametov, R.; Hornung, T.; Ahmad, Z.; Frasch, W.D. Anatomy of F1-ATPase Powered Rotation. Proc. Natl. Acad. Sci. USA 2014, 111, 3715–3720. [Google Scholar] [CrossRef] [Green Version]
- Spetzler, D.; Ishmukhametov, R.; Hornung, T.; Day, L.J.; Martin, J.; Frasch, W.D. Single Molecule Measurements of F1-ATPase Reveal an Interdependence between the Power Stroke and the Dwell Duration. Biochemistry 2009, 48, 7979–7985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omote, H.; Sambonmatsu, N.; Saito, K.; Sambongi, Y.; Iwamoto-Kihara, A.; Yanagida, T.; Wada, Y.; Futai, M. The γ-Subunit Rotation and Torque Generation in F1-ATPase from Wild-Type or Uncoupled Mutant Escherichia coli. Proc. Natl. Acad. Sci. USA 1999, 96, 7780–7784. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, R.; Hayashi, K.; Ueno, H.; Noji, H. Catalysis-Enhancement via Rotary Fluctuation of F1-ATPase. Biophys. J. 2013, 105, 2385–2391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Begum, R.; Powner, M.B.; Hudson, N.; Hogg, C.; Jeffery, G. Treatment with 670 Nm Light up Regulates Cytochrome C Oxidase Expression and Reduces Inflammation in an Age-Related Macular Degeneration Model. PLoS ONE 2013, 8, e57828. [Google Scholar] [CrossRef] [Green Version]
- Ahamed Basha, A.; Mathangi, D.C.; Shyamala, R. Effect of LED Photobiomodulation on Fluorescent Light Induced Changes in Cellular ATPases and Cytochrome c Oxidase Activity in Wistar Rat. Lasers Med. Sci. 2016, 31, 1803–1809. [Google Scholar] [CrossRef]
- Gkotsi, D.; Begum, R.; Salt, T.; Lascaratos, G.; Hogg, C.; Chau, K.-Y.; Schapira, A.H.V.; Jeffery, G. Recharging Mitochondrial Batteries in Old Eyes. Near Infra-Red Increases ATP. Exp. Eye Res. 2014, 122, 50–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong-Riley, M.T.T.; Liang, H.L.; Eells, J.T.; Chance, B.; Henry, M.M.; Buchmann, E.; Kane, M.; Whelan, H.T. Photobiomodulation Directly Benefits Primary Neurons Functionally Inactivated by Toxins: Role of cytochrome c oxidase *. J. Biol. Chem. 2005, 280, 4761–4771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Park, J.-S.; Deng, J.-H.; Bai, Y. Cytochrome c Oxidase Subunit IV Is Essential for Assembly and Respiratory Function of the Enzyme Complex. J. Bioenerg. Biomembr. 2006, 38, 283–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedlák, E.; Fabian, M.; Robinson, N.C.; Musatov, A. Ferricytochrome c Protects Mitochondrial Cytochrome c Oxidase against Hydrogen Peroxide-Induced Oxidative Damage. Free Radic. Biol. Med. 2010, 49, 1574–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musatov, A.; Robinson, N.C. Susceptibility of Mitochondrial Electron-Transport Complexes to Oxidative Damage. Focus on Cytochrome c Oxidase. Free Radic. Res. 2012, 46, 1313–1326. [Google Scholar] [CrossRef]
- Choksi, K.B.; Nuss, J.E.; Boylston, W.H.; Rabek, J.P.; Papaconstantinou, J. Age-Related Increases in Oxidatively Damaged Proteins of Mouse Kidney Mitochondrial Electron Transport Chain Complexes. Free Radic. Biol. Med. 2007, 43, 1423–1438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Marcillat, O.; Giulivi, C.; Ernster, L.; Davies, K.J. The Oxidative Inactivation of Mitochondrial Electron Transport Chain Components and ATPase. J. Biol. Chem. 1990, 265, 16330–16336. [Google Scholar] [CrossRef]
- Chen, J.; Schenker, S.; Frosto, T.A.; Henderson, G.I. Inhibition of Cytochrome c Oxidase Activity by 4-Hydroxynonenal (HNE). Role of HNE Adduct Formation with the Enzyme Subunits. Biochim. Biophys. Acta 1998, 1380, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Karu, T.I.; Pyatibrat, L.V.; Kolyakov, S.F.; Afanasyeva, N.I. Absorption Measurements of a Cell Monolayer Relevant to Phototherapy: Reduction of Cytochrome c Oxidase under near IR Radiation. J. Photochem. Photobiol. B 2005, 81, 98–106. [Google Scholar] [CrossRef]
- Sommer, A.P.; Haddad, M.K.; Fecht, H.-J. Light Effect on Water Viscosity: Implication for ATP Biosynthesis. Sci. Rep. 2015, 5, 12029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommer, A.P. Mitochondrial Cytochrome c Oxidase Is Not the Primary Acceptor for near Infrared Light-It Is Mitochondrial Bound Water: The Principles of Low-Level Light Therapy. Ann. Transl. Med. 2019, 7 (Suppl. 1), S13. [Google Scholar] [CrossRef] [PubMed]
- Sommer, A.P.; Schemmer, P.; Pavláth, A.E.; Försterling, H.-D.; Mester, Á.R.; Trelles, M.A. Quantum Biology in Low Level Light Therapy: Death of a Dogma. Ann. Transl. Med. 2020, 8, 440. [Google Scholar] [CrossRef] [PubMed]
- Hasinoff, B.B.; Davey, J.P. The Kinetics of the Aerobic Oxidation of Ferrocytochrome c by Cytochrome c Oxidase in Solvents of Increased Viscosity Are Partially Diffusion Controlled. Biochim. Biophys. Acta 1987, 892, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Kedia, R.V.; Hazzard, J.T.; Hurley, J.K.; Tollin, G.; Enemark, J.H. Effect of Solution Viscosity on Intramolecular Electron Transfer in Sulfite Oxidase. Biochemistry 2002, 41, 5816–5821. [Google Scholar] [CrossRef]
- Passarella, S.; Ostuni, A.; Atlante, A.; Quagliariello, E. Increase in the ADP/ATP Exchange in Rat Liver Mitochondria Irradiated in Vitro by Helium-Neon Laser. Biochem. Biophys. Res. Commun. 1988, 156, 978–986. [Google Scholar] [CrossRef]
- Karu, T.; Pyatibrat, L.; Kalendo, G. Irradiation with He Ne Laser Increases ATP Level in Cells Cultivated in Vitro. J. Photochem. Photobiol. B 1995, 27, 219–223. [Google Scholar] [CrossRef]
- Sommer, A.P.; Bieschke, J.; Friedrich, R.P.; Zhu, D.; Wanker, E.E.; Fecht, H.J.; Mereles, D.; Hunstein, W. 670 Nm Laser Light and EGCG Complementarily Reduce Amyloid-β Aggregates in Human Neuroblastoma Cells: Basis for Treatment of Alzheimer’s Disease? Photomed. Laser Surg. 2012, 30, 54–60. [Google Scholar] [CrossRef]
- Castellano-González, G.; Pichaud, N.; Ballard, J.W.O.; Bessede, A.; Marcal, H.; Guillemin, G.J. Epigallocatechin-3-Gallate Induces Oxidative Phosphorylation by Activating Cytochrome c Oxidase in Human Cultured Neurons and Astrocytes. Oncotarget 2016, 7, 7426–7440. [Google Scholar] [CrossRef] [Green Version]
- Reiter, R.J.; Rosales-Corral, S.; Tan, D.X.; Jou, M.J.; Galano, A.; Xu, B. Melatonin as a Mitochondria-Targeted Antioxidant: One of Evolution’s Best Ideas. Cell. Mol. Life Sci. 2017, 74, 3863–3881. [Google Scholar] [CrossRef] [PubMed]
- Petrosillo, G.; De Benedictis, V.; Ruggiero, F.M.; Paradies, G. Decline in Cytochrome c Oxidase Activity in Rat-Brain Mitochondria with Aging. Role of Peroxidized Cardiolipin and Beneficial Effect of Melatonin. J. Bioenerg. Biomembr. 2013, 45, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Agil, A.; Chayah, M.; Visiedo, L.; Navarro-Alarcon, M.; Rodríguez Ferrer, J.M.; Tassi, M.; Reiter, R.J.; Fernández-Vázquez, G. Melatonin Improves Mitochondrial Dynamics and Function in the Kidney of Zücker Diabetic Fatty Rats. J. Clin. Med. Res. 2020, 9, 2916. [Google Scholar] [CrossRef] [PubMed]
