
Differential Effects of Overexpression of Wild Type and Kinase-Dead MELK in Fibroblasts and Keratinocytes, Potential Implications for Skin Wound Healing and Cancer
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Fibroblasts
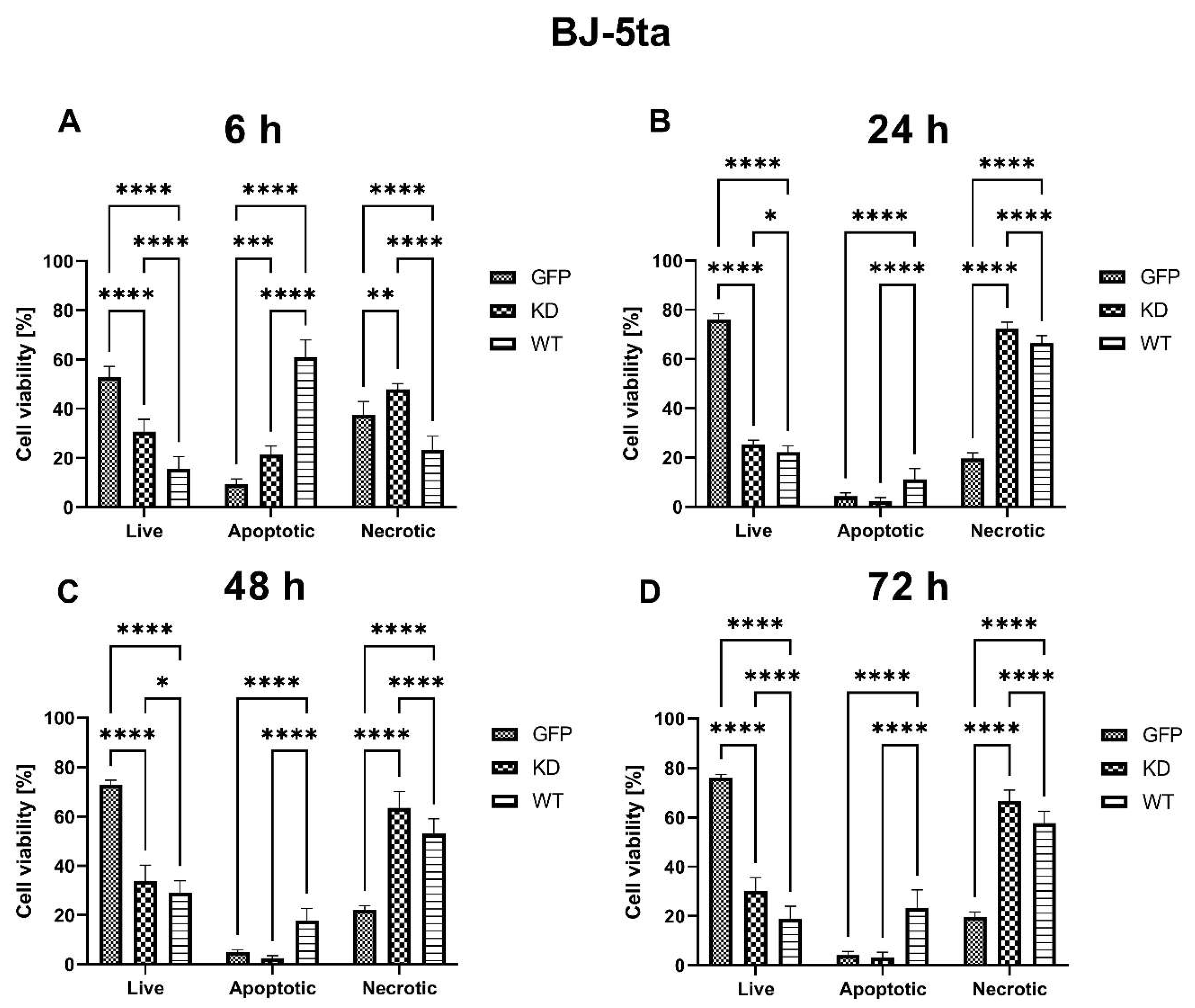
2.1.1. Effects of MELK Overexpression on BJ-5ta Cell Viability
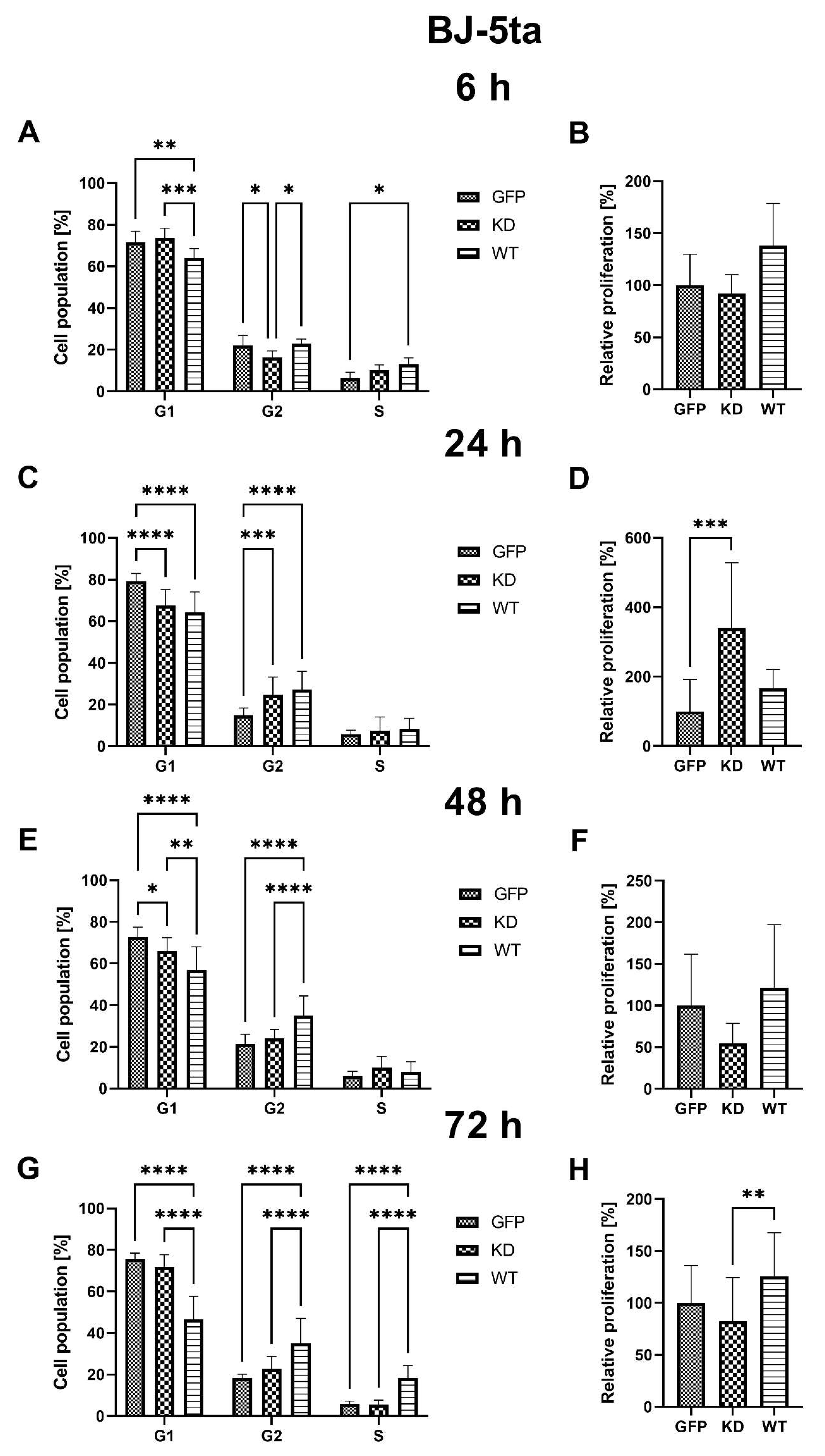
2.1.2. Effects of MELK Overexpression on BJ-5ta Cell Cycle Phase Distribution and Proliferation Rate
2.1.3. Effects of MELK Overexpression on Selected Signaling Pathways Activation
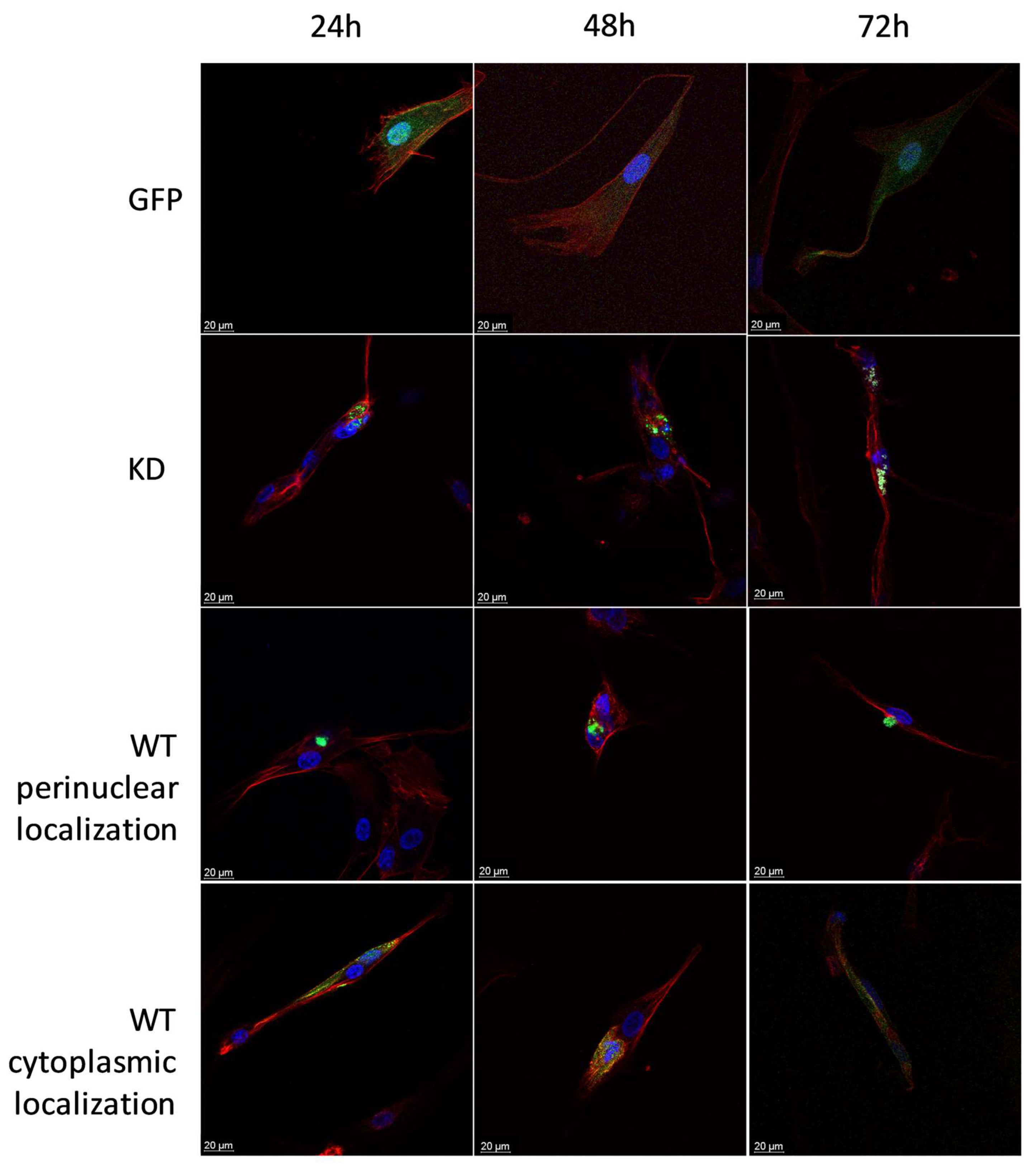
2.1.4. Intracellular Localization of MELK WT and MELK KD in BJ-5ta Cells
2.2. Keratinocytes
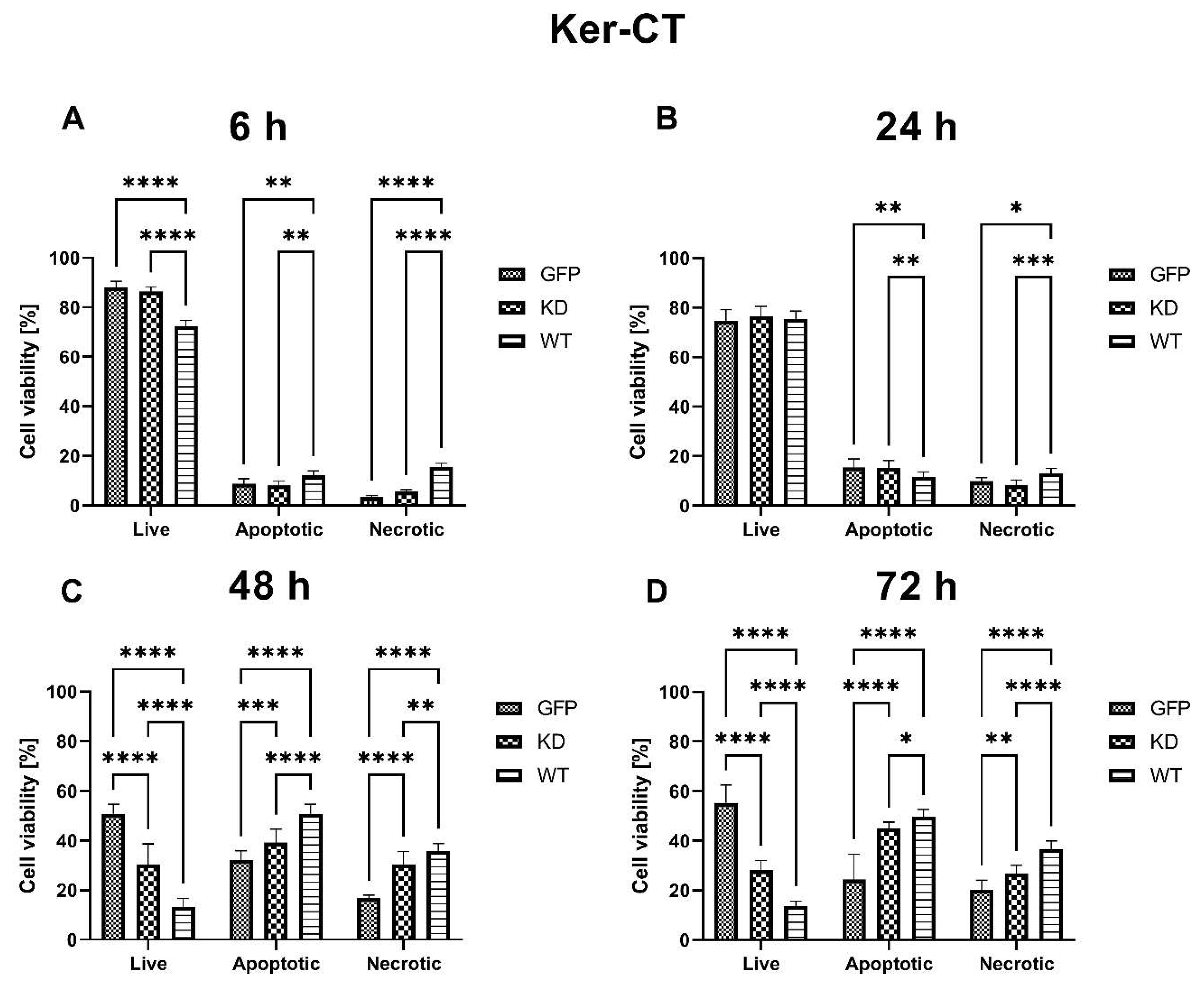
2.2.1. Effects of MELK Overexpression on Ker-CT Cell Viability
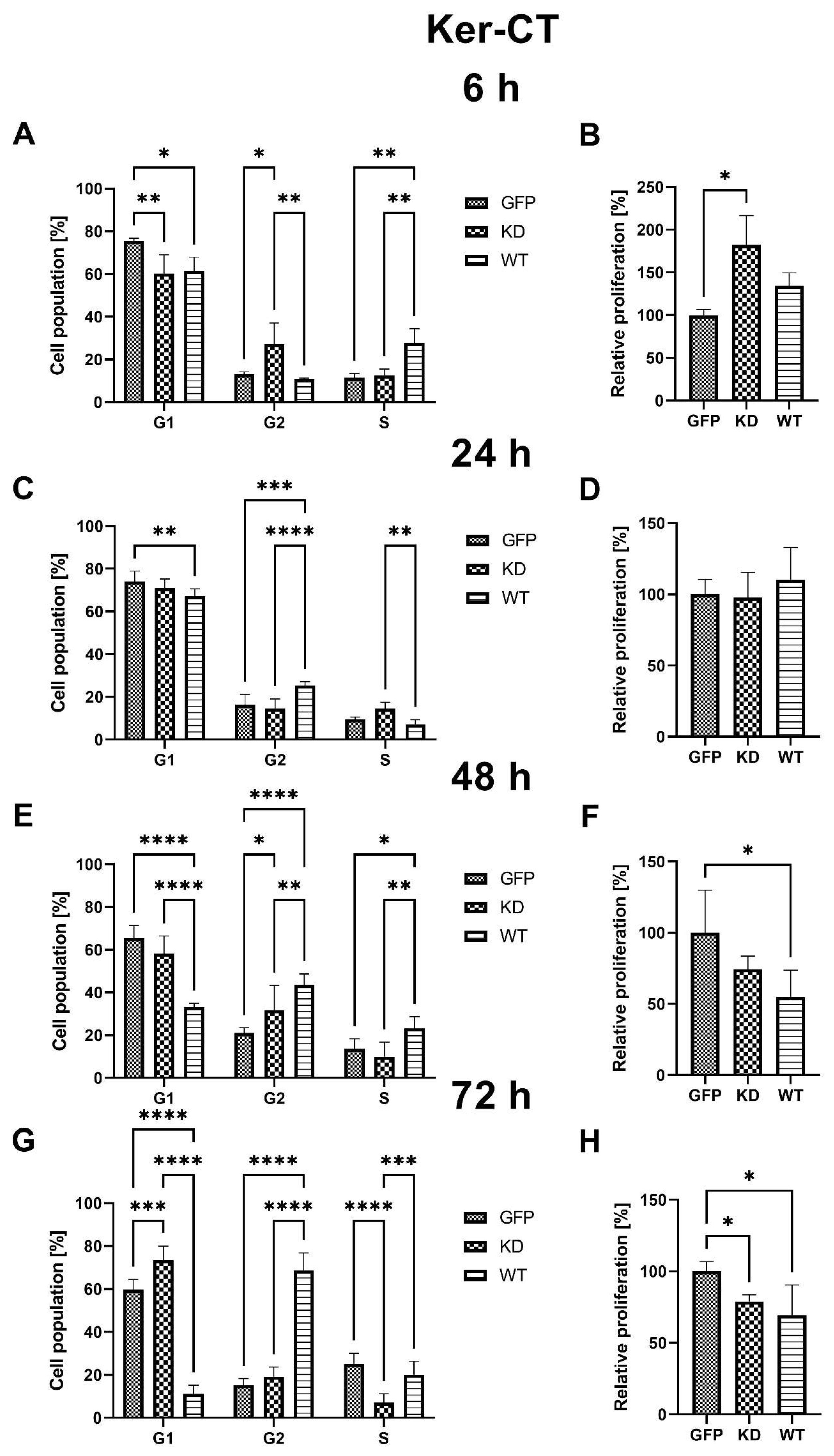
2.2.2. Effects of MELK Overexpression on Ker-CT Cell Cycle Phase Distribution and Proliferation Rate
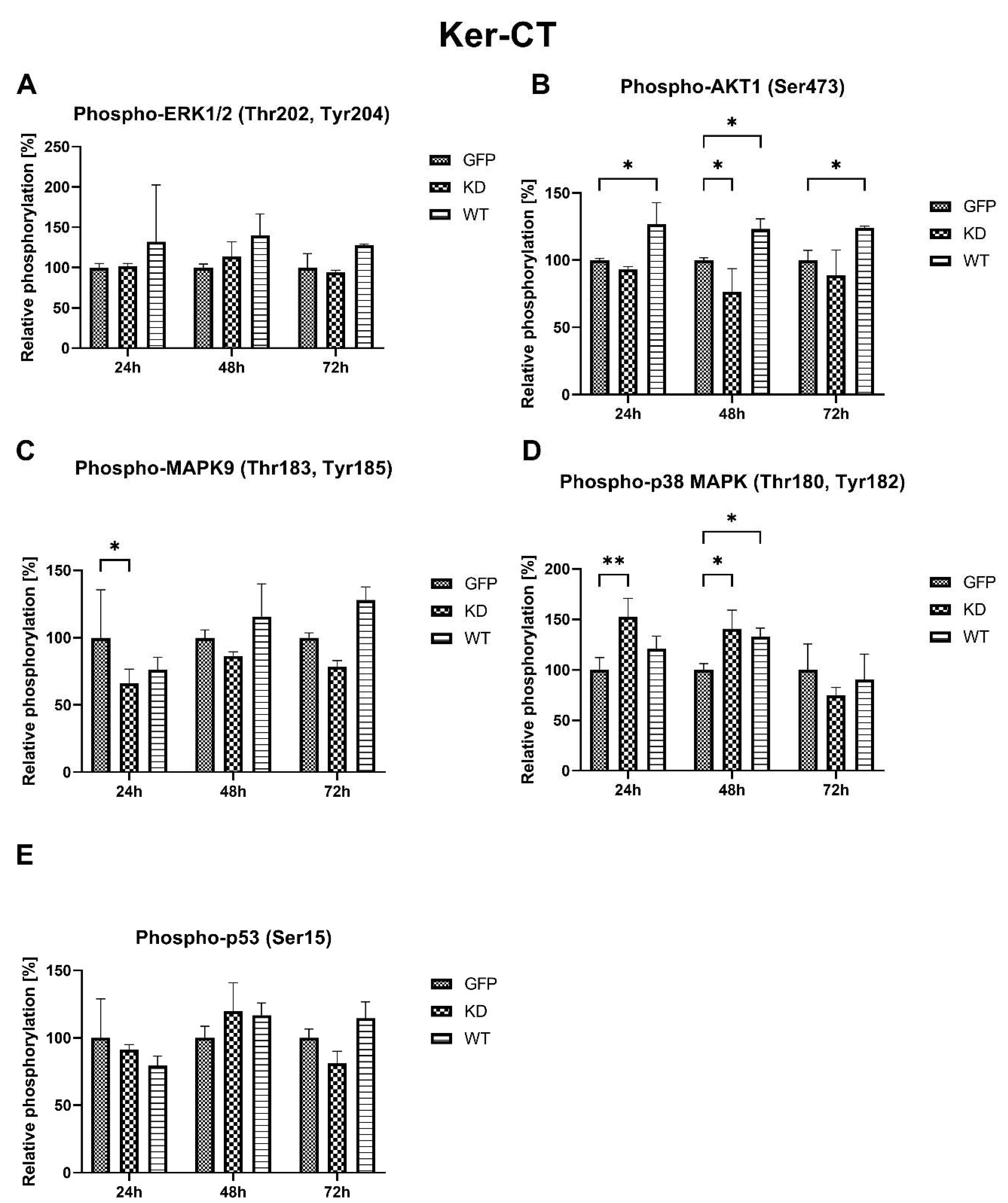
2.2.3. Effects of MELK Overexpression on Selected Signaling Pathways Activation
2.2.4. Intracellular Localization of MELK WT and MELK KD in Ker-CT Cells
3. Discussion
Limitations
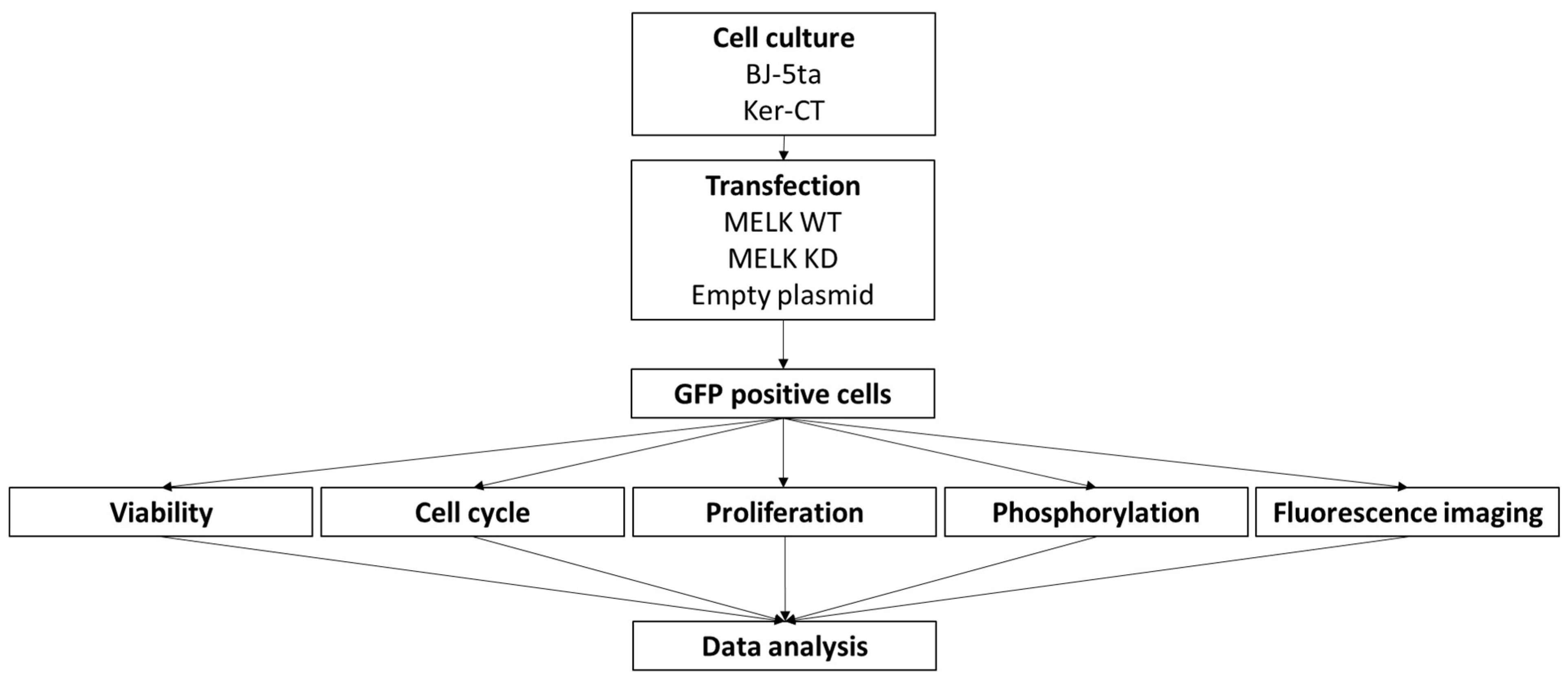
4. Materials and Methods
4.1. Cell Culture
4.2. Expression Constructs
4.3. Transfection
4.4. Cell Viability
4.5. Proliferation (Ki67 Assay) and Cell Cycle
4.6. Cell Signaling—Phosphorylation
4.7. Cell Imaging
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, G.; Yang, M.; Zuo, L.; Wang, M.-X. MELK as a Potential Target to Control Cell Proliferation in Triple-Negative Breast Cancer MDA-MB-231 Cells. Oncol. Lett. 2018, 15, 9934–9940. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, R.; Mohyeldin, A.; Thiel, J.; Kornblum, H.I.; Beullens, M.; Nakano, I. MELK—A Conserved Kinase: Functions, Signaling, Cancer, and Controversy. Clin. Transl. Med. 2015, 4, 11. [Google Scholar] [CrossRef] [PubMed]
