

Challenges to Cannabis sativa Production from Pathogens and Microbes—The Role of Molecular Diagnostics and Bioinformatics

Abstract
:1. Introduction
2. Results
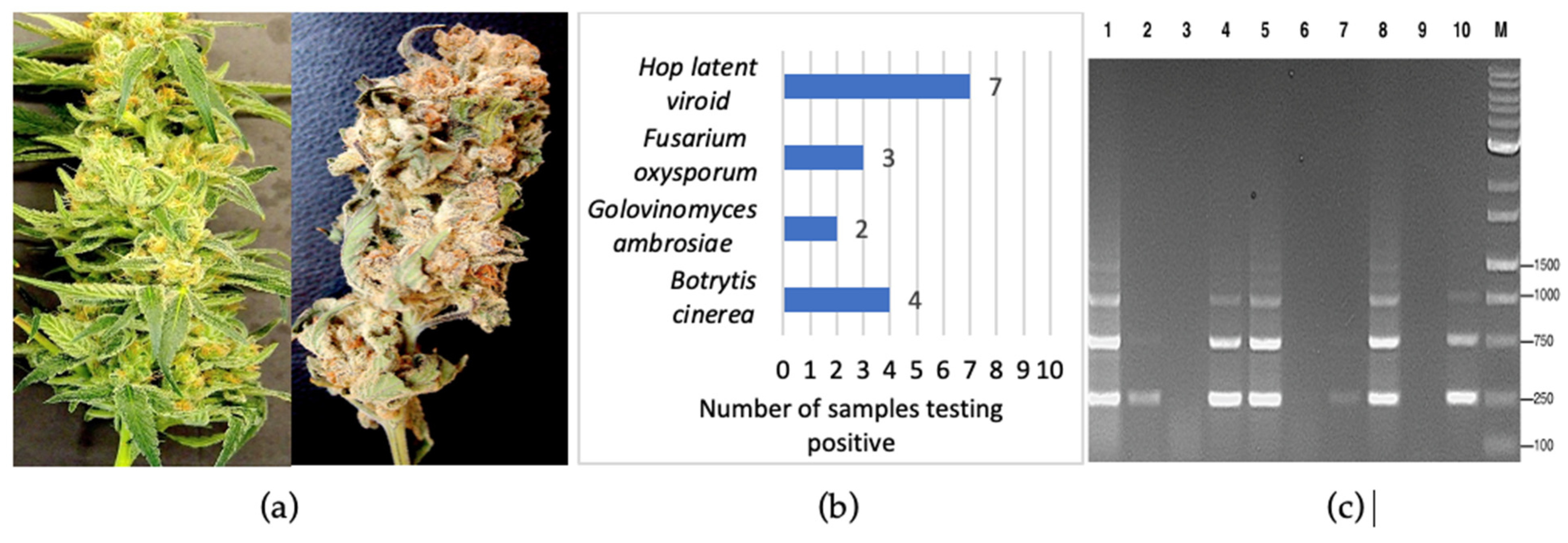