- Fernández Vázquez, G.; Reiter, R.J.; Agil, A. Melatonin Increases Brown Adipose Tissue Mass and Function in Zücker Diabetic Fatty Rats: Implications for Obesity Control. J. Pineal Res. 2018, 64, e12472. [Google Scholar] [CrossRef]
- Randi, E.B.; Zuhra, K.; Pecze, L.; Panagaki, T.; Szabo, C. Physiological Concentrations of Cyanide Stimulate Mitochondrial Complex IV and Enhance Cellular Bioenergetics. Proc. Natl. Acad. Sci. USA 2021, 118, e2026245118. [Google Scholar] [CrossRef]
- Pacher, P. Cyanide Emerges as an Endogenous Mammalian Gasotransmitter. Proc. Natl. Acad. Sci. USA 2021, 118, e2108040118. [Google Scholar] [CrossRef]
- Zuhra, K.; Szabo, C. The Two Faces of Cyanide: An Environmental Toxin and a Potential Novel Mammalian Gasotransmitter. FEBS J. 2022, 289, 2481–2515. [Google Scholar] [CrossRef]
- Nelson, L. Acute Cyanide Toxicity: Mechanisms and Manifestations. J. Emerg. Nurs. 2006, 32 (Suppl. 4), S8–S11. [Google Scholar] [CrossRef]
- Leavesley, H.B.; Li, L.; Prabhakaran, K.; Borowitz, J.L.; Isom, G.E. Interaction of Cyanide and Nitric Oxide with Cytochrome c Oxidase: Implications for Acute Cyanide Toxicity. Toxicol. Sci. 2008, 101, 101–111. [Google Scholar] [CrossRef] [Green Version]
- Gunasekar, P.G.; Borowitz, J.L.; Isom, G.E. Cyanide-Induced Generation of Oxidative Species: Involvement of Nitric Oxide Synthase and Cyclooxygenase-2. J. Pharmacol. Exp. Ther. 1998, 285, 236–241. [Google Scholar] [PubMed]
- Zhang, W.; Xu, K.; Yue, L.; Shao, Z.; Feng, Y.; Fang, M. Two-Dimensional Carbazole-Based Derivatives as Versatile Chemosensors for Colorimetric Detection of Cyanide and Two-Photon Fluorescence Imaging of Viscosity in Vitro. Dyes Pigments 2017, 137, 560–568. [Google Scholar] [CrossRef]
- Martín, M.; Macías, M.; León, J.; Escames, G.; Khaldy, H.; Acuña-Castroviejo, D. Melatonin Increases the Activity of the Oxidative Phosphorylation Enzymes and the Production of ATP in Rat Brain and Liver Mitochondria. Int. J. Biochem. Cell Biol. 2002, 34, 348–357. [Google Scholar] [CrossRef]
- Martín, M.; Macías, M.; Escames, G.; Reiter, R.J.; Agapito, M.T.; Ortiz, G.G.; Acuña-Castroviejo, D. Melatonin-Induced Increased Activity of the Respiratory Chain Complexes I and IV Can Prevent Mitochondrial Damage Induced by Ruthenium Red in Vivo. J. Pineal Res. 2000, 28, 242–248. [Google Scholar] [CrossRef] [Green Version]
- Jiang, G.-B.; Zheng, X.; Yao, J.-H.; Han, B.-J.; Li, W.; Wang, J.; Huang, H.-L.; Liu, Y.-J. Ruthenium(II) Polypyridyl Complexes Induce BEL-7402 Cell Apoptosis by ROS-Mediated Mitochondrial Pathway. J. Inorg. Biochem. 2014, 141, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Jiang, G.-B.; Yao, J.-H.; Wang, X.-Z.; Wang, J.; Han, B.-J.; Xie, Y.-Y.; Lin, G.-J.; Huang, H.-L.; Liu, Y.-J. Ruthenium(II) Complexes: DNA-Binding, Cytotoxicity, Apoptosis, Cellular Localization, Cell Cycle Arrest, Reactive Oxygen Species, Mitochondrial Membrane Potential and Western Blot Analysis. J. Photochem. Photobiol. B 2014, 140, 94–104. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, J.; Wan, J.; Liu, A.; Sun, J. Melatonin Regulates Aβ Production/clearance Balance and Aβ Neurotoxicity: A Potential Therapeutic Molecule for Alzheimer’s Disease. Biomed. Pharmacother. 2020, 132, 110887. [Google Scholar] [CrossRef] [PubMed]
- Cardinali, D.P. Melatonin: Clinical Perspectives in Neurodegeneration. Front. Endocrinol. 2019, 10, 480. [Google Scholar] [CrossRef]
- Vincent, B. Protective Roles of Melatonin against the Amyloid-Dependent Development of Alzheimer’s Disease: A Critical Review. Pharmacol. Res. 2018, 134, 223–237. [Google Scholar] [CrossRef]
- Wang, J.-Z.; Wang, Z.-F. Role of Melatonin in Alzheimer-like Neurodegeneration. Acta Pharmacol. Sin. 2006, 27, 41–49. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, V.; Pandi-Perumal, S.R.; Maestroni, G.J.; Esquifino, A.I.; Hardeland, R.; Cardinali, D.P. Role of Melatonin in Neurodegenerative Diseases. Neurotox. Res. 2005, 7, 293–318. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Mochizuki, H.; Ikeda, T.; Nihira, T.; Takasaki, J.-I.; Teplow, D.B.; Yamada, M. Effect of Melatonin on α-Synuclein Self-Assembly and Cytotoxicity. Neurobiol. Aging 2012, 33, 2172–2185. [Google Scholar] [CrossRef] [Green Version]
- Lin, A.M.Y.; Fang, S.F.; Chao, P.L.; Yang, C.H. Melatonin Attenuates Arsenite-Induced Apoptosis in Rat Brain: Involvement of Mitochondrial and Endoplasmic Reticulum Pathways and Aggregation of Alpha-Synuclein. J. Pineal Res. 2007, 43, 163–171. [Google Scholar] [CrossRef]
- Pappolla, M.; Bozner, P.; Soto, C.; Shao, H.; Robakis, N.K.; Zagorski, M.; Frangione, B.; Ghiso, J. Inhibition of Alzheimer Beta-Fibrillogenesis by Melatonin. J. Biol. Chem. 1998, 273, 7185–7188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skribanek, Z.; Baláspiri, L.; Mák, M. Interaction between Synthetic Amyloid-Beta-Peptide (1-40) and Its Aggregation Inhibitors Studied by Electrospray Ionization Mass Spectrometry. J. Mass Spectrom. 2001, 36, 1226–1229. [Google Scholar] [CrossRef] [PubMed]
- Poeggeler, B.; Miravalle, L.; Zagorski, M.G.; Wisniewski, T.; Chyan, Y.J.; Zhang, Y.; Shao, H.; Bryant-Thomas, T.; Vidal, R.; Frangione, B.; et al. Melatonin Reverses the Profibrillogenic Activity of Apolipoprotein E4 on the Alzheimer Amyloid Abeta Peptide. Biochemistry 2001, 40, 14995–15001. [Google Scholar] [CrossRef]
- Pappolla, M.A.; Matsubara, E.; Vidal, R.; Pacheco-Quinto, J.; Poeggeler, B.; Zagorski, M.; Sambamurti, K. Melatonin Treatment Enhances Aβ Lymphatic Clearance in a Transgenic Mouse Model of Amyloidosis. Curr. Alzheimer Res. 2018, 15, 637–642. [Google Scholar] [CrossRef]
- Matsubara, E.; Bryant-Thomas, T.; Pacheco Quinto, J.; Henry, T.L.; Poeggeler, B.; Herbert, D.; Cruz-Sanchez, F.; Chyan, Y.-J.; Smith, M.A.; Perry, G.; et al. Melatonin Increases Survival and Inhibits Oxidative and Amyloid Pathology in a Transgenic Model of Alzheimer’s Disease. J. Neurochem. 2003, 85, 1101–1108. [Google Scholar] [CrossRef]
- Quinn, J.; Kulhanek, D.; Nowlin, J.; Jones, R.; Praticò, D.; Rokach, J.; Stackman, R. Chronic Melatonin Therapy Fails to Alter Amyloid Burden or Oxidative Damage in Old Tg2576 Mice: Implications for Clinical Trials. Brain Res. 2005, 1037, 209–213. [Google Scholar] [CrossRef]
- Lahiri, D.K.; Chen, D.; Ge, Y.-W.; Bondy, S.C.; Sharman, E.H. Dietary Supplementation with Melatonin Reduces Levels of Amyloid Beta-Peptides in the Murine Cerebral Cortex. J. Pineal Res. 2004, 36, 224–231. [Google Scholar] [CrossRef]
- Ali, T.; Kim, M.O. Melatonin Ameliorates Amyloid Beta-Induced Memory Deficits, Tau Hyperphosphorylation and Neurodegeneration via PI3/Akt/GSk3β Pathway in the Mouse Hippocampus. J. Pineal Res. 2015, 59, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Luengo, E.; Buendia, I.; Fernández-Mendívil, C.; Trigo-Alonso, P.; Negredo, P.; Michalska, P.; Hernández-García, B.; Sánchez-Ramos, C.; Bernal, J.A.; Ikezu, T.; et al. Pharmacological Doses of Melatonin Impede Cognitive Decline in Tau-Related Alzheimer Models, Once Tauopathy Is Initiated, by Restoring the Autophagic Flux. J. Pineal Res. 2019, 67, e12578. [Google Scholar] [CrossRef]