- Beke, L.; Kig, C.; Linders, J.T.M.; Boens, S.; Boeckx, A.; van Heerde, E.; Parade, M.; De Bondt, A.; Van den Wyngaert, I.; Bashir, T.; et al. MELK-T1, a Small-Molecule Inhibitor of Protein Kinase MELK, Decreases DNA-Damage Tolerance in Proliferating Cancer Cells. Biosci. Rep. 2015, 35, e00267. [Google Scholar] [CrossRef] [PubMed]
- Badouel, C.; Chartrain, I.; Blot, J.; Tassan, J.-P. Maternal Embryonic Leucine Zipper Kinase Is Stabilized in Mitosis by Phosphorylation and Is Partially Degraded upon Mitotic Exit. Exp. Cell. Res. 2010, 316, 2166–2173. [Google Scholar] [CrossRef]
- Blot, J.; Chartrain, I.; Roghi, C.; Philippe, M.; Tassan, J.-P. Cell Cycle Regulation of PEg3, a New Xenopus Protein Kinase of the KIN1/PAR-1/MARK Family. Dev. Biol. 2002, 241, 327–338. [Google Scholar] [CrossRef]
- Ye, J.; Deng, W.; Zhong, Y.; Liu, H.; Guo, B.; Qin, Z.; Li, P.; Zhong, X.; Wang, L. MELK Predicts Poor Prognosis and Promotes Metastasis in Esophageal Squamous Cell Carcinoma via Activating the NF-κB Pathway. Int. J. Oncol. 2022, 61, 94. [Google Scholar] [CrossRef]
- MacCarthy-Morrogh, L.; Martin, P. The Hallmarks of Cancer Are Also the Hallmarks of Wound Healing. Sci. Signal. 2020, 13, eaay8690. [Google Scholar] [CrossRef]
- Amiri, N.; Golin, A.P.; Jalili, R.B.; Ghahary, A. Roles of Cutaneous Cell-Cell Communication in Wound Healing Outcome: An Emphasis on Keratinocyte-Fibroblast Crosstalk. Exp. Dermatol. 2022, 31, 475–484. [Google Scholar] [CrossRef]
- Rodrigues, M.; Kosaric, N.; Bonham, C.A.; Gurtner, G.C. Wound Healing: A Cellular Perspective. Physiol. Rev. 2019, 99, 665–706. [Google Scholar] [CrossRef]