2.1. Detection of Fungal and Oomycete Pathogens on Cannabis
2.1.1. Symptoms and Pathogen Isolation
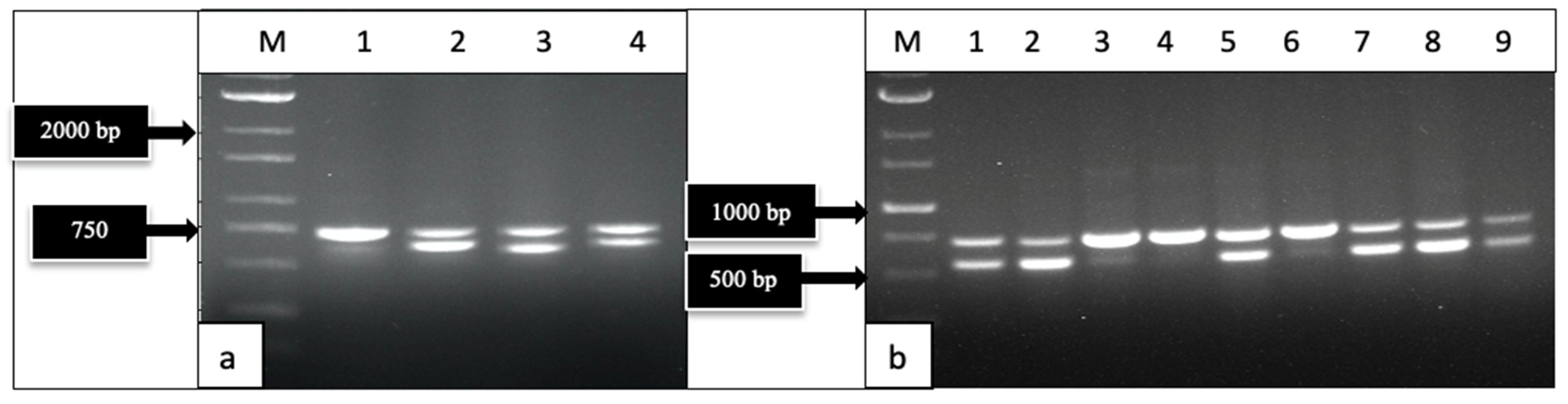
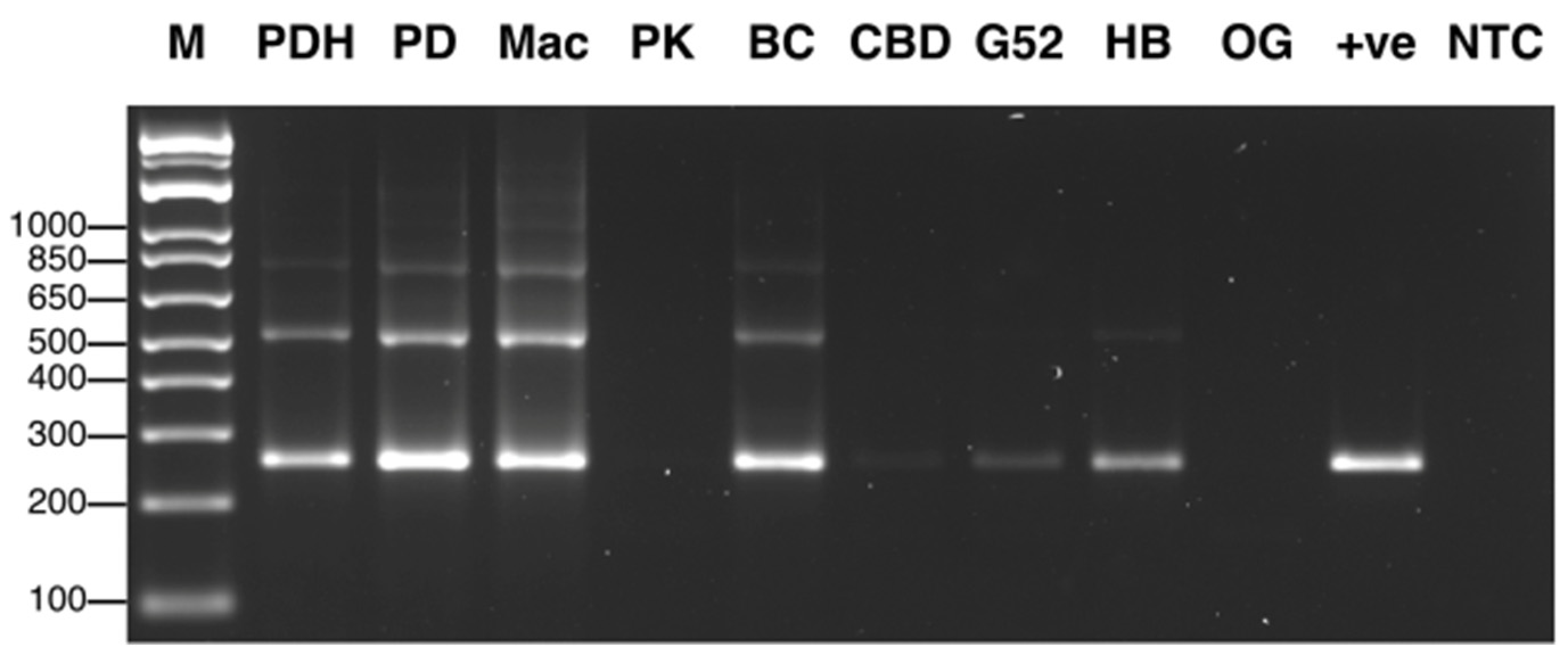
2.1.2. Molecular Detection by PCR
2.2. Detection of Viral Pathogens on Cannabis