- Balmik, A.A.; Das, R.; Dangi, A.; Gorantla, N.V.; Marelli, U.K.; Chinnathambi, S. Melatonin Interacts with Repeat Domain of Tau to Mediate Disaggregation of Paired Helical Filaments. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129467. [Google Scholar] [CrossRef]
- Das, R.; Balmik, A.A.; Chinnathambi, S. Effect of Melatonin on Tau Aggregation and Tau-Mediated Cell Surface Morphology. Int. J. Biol. Macromol. 2020, 152, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Nussinov, R. Salt Bridge Stability in Monomeric Proteins. J. Mol. Biol. 1999, 293, 1241–1255. [Google Scholar] [CrossRef]
- Xu, D.; Tsai, C.J.; Nussinov, R. Hydrogen Bonds and Salt Bridges across Protein-Protein Interfaces. Protein Eng. 1997, 10, 999–1012. [Google Scholar] [CrossRef] [Green Version]
- Musafia, B.; Buchner, V.; Arad, D. Complex Salt Bridges in Proteins: Statistical Analysis of Structure and Function. J. Mol. Biol. 1995, 254, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Tarus, B.; Straub, J.E.; Thirumalai, D. Dynamics of Asp23-Lys28 Salt-Bridge Formation in Abeta10-35 Monomers. J. Am. Chem. Soc. 2006, 128, 16159–16168. [Google Scholar] [CrossRef]
- Zwier, T.S. The spectroscopy of solvation in hydrogen-bonded aromatic clusters. Annu. Rev. Phys. Chem. 1996, 47, 205–241. [Google Scholar] [CrossRef]
- Zhu, L.; Gong, Y.; Lju, H.; Sun, G.; Zhang, Q.; Qian, Z. Mechanisms of Melatonin Binding and Destabilizing the Protofilament and Filament of Tau R3–R4 Domains Revealed by Molecular Dynamics Simulation. Phys. Chem. Chem. Phys. 2021, 23, 20615–20626. [Google Scholar] [CrossRef]
- Köpke, E.; Tung, Y.C.; Shaikh, S.; Alonso, A.C.; Iqbal, K.; Grundke-Iqbal, I. Microtubule-Associated Protein Tau. Abnormal Phosphorylation of a Non-Paired Helical Filament Pool in Alzheimer Disease. J. Biol. Chem. 1993, 268, 24374–24384. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Z.; Gong, C.X.; Zaidi, T.; Grundke-Iqbal, I.; Iqbal, K. Dephosphorylation of Alzheimer Paired Helical Filaments by Protein Phosphatase-2A and -2B. J. Biol. Chem. 1995, 270, 4854–4860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.Z.; Grundke-Iqbal, I.; Iqbal, K. Restoration of Biological Activity of Alzheimer Abnormally Phosphorylated Tau by Dephosphorylation with Protein Phosphatase-2A, -2B and -1. Brain Res. Mol. Brain Res. 1996, 38, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Poppek, D.; Keck, S.; Ermak, G.; Jung, T.; Stolzing, A.; Ullrich, O.; Davies, K.J.A.; Grune, T. Phosphorylation Inhibits Turnover of the Tau Protein by the Proteasome: Influence of RCAN1 and Oxidative Stress. Biochem. J 2006, 400, 511–520. [Google Scholar] [CrossRef] [Green Version]
- Bloom, G.S. Amyloid-β and Tau: The Trigger and Bullet in Alzheimer Disease Pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef] [Green Version]
- Cline, E.N.; Bicca, M.A.; Viola, K.L.; Klein, W.L. The Amyloid-β Oligomer Hypothesis: Beginning of the Third Decade. J. Alzheimer’s Dis. 2018, 64, S567–S610. [Google Scholar] [CrossRef] [Green Version]
- Kametani, F.; Hasegawa, M. Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 25. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.-Q.; Mobley, W.C. Alzheimer Disease Pathogenesis: Insights From Molecular and Cellular Biology Studies of Oligomeric Aβ and Tau Species. Front. Neurosci. 2019, 13, 659. [Google Scholar] [CrossRef]
- Chen, X.; Chen, M.; Schafer, N.P.; Wolynes, P.G. Exploring the Interplay between Fibrillization and Amorphous Aggregation Channels on the Energy Landscapes of Tau Repeat Isoforms. Proc. Natl. Acad. Sci. USA 2020, 117, 4125–4130. [Google Scholar] [CrossRef] [PubMed]
- Ruben, G.C.; Ciardelli, T.L.; Grundke-Iqbal, I.; Iqbal, K. Alzheimer Disease Hyperphosphorylated Tau Aggregates Hydrophobically. Synapse 1997, 27, 208–229. [Google Scholar] [CrossRef]
- Wegmann, S.; Eftekharzadeh, B.; Tepper, K.; Zoltowska, K.M.; Bennett, R.E.; Dujardin, S.; Laskowski, P.R.; MacKenzie, D.; Kamath, T.; Commins, C.; et al. Tau Protein Liquid-Liquid Phase Separation Can Initiate Tau Aggregation. EMBO J. 2018, 37, e98049. [Google Scholar] [CrossRef] [PubMed]
- Mandl, I.; Grauer, A.; Neuberg, C. Solubilization of Insoluble Matter in Nature; I. The Part Played by Salts of Adenosinetriphosphate. Biochim. Biophys. Acta 1952, 8, 654–663. [Google Scholar] [CrossRef] [PubMed]
- Hayes, M.H.; Peuchen, E.H.; Dovichi, N.J.; Weeks, D.L. Dual Roles for ATP in the Regulation of Phase Separated Protein Aggregates in Xenopus Oocyte Nucleoli. Elife 2018, 7, e35224. [Google Scholar] [CrossRef]
- Patel, A.; Malinovska, L.; Saha, S.; Wang, J.; Alberti, S.; Krishnan, Y.; Hyman, A.A. ATP as a Biological Hydrotrope. Science 2017, 356, 753–756. [Google Scholar] [CrossRef]
- Kurisaki, I.; Tanaka, S. ATP Converts Aβ42 Oligomer into Off-Pathway Species by Making Contact with Its Backbone Atoms Using Hydrophobic Adenosine. J. Phys. Chem. B 2019, 123, 9922–9933. [Google Scholar] [CrossRef] [PubMed]
- Mehringer, J.; Do, T.-M.; Touraud, D.; Hohenschutz, M.; Khoshsima, A.; Horinek, D.; Kunz, W. Hofmeister versus Neuberg: Is ATP Really a Biological Hydrotrope? Cell Rep. Phys. Sci. 2021, 2, 100343. [Google Scholar] [CrossRef]
- Zhang, C.; Rissman, R.A.; Feng, J. Characterization of ATP Alternations in an Alzheimer’s Disease Transgenic Mouse Model. J. Alzheimer’s Dis. 2015, 44, 375–378. [Google Scholar] [CrossRef] [Green Version]
- Pal, S.; Paul, S. ATP Controls the Aggregation of Aβ16-22 Peptides. J. Phys. Chem. B 2020, 124, 210–223. [Google Scholar] [CrossRef]
- Oro, J. Mechanism of Synthesis of Adenine from Hydrogen Cyanide under Possible Primitive Earth Conditions. Nature 1961, 191, 1193–1194. [Google Scholar] [CrossRef]
- Newby, A.C. Adenosine and the Concept of “retaliatory Metabolites”. Trends Biochem. Sci. 1984, 9, 42–44. [Google Scholar] [CrossRef]
- Cate, J.H.; Gooding, A.R.; Podell, E.; Zhou, K.; Golden, B.L.; Szewczak, A.A.; Kundrot, C.E.; Cech, T.R.; Doudna, J.A. RNA Tertiary Structure Mediation by Adenosine Platforms. Science 1996, 273, 1696–1699. [Google Scholar] [CrossRef] [Green Version]
- Ren, C.-L.; Shan, Y.; Zhang, P.; Ding, H.-M.; Ma, Y.-Q. Uncovering the Molecular Mechanism for Dual Effect of ATP on Phase Separation in FUS Solution. Sci. Adv. 2022, 8, eabo7885. [Google Scholar] [CrossRef]
- Kang, J.; Lim, L.; Song, J. ATP Enhances at Low Concentrations but Dissolves at High Concentrations Liquid-Liquid Phase Separation (LLPS) of ALS/FTD-Causing FUS. Biochem. Biophys. Res. Commun. 2018, 504, 545–551. [Google Scholar] [CrossRef]