- Landén, N.X.; Li, D.; Ståhle, M. Transition from Inflammation to Proliferation: A Critical Step during Wound Healing. Cell. Mol. Life Sci. 2016, 73, 3861–3885. [Google Scholar] [CrossRef]
- Le Page, Y.; Chartrain, I.; Badouel, C.; Tassan, J.-P. A Functional Analysis of MELK in Cell Division Reveals a Transition in the Mode of Cytokinesis during Xenopus Development. J. Cell Sci. 2011, 124, 958–968. [Google Scholar] [CrossRef]
- Marie, S.K.N.; Okamoto, O.K.; Uno, M.; Hasegawa, A.P.G.; Oba-Shinjo, S.M.; Cohen, T.; Camargo, A.A.; Kosoy, A.; Carlotti, C.G.; Toledo, S.; et al. Maternal Embryonic Leucine Zipper Kinase Transcript Abundance Correlates with Malignancy Grade in Human Astrocytomas. Int. J. Cancer 2008, 122, 807–815. [Google Scholar] [CrossRef]
- Janostiak, R.; Rauniyar, N.; Lam, T.T.; Ou, J.; Zhu, L.J.; Green, M.R.; Wajapeyee, N. MELK Promotes Melanoma Growth by Stimulating the NF-ΚB Pathway. Cell Rep. 2017, 21, 2829–2841. [Google Scholar] [CrossRef]
- Lin, M.-L.; Park, J.-H.; Nishidate, T.; Nakamura, Y.; Katagiri, T. Involvement of Maternal Embryonic Leucine Zipper Kinase (MELK) in Mammary Carcinogenesis through Interaction with Bcl-G, a pro-Apoptotic Member of the Bcl-2 Family. Breast Cancer Res. 2007, 9, R17. [Google Scholar] [CrossRef]
- Kuner, R.; Fälth, M.; Pressinotti, N.C.; Brase, J.C.; Puig, S.B.; Metzger, J.; Gade, S.; Schäfer, G.; Bartsch, G.; Steiner, E.; et al. The Maternal Embryonic Leucine Zipper Kinase (MELK) Is Upregulated in High-Grade Prostate Cancer. J. Mol. Med. 2013, 91, 237–248. [Google Scholar] [CrossRef]
- Zhang, H.; Wei, P.; Lv, W.; Han, X.; Yang, J.; Qin, S.; Zhang, Y. MELK Is Upregulated in Advanced Clear Cell Renal Cell Carcinoma and Promotes Disease Progression by Phosphorylating PRAS40. Cell Transpl. 2019, 28, 37S–50S. [Google Scholar] [CrossRef]