2.2.1. Symptoms
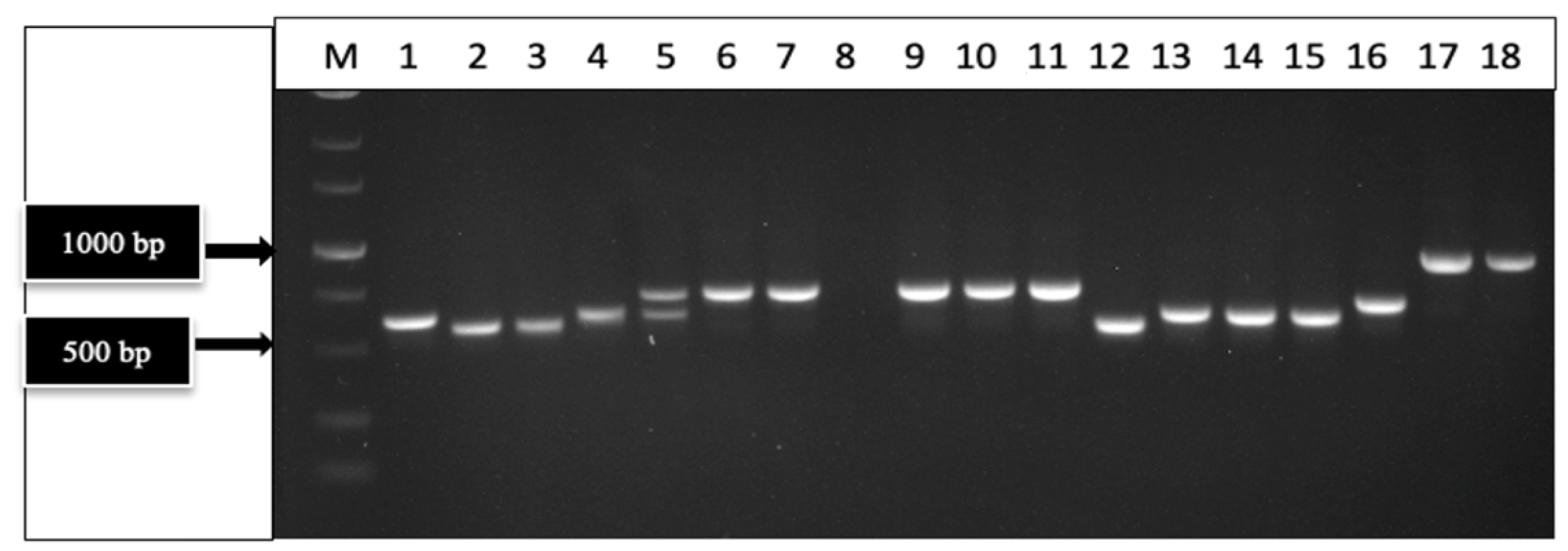
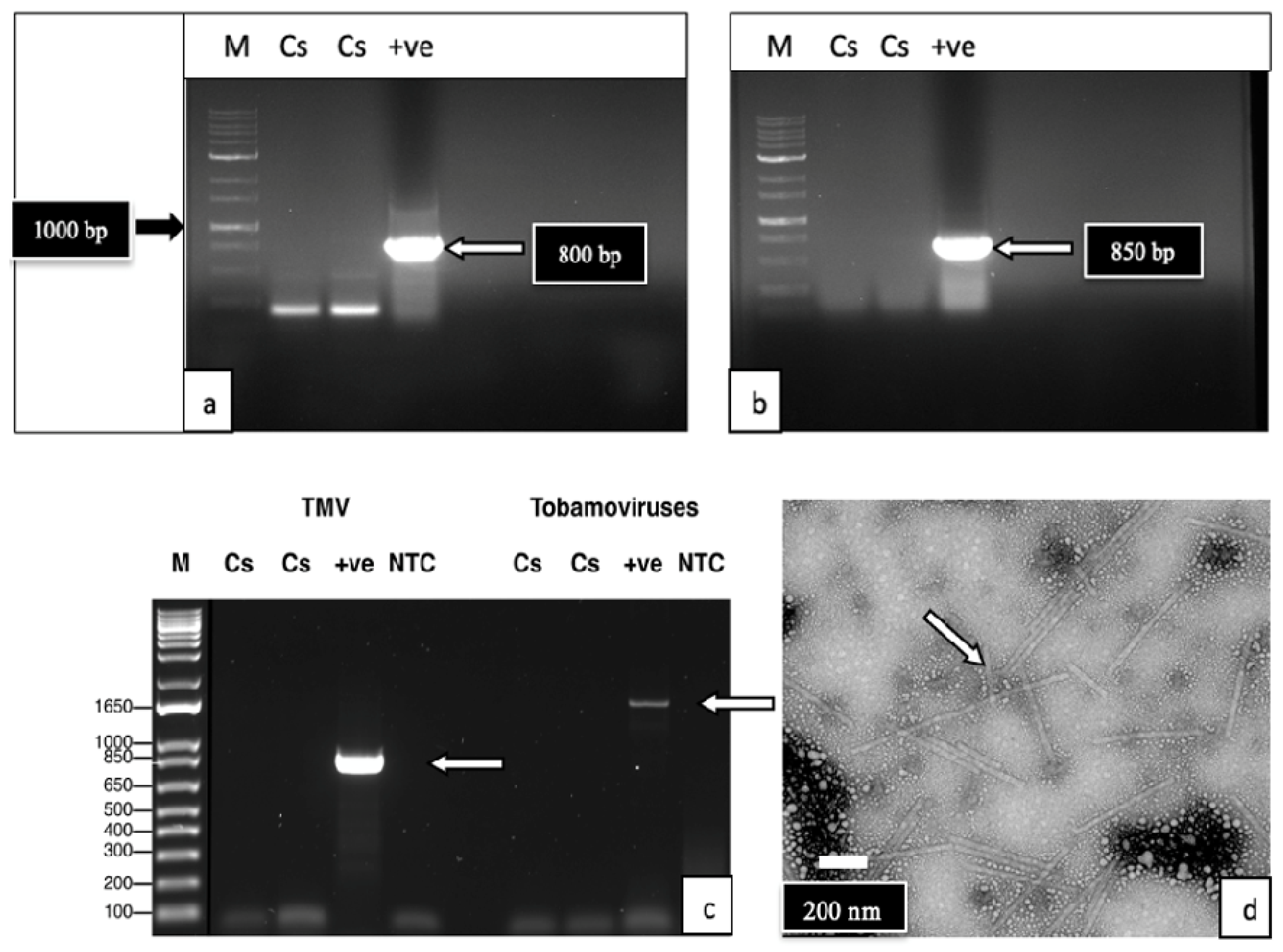
2.2.2. PCR with Broad-Spectrum and Specific Primer Sets
2.2.3. Transmission Electron Microscopy (TEM)
2.2.4. Host Range Studies