- Henninger, J.E.; Oksuz, O.; Shrinivas, K.; Sagi, I.; LeRoy, G.; Zheng, M.M.; Andrews, J.O.; Zamudio, A.V.; Lazaris, C.; Hannett, N.M.; et al. RNA-Mediated Feedback Control of Transcriptional Condensates. Cell 2021, 184, 207–225.e24. [Google Scholar] [CrossRef] [PubMed]
- Maharana, S.; Wang, J.; Papadopoulos, D.K.; Richter, D.; Pozniakovsky, A.; Poser, I.; Bickle, M.; Rizk, S.; Guillén-Boixet, J.; Franzmann, T.M.; et al. RNA Buffers the Phase Separation Behavior of Prion-like RNA Binding Proteins. Science 2018, 360, 918–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aida, H.; Shigeta, Y.; Harada, R. The Role of ATP in Solubilizing RNA-Binding Protein Fused in Sarcoma. Proteins 2022, 90, 1606–1612. [Google Scholar] [CrossRef]
- Coskuner, O.; Murray, I.V.J. Adenosine Triphosphate (ATP) Reduces Amyloid-β Protein Misfolding in Vitro. J. Alzheimer’s Dis. 2014, 41, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Cohen, T.; Frydman-Marom, A.; Rechter, M.; Gazit, E. Inhibition of Amyloid Fibril Formation and Cytotoxicity by Hydroxyindole Derivatives. Biochemistry 2006, 45, 4727–4735. [Google Scholar] [CrossRef] [PubMed]
- Morshedi, D.; Rezaei-Ghaleh, N.; Ebrahim-Habibi, A.; Ahmadian, S.; Nemat-Gorgani, M. Inhibition of Amyloid Fibrillation of Lysozyme by Indole Derivatives—Possible Mechanism of Action. FEBS J. 2007, 274, 6415–6425. [Google Scholar] [CrossRef]
- Swan, I.D. The Inhibition of Hen Egg-White Lysozyme by Imidazole and Indole Derivatives. J. Mol. Biol. 1972, 65, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Di Bella, G.; Mascia, F.; Gualano, L.; Di Bella, L. Melatonin Anticancer Effects: Review. Int. J. Mol. Sci. 2013, 14, 2410–2430. [Google Scholar] [CrossRef] [Green Version]
- Di Bella, G.; Gualano, L.; Di Bella, L. Melatonin with Adenosine Solubilized in Water and Stabilized with Glycine for Oncological Treatment—Technical Preparation, Effectivity and Clinical Findings. Neuro Endocrinol. Lett. 2017, 38, 465–474. [Google Scholar]
- Todisco, M. Effectiveness of a Treatment Based on Melatonin in Five Patients with Systemic Sclerosis. Am. J. Ther. 2006, 13, 84–87. [Google Scholar] [CrossRef]
- Todisco, M.; Rossi, N. Melatonin for Refractory Idiopathic Thrombocytopenic Purpura: A Report of 3 Cases. Am. J. Ther. 2002, 9, 524–526. [Google Scholar] [CrossRef] [PubMed]
- Pattanayak, G.K.; Liao, Y.; Wallace, E.W.J.; Budnik, B.; Drummond, D.A.; Rust, M.J. Daily Cycles of Reversible Protein Condensation in Cyanobacteria. Cell Rep. 2020, 32, 108032. [Google Scholar] [CrossRef] [PubMed]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pathological Overproduction: The Bad Side of Adenosine. Br. J. Pharmacol. 2017, 174, 1945–1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, H. Extracellular Metabolism of ATP and Other Nucleotides. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2000, 362, 299–309. [Google Scholar] [CrossRef]
- Fredholm, B.B. Physiological and Pathophysiological Roles of Adenosine. Sleep Biol. Rhythms 2011, 9, 24–28. [Google Scholar] [CrossRef]
- Chang, C.-P.; Wu, K.-C.; Lin, C.-Y.; Chern, Y. Emerging Roles of Dysregulated Adenosine Homeostasis in Brain Disorders with a Specific Focus on Neurodegenerative Diseases. J. Biomed. Sci. 2021, 28, 70. [Google Scholar] [CrossRef]
- Antonioli, L.; Blandizzi, C.; Pacher, P.; Haskó, G. Immunity, Inflammation and Cancer: A Leading Role for Adenosine. Nat. Rev. Cancer 2013, 13, 842–857. [Google Scholar] [CrossRef]
- Morandi, F.; Horenstein, A.L.; Rizzo, R.; Malavasi, F. The Role of Extracellular Adenosine Generation in the Development of Autoimmune Diseases. Mediat. Inflamm. 2018, 2018, 7019398. [Google Scholar] [CrossRef] [PubMed]
- Blay, J.; White, T.D.; Hoskin, D.W. The Extracellular Fluid of Solid Carcinomas Contains Immunosuppressive Concentrations of Adenosine. Cancer Res. 1997, 57, 2602–2605. [Google Scholar]
- Rudolphi, K.A.; Schubert, P. Adenosine and Brain Ischemia. In Adenosine and Adenine Nucleotides: From Molecular Biology to Integrative Physiology; Belardinelli, L., Pelleg, A., Eds.; Springer: Boston, MA, USA, 1995; pp. 391–397. [Google Scholar] [CrossRef]
- Tan, D.-X.; Hardeland, R. The Reserve/Maximum Capacity of Melatonin’s Synthetic Function for the Potential Dimorphism of Melatonin Production and Its Biological Significance in Mammals. Molecules 2021, 26, 7302. [Google Scholar] [CrossRef] [PubMed]
- Löfgren, L.; Pehrsson, S.; Hägglund, G.; Tjellström, H.; Nylander, S. Accurate Measurement of Endogenous Adenosine in Human Blood. PLoS ONE 2018, 13, e0205707. [Google Scholar] [CrossRef] [Green Version]
- Waldhauser, F.; Weiszenbacher, G.; Tatzer, E.; Gisinger, B.; Waldhauser, M.; Schemper, M.; Frisch, H. Alterations in Nocturnal Serum Melatonin Levels in Humans with Growth and Aging. J. Clin. Endocrinol. Metab. 1988, 66, 648–652. [Google Scholar] [CrossRef] [PubMed]
- Kennaway, D.J.; Lushington, K.; Dawson, D.; Lack, L.; van den Heuvel, C.; Rogers, N. Urinary 6-Sulfatoxymelatonin Excretion and Aging: New Results and a Critical Review of the Literature. J. Pineal Res. 1999, 27, 210–220. [Google Scholar] [CrossRef]
- Waldhauser, F.; Kovács, J.; Reiter, E. Age-Related Changes in Melatonin Levels in Humans and Its Potential Consequences for Sleep Disorders. Exp. Gerontol. 1998, 33, 759–772. [Google Scholar] [CrossRef]
- Waldhauser, F.; Weiszenbacher, G.; Frisch, H.; Zeitlhuber, U.; Waldhauser, M.; Wurtman, R.J. Fall in Nocturnal Serum Melatonin during Prepuberty and Pubescence. Lancet 1984, 1, 362–365. [Google Scholar] [CrossRef]
- Burch, J.B.; Reif, J.S.; Yost, M.G.; Keefe, T.J.; Pitrat, C.A. Reduced Excretion of a Melatonin Metabolite in Workers Exposed to 60 Hz Magnetic Fields. Am. J. Epidemiol. 1999, 150, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Prayag, A.S.; Najjar, R.P.; Gronfier, C. Melatonin Suppression Is Exquisitely Sensitive to Light and Primarily Driven by Melanopsin in Humans. J. Pineal Res. 2019, 66, e12562. [Google Scholar] [CrossRef]
- Bojkowski, C.J.; Aldhous, M.E.; English, J.; Franey, C.; Poulton, A.L.; Skene, D.J.; Arendt, J. Suppression of Nocturnal Plasma Melatonin and 6-Sulphatoxymelatonin by Bright and Dim Light in Man. Horm. Metab. Res. 1987, 19, 437–440. [Google Scholar] [CrossRef]
- Tan, D.-X.; Reiter, R.J.; Manchester, L.C.; Yan, M.-T.; El-Sawi, M.; Sainz, R.M.; Mayo, J.C.; Kohen, R.; Allegra, M.; Hardeland, R. Chemical and Physical Properties and Potential Mechanisms: Melatonin as a Broad Spectrum Antioxidant and Free Radical Scavenger. Curr. Top. Med. Chem. 2002, 2, 181–197. [Google Scholar] [CrossRef] [Green Version]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 5957, Adenosine-5′-Triphosphate; National Center for Biotechnology Information: Bethesda, MD, USA, 2023. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Adenosine-5_-triphosphate (accessed on 5 January 2023).