- Nakano, I.; Masterman-Smith, M.; Saigusa, K.; Paucar, A.A.; Horvath, S.; Shoemaker, L.; Watanabe, M.; Negro, A.; Bajpai, R.; Howes, A.; et al. Maternal Embryonic Leucine Zipper Kinase Is a Key Regulator of the Proliferation of Malignant Brain Tumors, Including Brain Tumor Stem Cells. J. Neurosci. Res. 2008, 86, 48–60. [Google Scholar] [CrossRef]
- Pickard, M.R.; Green, A.R.; Ellis, I.O.; Caldas, C.; Hedge, V.L.; Mourtada-Maarabouni, M.; Williams, G.T. Dysregulated Expression of Fau and MELK Is Associated with Poor Prognosis in Breast Cancer. Breast Cancer Res. 2009, 11, R60. [Google Scholar] [CrossRef]
- Ren, L.; Deng, B.; Saloura, V.; Park, J.-H.; Nakamura, Y. MELK Inhibition Targets Cancer Stem Cells through Downregulation of SOX2 Expression in Head and Neck Cancer Cells. Oncol. Rep. 2019, 41, 2540–2548. [Google Scholar] [CrossRef]
- Lin, A.; Giuliano, C.J.; Sayles, N.M.; Sheltzer, J.M. CRISPR/Cas9 Mutagenesis Invalidates a Putative Cancer Dependency Targeted in on-Going Clinical Trials. eLife 2017, 6, 4179. [Google Scholar] [CrossRef]
- McDonald, I.M.; Graves, L.M. Enigmatic MELK: The Controversy Surrounding Its Complex Role in Cancer. J. Biol. Chem. 2020, 295, 8195–8203. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, B.B.; Li, J.; Roberts, T.M.; Zhao, J.J. A Conditional Dependency on MELK for the Proliferation of Triple-Negative Breast Cancer Cells. iScience 2018, 9, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Davezac, N.; Baldin, V.; Blot, J.; Ducommun, B.; Tassan, J.-P. Human PEg3 Kinase Associates with and Phosphorylates CDC25B Phosphatase: A Potential Role for PEg3 in Cell Cycle Regulation. Oncogene 2002, 21, 7630–7641. [Google Scholar] [CrossRef] [PubMed]