- National Center for Biotechnology Information. PubChem Compound Summary for CID 896, Melatonin; National Center for Biotechnology Information: Bethesda, MD, USA, 2023. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Melatonin (accessed on 7 January 2023).
- Ishii, M.; Wang, G.; Racchumi, G.; Dyke, J.P.; Iadecola, C. Transgenic Mice Overexpressing Amyloid Precursor Protein Exhibit Early Metabolic Deficits and a Pathologically Low Leptin State Associated with Hypothalamic Dysfunction in Arcuate Neuropeptide Y Neurons. J. Neurosci. 2014, 34, 9096–9106. [Google Scholar] [CrossRef]
- Feng, Z.; Qin, C.; Chang, Y.; Zhang, J.-T. Early Melatonin Supplementation Alleviates Oxidative Stress in a Transgenic Mouse Model of Alzheimer’s Disease. Free. Radic. Biol. Med. 2006, 40, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Bachmanov, A.A.; Reed, D.R.; Beauchamp, G.K.; Tordoff, M.G. Food Intake, Water Intake, and Drinking Spout Side Preference of 28 Mouse Strains. Behav. Genet. 2002, 32, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Gould, S.J. Allometry in Primates, with Emphasis on Scaling and the Evolution of the Brain. Contrib. Primatol. 1975, 5, 244–292. [Google Scholar] [PubMed]
- Mirth, C.K.; Frankino, W.A.; Shingleton, A.W. Allometry and Size Control: What Can Studies of Body Size Regulation Teach Us about the Evolution of Morphological Scaling Relationships? Curr. Opin. Insect Sci. 2016, 13, 93–98. [Google Scholar] [CrossRef] [Green Version]
- Galilei, G. Dialogues Concerning Two New Sciences, 1637; Crew, H.; De Salvio, A., Translators; Macmillan: New York, NY, USA, 1914. [Google Scholar]
- Boxenbaum, H. Interspecies Scaling, Allometry, Physiological Time, and the Ground Plan of Pharmacokinetics. J. Pharmacokinet. Pharmacodyn. 1982, 10, 201–227. [Google Scholar] [CrossRef] [PubMed]
- Boxenbaum, H.; Fertig, J.B. Scaling of Antipyrine Intrinsic Clearance of Unbound Drug in 15 Mammalian Species. Eur. J. Drug Metab. Pharmacokinet. 1984, 9, 177–183. [Google Scholar] [CrossRef]
- Boxenbaum, H. Interspecies Pharmacokinetic Scaling and the Evolutionary-Comparative Paradigm. Drug Metab. Rev. 1984, 15, 1071–1121. [Google Scholar] [CrossRef]
- Ritschel, W.A.; Vachharajani, N.N.; Johnson, R.D.; Hussain, A.S. The Allometric Approach for Interspecies Scaling of Pharmacokinetic Parameters. Comp. Biochem. Physiol. Part C 1992, 103, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Boxenbaum, H.; DiLea, C. First-Time-in-Human Dose Selection: Allometric Thoughts and Perspectives. J. Clin. Pharmacol. 1995, 35, 957–966. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.M.; Hayton, W.L. Allometric Scaling of Xenobiotic Clearance: Uncertainty versus Universality. AAPS PharmSci 2001, 3, E29. [Google Scholar] [CrossRef] [PubMed]
- Gibson, S.; Numa, A. The Importance of Metabolic Rate and the Folly of Body Surface Area Calculations. Anaesthesia 2003, 58, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Savage, V.M.; Gillooly, J.F.; Woodruff, W.H.; West, G.B.; Allen, A.P.; Enquist, B.J.; Brown, J.H. The Predominance of Quarter-Power Scaling in Biology. Funct. Ecol. 2004, 18, 257–282. [Google Scholar] [CrossRef]
- Glazier, D.S. Beyond the “3/4-Power Law”: Variation in the Intra- and Interspecific Scaling of Metabolic Rate in Animals. Biol. Rev. Camb. Philos. Soc. 2005, 80, 611–662. [Google Scholar] [CrossRef]
- West, G.B.; Brown, J.H. The Origin of Allometric Scaling Laws in Biology from Genomes to Ecosystems: Towards a Quantitative Unifying Theory of Biological Structure and Organization. J. Exp. Biol. 2005, 208 Pt 9, 1575–1592. [Google Scholar] [CrossRef] [Green Version]
- Rhomberg, L.R.; Lewandowski, T.A. Methods for Identifying a Default Cross-Species Scaling Factor. Hum. Ecol. Risk Assess. Int. J. 2006, 12, 1094–1127. [Google Scholar] [CrossRef]
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose Translation from Animal to Human Studies Revisited. FASEB J. 2008, 22, 659–661. [Google Scholar] [CrossRef] [Green Version]
- White, C.R.; Kearney, M.R. Metabolic Scaling in Animals: Methods, Empirical Results, and Theoretical Explanations. Compr. Physiol. 2014, 4, 231–256. [Google Scholar] [CrossRef]
- Glazier, D.S. Effects of Contingency versus Constraints on the Body-Mass Scaling of Metabolic Rate. Challenges 2018, 9, 4. [Google Scholar] [CrossRef] [Green Version]
- Li, X.-X.; He, J.-H. Along the evolution process kleiber’s 3/4 law makes way for rubner’s surface law: A fractal approach. Fractals 2019, 27, 1950015. [Google Scholar] [CrossRef]
- Dodds, P.S.; Rothman, D.H.; Weitz, J.S. Re-Examination of the “3/4-Law” of Metabolism. J. Theor. Biol. 2001, 209, 9–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, C.R.; Seymour, R.S. Mammalian Basal Metabolic Rate Is Proportional to Body mass2/3. Proc. Natl. Acad. Sci. USA 2003, 100, 4046–4049. [Google Scholar] [CrossRef] [Green Version]
- White, C.R.; Cassey, P.; Blackburn, T.M. Allometric Exponents Do Not Support a Universal Metabolic Allometry. Ecology 2007, 88, 315–323. [Google Scholar] [CrossRef]
- Banavar, J.R.; Moses, M.E.; Brown, J.H.; Damuth, J.; Rinaldo, A.; Sibly, R.M.; Maritan, A. A General Basis for Quarter-Power Scaling in Animals. Proc. Natl. Acad. Sci. USA 2010, 107, 15816–15820. [Google Scholar] [CrossRef] [Green Version]
- Dreyer, G. The normal basal metabolism in man: And its relation to the size of the body and age, expressed in simple formulæ. Lancet 1920, 196, 289–291. [Google Scholar] [CrossRef] [Green Version]
- Kleiber, M. Body Size and Metabolism. Hilgardia 1932, 6, 315–353. [Google Scholar] [CrossRef] [Green Version]
- Nair, A.B.; Jacob, S. A Simple Practice Guide for Dose Conversion between Animals and Human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reading, B.D.; Freeman, B. Simple Formula for the Surface Area of the Body and a Simple Model for Anthropometry. Clin. Anat. 2005, 18, 126–130. [Google Scholar] [CrossRef]
- Cheung, M.C.; Spalding, P.B.; Gutierrez, J.C.; Balkan, W.; Namias, N.; Koniaris, L.G.; Zimmers, T.A. Body Surface Area Prediction in Normal, Hypermuscular, and Obese Mice. J. Surg. Res. 2009, 153, 326–331. [Google Scholar] [CrossRef]
- Gouma, E.; Simos, Y.; Verginadis, I.; Lykoudis, E.; Evangelou, A.; Karkabounas, S. A Simple Procedure for Estimation of Total Body Surface Area and Determination of a New Value of Meeh’s Constant in Rats. Lab. Anim. 2012, 46, 40–45. [Google Scholar] [CrossRef]
- Toutain, P.L.; Bousquet-Mélou, A. Bioavailability and Its Assessment. J. Vet. Pharmacol. Ther. 2004, 27, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Waldhauser, F.; Waldhauser, M.; Lieberman, H.R.; Deng, M.H.; Lynch, H.J.; Wurtman, R.J. Bioavailability of Oral Melatonin in Humans. Neuroendocrinology 1984, 39, 307–313. [Google Scholar] [CrossRef]
- Zetner, D.; Andersen, L.P.K.; Alder, R.; Jessen, M.L.; Tolstrup, A.; Rosenberg, J. Pharmacokinetics and Safety of Intravenous, Intravesical, Rectal, Transdermal, and Vaginal Melatonin in Healthy Female Volunteers: A Cross-Over Study. Pharmacology 2021, 106, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Lane, E.A.; Moss, H.B. Pharmacokinetics of Melatonin in Man: First Pass Hepatic Metabolism. J. Clin. Endocrinol. Metab. 1985, 61, 1214–1216. [Google Scholar] [CrossRef]
- DeMuro, R.L.; Nafziger, A.N.; Blask, D.E.; Menhinick, A.M.; Bertino, J.S., Jr. The Absolute Bioavailability of Oral Melatonin. J. Clin. Pharmacol. 2000, 40, 781–784. [Google Scholar] [CrossRef] [PubMed]
- Harpsøe, N.G.; Andersen, L.P.H.; Gögenur, I.; Rosenberg, J. Clinical Pharmacokinetics of Melatonin: A Systematic Review. Eur. J. Clin. Pharmacol. 2015, 71, 901–909. [Google Scholar] [CrossRef]
- Andersen, L.P.H.; Werner, M.U.; Rosenkilde, M.M.; Harpsøe, N.G.; Fuglsang, H.; Rosenberg, J.; Gögenur, I. Pharmacokinetics of Oral and Intravenous Melatonin in Healthy Volunteers. BMC Pharmacol. Toxicol. 2016, 17, 8. [Google Scholar] [CrossRef] [Green Version]
- Di, W.L.; Kadva, A.; Johnston, A.; Silman, R. Variable Bioavailability of Oral Melatonin. N. Engl. J. Med. 1997, 336, 1028–1029. [Google Scholar] [CrossRef]
- Bagci, S.; Horoz, Ö.Ö.; Yildizdas, D.; Reinsberg, J.; Bartmann, P.; Müller, A. Melatonin Status in Pediatric Intensive Care Patients with Sepsis. Pediatr. Crit. Care Med. 2012, 13, e120–e123. [Google Scholar] [CrossRef]
- Tordjman, S.; Anderson, G.M.; Pichard, N.; Charbuy, H.; Touitou, Y. Nocturnal Excretion of 6-Sulphatoxymelatonin in Children and Adolescents with Autistic Disorder. Biol. Psychiatry 2005, 57, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Tordjman, S.; Anderson, G.M.; Bellissant, E.; Botbol, M.; Charbuy, H.; Camus, F.; Graignic, R.; Kermarrec, S.; Fougerou, C.; Cohen, D.; et al. Day and Nighttime Excretion of 6-Sulphatoxymelatonin in Adolescents and Young Adults with Autistic Disorder. Psychoneuroendocrinology 2012, 37, 1990–1997. [Google Scholar] [CrossRef]
- Kennaway, D.J.; Flanagan, D.E.; Moore, V.M.; Cockington, R.A.; Robinson, J.S.; Phillips, D.I. The Impact of Fetal Size and Length of Gestation on 6-Sulphatoxymelatonin Excretion in Adult Life. J. Pineal Res. 2001, 30, 188–192. [Google Scholar] [CrossRef]
- Girotti, L.; Lago, M.; Ianovsky, O.; Carbajales, J.; Elizari, M.V.; Brusco, L.I.; Cardinali, D.P. Low Urinary 6-Sulphatoxymelatonin Levels in Patients with Coronary Artery Disease. J. Pineal Res. 2000, 29, 138–142. [Google Scholar] [CrossRef]
- Yeleswaram, K.; McLaughlin, L.G.; Knipe, J.O.; Schabdach, D. Pharmacokinetics and Oral Bioavailability of Exogenous Melatonin in Preclinical Animal Models and Clinical Implications. J. Pineal Res. 1997, 22, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Kopin, I.J.; Pare, C.M.; Axelrod, J.; Weissbach, H. The Fate of Melatonin in Animals. J. Biol. Chem. 1961, 236, 3072–3075. [Google Scholar] [CrossRef]
- Raynaud, F.; Mauviard, F.; Geoffriau, M.; Claustrat, B.; Pevet, P. Plasma 6-Hydroxymelatonin, 6-Sulfatoxymelatonin and Melatonin Kinetics after Melatonin Administration to Rats. Biol. Signals 1993, 2, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.; Kumar, A.; Saha, N.; Chaudhury, N.K. PK-PD Based Optimal Dose and Time for Orally Administered Supra-Pharmacological Dose of Melatonin to Prevent Radiation Induced Mortality in Mice. Life Sci. 2019, 219, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Galley, H.F.; Allen, L.; Colin, P.J.; Galt, S.P.; Webster, N.R. Dose Assessment of Melatonin in Sepsis (DAMSEL2) Study: Pharmacokinetics of Two Doses of Oral Melatonin in Patients with Sepsis. J. Pineal Res. 2022, 73, e12830. [Google Scholar] [CrossRef]
- Mao, S.; Chen, J.; Wei, Z.; Liu, H.; Bi, D. Intranasal Administration of Melatonin Starch Microspheres. Int. J. Pharm. 2004, 272, 37–43. [Google Scholar] [CrossRef]
- Priprem, A.; Johns, J.R.; Limsitthichaikoon, S.; Limphirat, W.; Mahakunakorn, P.; Johns, N.P. Intranasal Melatonin Nanoniosomes: Pharmacokinetic, Pharmacodynamics and Toxicity Studies. Ther. Deliv. 2017, 8, 373–390. [Google Scholar] [CrossRef]