- Gray, D.; Jubb, A.M.; Hogue, D.; Dowd, P.; Kljavin, N.; Yi, S.; Bai, W.; Frantz, G.; Zhang, Z.; Koeppen, H.; et al. Maternal Embryonic Leucine Zipper Kinase/Murine Protein Serine-Threonine Kinase 38 Is a Promising Therapeutic Target for Multiple Cancers. Cancer Res. 2005, 65, 9751–9761. [Google Scholar] [CrossRef] [PubMed]
- Mollereau, B.; Ma, D. The P53 Control of Apoptosis and Proliferation: Lessons from Drosophila. Apoptosis 2014, 19, 1421–1429. [Google Scholar] [CrossRef]
- Seong, H.-A.; Ha, H. Murine Protein Serine-Threonine Kinase 38 Activates P53 Function through Ser15 Phosphorylation. J. Biol. Chem. 2012, 287, 20797–20810. [Google Scholar] [CrossRef]
- Miller, I.; Min, M.; Yang, C.; Tian, C.; Gookin, S.; Carter, D.; Spence, S.L. Ki67 Is a Graded Rather than a Binary Marker of Proliferation versus Quiescence. Cell. Rep. 2018, 24, 1105–1112.e5. [Google Scholar] [CrossRef]
- Tipton, A.R.; Wang, K.; Oladimeji, P.; Sufi, S.; Gu, Z.; Liu, S.-T. Identification of Novel Mitosis Regulators through Data Mining with Human Centromere/Kinetochore Proteins as Group Queries. BMC Cell. Biol. 2012, 13, 15. [Google Scholar] [CrossRef]
- Katayama, K.; Fujita, N.; Tsuruo, T. Akt/Protein Kinase B-Dependent Phosphorylation and Inactivation of WEE1Hu Promote Cell Cycle Progression at G2/M Transition. Mol. Cell. Biol. 2005, 25, 5725–5737. [Google Scholar] [CrossRef]
- Anai, M.; Shojima, N.; Katagiri, H.; Ogihara, T.; Sakoda, H.; Onishi, Y.; Ono, H.; Fujishiro, M.; Fukushima, Y.; Horike, N.; et al. A Novel Protein Kinase B (PKB)/AKT-Binding Protein Enhances PKB Kinase Activity and Regulates DNA Synthesis. J. Biol. Chem. 2005, 280, 18525–18535. [Google Scholar] [CrossRef]