- Maeda, H.; Ogawa, Y.; Nakayama, H. Inclusion Complexes of Melatonin with Modified Cyclodextrins. J. Incl. Phenom. Macrocycl. Chem. 2014, 78, 217–224. [Google Scholar] [CrossRef]
- Sakellaropoulou, A.; Siamidi, A.; Vlachou, M. Melatonin/Cyclodextrin Inclusion Complexes: A Review. Molecules 2022, 27, 445. [Google Scholar] [CrossRef]
- Barchas, J.; DaCosta, F.; Spector, S. Acute Pharmacology of Melatonin. Nature 1967, 214, 919–920. [Google Scholar] [CrossRef]
- Boutin, J.A.; Jockers, R. Melatonin Controversies, an Update. J. Pineal Res. 2021, 70, e12702. [Google Scholar] [CrossRef] [PubMed]
- Cai, R.; Yang, H.; He, J.; Zhu, W. The Effects of Magnetic Fields on Water Molecular Hydrogen Bonds. J. Mol. Struct. 2009, 938, 15–19. [Google Scholar] [CrossRef]
- Menczel Schrire, Z.; Gordon, C.J.; Palmer, J.R.; Murray, J.; Hickie, I.; Rogers, N.L.; Lewis, S.J.; Terpening, Z.; Pye, J.E.; Naismith, S.L.; et al. Actigraphic and Melatonin Alignment in Older Adults with Varying Dementia Risk. Chronobiol. Int. 2022, 1–12. [Google Scholar] [CrossRef]
- Glacet, R.; Reynaud, E.; Robin-Choteau, L.; Reix, N.; Hugueny, L.; Ruppert, E.; Geoffroy, P.A.; Kilic-Huck, Ü.; Comtet, H.; Bourgin, P. A Comparison of Four Methods to Estimate Dim Light Melatonin Onset: A Repeatability and Agreement Study. Chronobiol. Int. 2022, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Irwin, M.R.; Vitiello, M.V. Implications of Sleep Disturbance and Inflammation for Alzheimer’s Disease Dementia. Lancet Neurol. 2019, 18, 296–306. [Google Scholar] [CrossRef] [PubMed]



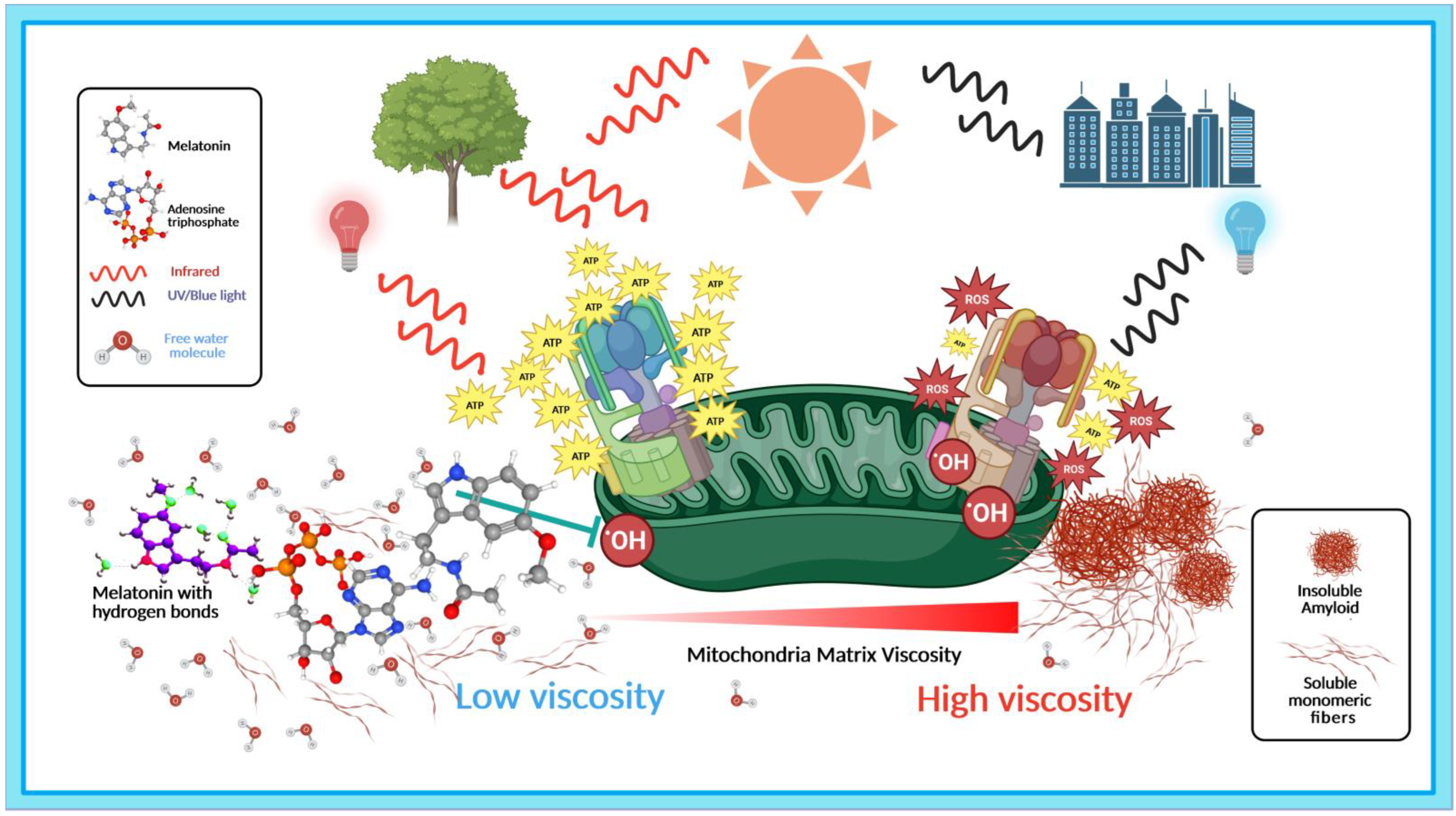{kind=link}
{kind=link}
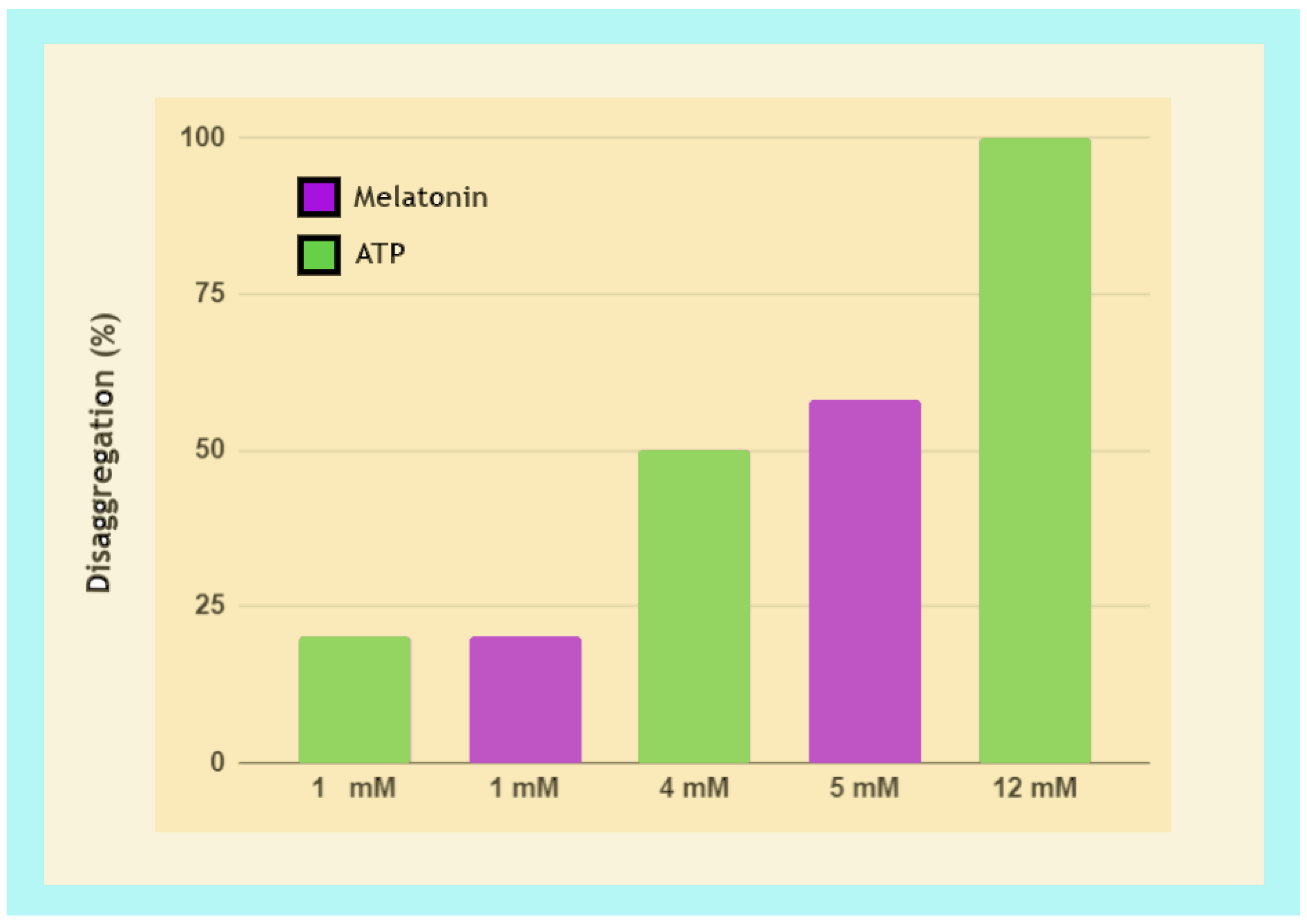{kind=link}
| Wavelength | Model/Cell Line/Device | Duration/Intensity | Results | Ref. |
|---|---|---|---|---|
| 670 nm | APP/PS1 AD transgenic mice/transcranial LED | 90 s (4 Joule/cm2)/day × 20 over 4 wks | Attenuated cerebellar cortex Aβ deposition, fibril formation. | [207] |
| 670 nm | K3 tau, APP/PS1 AD transgenic mice/transcranial LED | 90 s (4 Joule/cm2)/day × 20 over 4 wks | Neocortex and hippocampus of K3 and APP/PS1 mice showed reduction in tau/fibril formation and size/number of Aβ, respectively. | [208] |
| 670 nm | C57BL/6, transgenic 2576 mice/transcranial LED | 90 s (4 Joule/cm2)/day × 20 over 4 wks | All mice showed reduced Aβ oligomer binding at CNS synapses. | [209] |
| 670 nm | h tau, 3xTgAD mice/transcranial LED | 90 s (4 Joule/cm2)/day × 20 over 4 wks | Reduced toxic tau oligomers, improved memory deficits, upregulated clearance of misfolded proteins in both models | [210] |
| 808 nm | Aβ-treated microglia cells from health mice/diode laser | 5 min (9 Joule/cm2) | Exceeded control cell ATP production after 24 h by 155%, suppressed ROS production promoting neuronal survival. | [211] |
| 810 nm | 8 patients diagnosed with dementia/transcranial+transnasal LED | 20 min (pulsed at 40 Hz at 50% duty cycle), 3 times/wk for 12 consecutive wks | Significant score improvements in ADAS-cog (13.8%) NPI-FS (61.4%) compared to baseline 1. | [212] |
| 1060–1080 nm | 11 patients with dementia/transcranial LED helmet | 6 min (1100 LEDs pulsed at 10 Hz at 50% duty cycle)/day × 28 consecutive days | Improved executive functioning in clock drawing, immediate recall, praxis memory, visual attention, and task switching. | [213] |
| 1060–1080 nm | 60 patients with mild to moderate dementia/transcranial LED helmet | 2 × 6 min (23.1 mW/cm2)/day × 8 consecutive weeks | Improved cognitive functions, auditory and verbal learning, processing speed, mood, energy, and sleep. | [214] |
| 1060 nm | 27 healthy participants aged 45+/transcranial LED helmet | 2 × 6 min (12 mW/cm2)/day × 28 minimum | Significant improvements in motor function, memory performance, and processing speed. | [215] |
| 1040–1090 nm | APP/PS1 AD double-transgenic mice/LED irradiation | 6 min (15 mW/cm2)/day × 55 with a 28-day suspension after day 40 | Improvement in memory, spatial learning ability, and modest plaque reduction; suspension period indicated treatment effects were transient. | [216] |
| 500 nm/800 nm/3–25 µm | APP/PS1 AD double-transgenic mice/LED irradiation | 60 min (0.13 mW/cm2)/day × 45 | FIR (3–25 µm) enhanced Aβ phagocytosis via increased ATP production and attenuated cognitive dysfunction compared to other wavelengths tested. | [217] |
| Melatonin Dosage/Duration | Study Design | Results | Ref. |
|---|---|---|---|
| 25 µM, 250 µM, 2.5 mM | In vitro α-Synuclein peptide aggregation | Blocked α-Syn fibril formation and destabilized preformed fibrils in a dose- and time-dependent manner; increased viability of primary mixed neurons treated with α-Syn to ~97% in a time-dependent manner. | [356] |
| 10 mg/kg (IP) × 5/day for 2 days, then × 2/day for 5 days | Arsenite-induced oxidative injury in substantial nigra of adult male rats | Attenuated arsenite-induced α-Syn aggregation, lipid peroxidation, and glutathione depletion. | [357] |
| 100 µM melatonin | Aβ peptides (1-40) and (1-42) β-sheet/fibril formation | Progressive reduction in Aβ1-40 β-sheet structures to 24% after 24 h incubation; immediate reduction in Aβ1-42 β-sheet structures from 89% to 65%, decreasing to 59% after 4 h. | [358] |
| Melatonin dissolved in 2 mM ammonium acetate | Aβ peptide (1-40) β-sheet/fibril formation | Inhibited β-sheet formation by targeting hydrophobic Aβ-peptide segment (29-40) intermolecular activities. | [359] |
| 1 mM melatonin | Aβ1-40 peptide, profibrillogenic apoE4/apoE | Melatonin alone delayed fibril formation from 24 h up to 72 h. Combined with either apoE4 or apoE3, inhibition remained effective at termination of experiment. | [360] |