- Engelbrecht, A.-M.; Niesler, C.; Page, C.; Lochner, A. P38 and JNK Have Distinct Regulatoryfunctions on the Development of Apoptosisduring Simulated Ischaemia and Reperfusionin Neonatal Cardiomyocytes. Basic. Res. Cardiol. 2004, 99, 338–350. [Google Scholar] [CrossRef]
- Azijli, K.; Yuvaraj, S.; van Roosmalen, I.; Flach, K.; Giovannetti, E.; Peters, G.J.; de Jong, S.; Kruyt, F.A.E. MAPK P38 and JNK Have Opposing Activities on TRAIL-Induced Apoptosis Activation in NSCLC H460 Cells That Involves RIP1 and Caspase-8 and Is Mediated by Mcl-1. Apoptosis 2013, 18, 851–860. [Google Scholar] [CrossRef]
- Nagata, Y.; Todokoro, K. Requirement of Activation of JNK and P38 for Environmental Stress-Induced Erythroid Differentiation and Apoptosis and of Inhibition of ERK for Apoptosis. Blood 1999, 94, 853–863. [Google Scholar] [CrossRef]
- Jung, H.; Seong, H.-A.; Ha, H. Murine Protein Serine/Threonine Kinase 38 Activates Apoptosis Signal-Regulating Kinase 1 via Thr838 Phosphorylation. J. Biol. Chem. 2008, 283, 34541–34553. [Google Scholar] [CrossRef]
- Morgan, M.J.; Kim, Y.-S.; Liu, Z. TNFα and Reactive Oxygen Species in Necrotic Cell Death. Cell Res. 2008, 18, 343–349. [Google Scholar] [CrossRef]
- Ventura, J.-J.; Cogswell, P.; Flavell, R.A.; Baldwin, A.S.; Davis, R.J. JNK Potentiates TNF-Stimulated Necrosis by Increasing the Production of Cytotoxic Reactive Oxygen Species. Genes Dev. 2004, 18, 2905–2915. [Google Scholar] [CrossRef]
- Lu, Q.; Rau, T.F.; Harris, V.; Johnson, M.; Poulsen, D.J.; Black, S.M. Increased P38 Mitogen-Activated Protein Kinase Signaling Is Involved in the Oxidative Stress Associated with Oxygen and Glucose Deprivation in Neonatal Hippocampal Slice Cultures. Eur. J. Neurosci. 2011, 34, 1093–1101. [Google Scholar] [CrossRef]
- Sakon, S.; Xue, X.; Takekawa, M.; Sasazuki, T.; Okazaki, T.; Kojima, Y.; Piao, J.-H.; Yagita, H.; Okumura, K.; Doi, T.; et al. NF-ΚB Inhibits TNF-Induced Accumulation of ROS That Mediate Prolonged MAPK Activation and Necrotic Cell Death. EMBO J. 2003, 22, 3898–3909. [Google Scholar] [CrossRef]
- Nikoloudaki, G.; Brooks, S.; Peidl, A.P.; Tinney, D.; Hamilton, D.W. JNK Signaling as a Key Modulator of Soft Connective Tissue Physiology, Pathology, and Healing. Int. J. Mol. Sci. 2020, 21, 1015. [Google Scholar] [CrossRef]
- Sharma, G.-D.; He, J.; Bazan, H.E.P. P38 and ERK1/2 Coordinate Cellular Migration and Proliferation in Epithelial Wound Healing: Evidence of Cross-Talk Activation between MAP Kinase Cascades. J. Biol. Chem. 2003, 278, 21989–21997. [Google Scholar] [CrossRef]
- Scepanovic, G.; Hunter, M.V.; Kafri, R.; Fernandez-Gonzalez, R. P38-Mediated Cell Growth and Survival Drive Rapid Embryonic Wound Repair. Cell Rep. 2021, 37, 109874. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Nadelman, C.; Henry, G.; Fan, J.; Muellenhoff, M.; Medina, E.; Gratch, N.S.; Chen, M.; Han, J.; Woodley, D. The P38-MAPK/SAPK Pathway Is Required for Human Keratinocyte Migration on Dermal Collagen. J. Investig. Dermatol. 2001, 117, 1601–1611. [Google Scholar] [CrossRef] [PubMed]
- Dekoninck, S.; Blanpain, C. Stem Cell Dynamics, Migration and Plasticity during Wound Healing. Nat. Cell Biol. 2019, 21, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, S.Y.; Adler, V.; Pincus, M.R.; Ronai, Z. MEKK1/JNK Signaling Stabilizes and Activates P53. Proc. Natl. Acad. Sci. USA 1998, 95, 10541–10546. [Google Scholar] [CrossRef]