| 2 mg/mL in drinking water starting at age 4 months until euthanasia | Transgenic Tg2576 AD mice, terminated at 4 months 1 wk or 15.5 months | The brains of animals treated with melatonin terminated at 15.5 months exhibited dramatic decline in oligomeric Aβ40 together with a significant increase in soluble monomeric Aβ40, and a decreasing trend in Aβ42 compared to untreated mice at same age. Melatonin prolonged survival rates of 15.5-month mice to levels attained by non-transgenic mice. | [361] |
| 2 mg/mL in drinking water starting at age 4 months until euthanasia at 15.5 months | Transgenic Tg2576 AD mice | Increased survival in treated mice (3 deaths/41 survivals) compared to untreated (13 deaths/31 survivals). | [362] |
| 0.5 mg/mL in drinking water starting at age 4 months | Transgenic Tg2576 AD mice | Striking reductions in Aβ levels in brain tissues of treated mice at 8, 9.5, 11, and 15.5 months. | [362] |
| 16 µg/mL in drinking water starting at age 14 months | Transgenic Tg2576 AD mice | Melatonin treatment failed to reduce brain Aβ levels or even oxidative damage. | [363] |
| 40-ppm (w/w) in pelleted minimal basal diet | Male B6C3F1 mice aged 6, 12, and 27 months | Significant reduction in Aβ in brain cortex tissues: 57% in Aβ40 and 73% in Aβ42; increased melatonin levels in cerebral cortex in all 3 treated age groups (12 > 6 > 27 mos) compared to untreated. | [364] |
| 10 mg/kg (IP) daily for 3 weeks | Male wild-type C57BL/6N mice (8 wks old) injected with Aβ1-42 peptide | Melatonin treatment reversed Aβ1-42-induced synaptic disorder, memory deficit, and prevented Aβ1-42-induced apoptosis, neurodegeneration, and tau phosphorylation. | [365] |
| 10 mg/kg in drinking water from day 7 after tauopathy induction to day 28 at termination | 4-month-old C57BL/6J mice injected with human tau mutation P301L (AAV-hTau) | Increased ROS and tau hyperphosphorylation starting at day 7 precedes cognitive decline; melatonin-treated animals showed reduced memory impairment, tau hyperphosphorylation, ROS, and neuroinflammation. | [366] |
| 10 μmol/L | Ex vivo brain slices from 3-month-old SD rats exposed to okadaic acid to induce tau hyperphosphorylation | Melatonin reduced tau hyperphosphorylation and ROS to control levels in OA-treated brain slices. | [366] |
| 100 μM–5000 μM | Aggregation/disaggregation of repeat domain Tau (K18wt) | Pre-formed tau fibril disaggregation was dose-dependent: 14% with 100 μM, 54% with 5000 μM. | [367] |
| 200–5000 μM | Aggregation/Disaggregation full-length tau (hTau40wt) | Tau treated with 200 μM melatonin showed no change in morphology compared to controls; 5000 μM melatonin treatment did not prevent aggregation but disaggregated tau fibrils into broken filaments. | [368] |
| Study Design/Total Daily Dose/Duration/Ref. | Results | (A) HED Daily Total (mg/kg) Scaled to Mb3/4 | (B) Dose (A) Adjusted by Bioavailability | (C) Dose (A) Adjusted by Enhanced Bioavailability |
|---|---|---|---|---|
| 2 mg/mL in drinking water, Tg2576 AD mice/266.66 mg/kg/11.5 mos starting at 4 mos old/[361,362] | Striking reductions in Aβ aggregates at all ages during treatment; dramatic extension of survival of AD mice to levels similar to wild types. | 2499 mg (35.7 mg/kg) | 4831 mg (69 mg/kg) | 10,621 mg (151.73 mg/kg) |
| 0.5 mg/mL in drinking water, Tg2576 AD mice/66.66 mg/kg/11.5 mos starting at 4 mos old/[362] | Striking reductions in Aβ levels in brain tissues of treated mice at 8, 9.5, 11, and 15.5 months. | 625 mg (8.928 mg/kg) | 1208 mg (17.26 mg/kg) | 2656 mg (37.94 mg/kg) |
| 0.016 mg/mL in drinking water, Tg2576 AD mice/2.13 mg/kg/10 wks starting at age 14 mos old/[363] | Failed to reduce brain Aβ levels, unable to reverse oxidative damage. | 19.96 mg (0.285 mg/kg) | 38.58 mg (0.55 mg/kg) | 84.83 mg (1.21 mg/kg) |
| 10 mg/kg in drinking water, healthy, normal C57BL/6J mice/14 days after tauopathy initiation/[366] | Reduced memory impairment, tau hyperphosphorylation, ROS, and neuroinflammation. | 96.23 mg (1.375 mg/kg) | 186.0 mg (2.66 mg/kg) | 408.98 mg (5.84 mg/kg) |
| 40 ppm in food pellets, healthy, normal B6C3F1 mice/7.2 mg/kg/11 weeks different age groups/[364] | Significant reduction in Aβ peptides in brain cortex tissues: 57% in Aβ40 and 73% in Aβ42; increased melatonin levels in cerebral cortex in all 3 treated age groups (12 > 6 > 27 mos) compared to untreated. | 69.29 mg (0.99 mg/kg) | 133.94 mg (1.91 mg/kg) | Not applicable |
| 10 mg/kg IP injection, C57BL/6J mice treated with Aβ1-42 peptide/daily IP injections for 3 wks/[365] | Reversed Aβ1-42-induced synaptic disorder, memory deficit; prevented Aβ1-42-induced apoptosis, neurodegeneration, and tau phosphorylation. | 98.55 mg (1.41 mg/kg) | 486.15 mg (6.95 mg/kg) | Not applicable |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loh, D.; Reiter, R.J. Light, Water, and Melatonin: The Synergistic Regulation of Phase Separation in Dementia. Int. J. Mol. Sci. 2023, 24, 5835. https://doi.org/10.3390/ijms24065835
Loh D, Reiter RJ. Light, Water, and Melatonin: The Synergistic Regulation of Phase Separation in Dementia. International Journal of Molecular Sciences. 2023; 24(6):5835. https://doi.org/10.3390/ijms24065835
Chicago/Turabian StyleLoh, Doris, and Russel J. Reiter. 2023. "Light, Water, and Melatonin: The Synergistic Regulation of Phase Separation in Dementia" International Journal of Molecular Sciences 24, no. 6: 5835. https://doi.org/10.3390/ijms24065835