- Lee, K.M.; Choi, K.H.; Ouellette, M.M. Use of Exogenous HTERT to Immortalize Primary Human Cells. Cytotechnology 2004, 45, 33–38. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Y.-M.; Baitsch, L.; Huang, A.; Xiang, Y.; Tong, H.; Lako, A.; Von, T.; Choi, C.; Lim, E.; et al. Correction: MELK Is an Oncogenic Kinase Essential for Mitotic Progression in Basal-like Breast Cancer Cells. eLife 2018, 7, e36414. [Google Scholar] [CrossRef]
- Szymański, Ł.; Ciepielak, M.; Cios, A.; Palusińska, M.; Stankiewicz, W.; Lewicki, S. Effects of 445 Nm, 520 Nm, and 638 Nm Laser Irradiation on the Dermal Cells. Int. J. Mol. Sci. 2021, 22, 11605. [Google Scholar] [CrossRef]
- Froger, A.; Hall, J.E. Transformation of Plasmid DNA into E. Coli Using the Heat Shock Method. J. Vis. Exp. 2007, 6, 253. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szymański, Ł.; Lieto, K.; Zdanowski, R.; Lewicki, S.; Tassan, J.-P.; Kubiak, J.Z. Differential Effects of Overexpression of Wild Type and Kinase-Dead MELK in Fibroblasts and Keratinocytes, Potential Implications for Skin Wound Healing and Cancer. Int. J. Mol. Sci. 2023, 24, 8089. https://doi.org/10.3390/ijms24098089
Szymański Ł, Lieto K, Zdanowski R, Lewicki S, Tassan J-P, Kubiak JZ. Differential Effects of Overexpression of Wild Type and Kinase-Dead MELK in Fibroblasts and Keratinocytes, Potential Implications for Skin Wound Healing and Cancer. International Journal of Molecular Sciences. 2023; 24(9):8089. https://doi.org/10.3390/ijms24098089
Chicago/Turabian StyleSzymański, Łukasz, Krystyna Lieto, Robert Zdanowski, Sławomir Lewicki, Jean-Pierre Tassan, and Jacek Z. Kubiak. 2023. "Differential Effects of Overexpression of Wild Type and Kinase-Dead MELK in Fibroblasts and Keratinocytes, Potential Implications for Skin Wound Healing and Cancer" International Journal of Molecular Sciences 24, no. 9: 8089. https://doi.org/10.3390/ijms24098089