2.3. High-Throughput Sequencing (HTS)
2.4. Molecular Assays for Viroid and Other Viral Pathogens
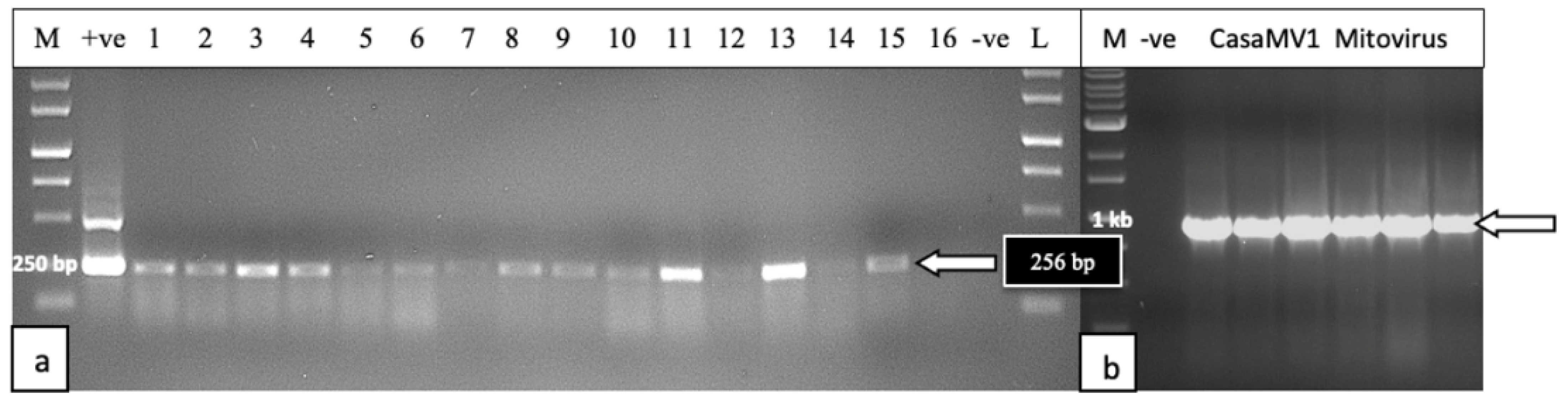
2.4.1. RNA Extractions, cDNA Preparation, and RT-PCR
Hop Latent Viroid
Mitovirus
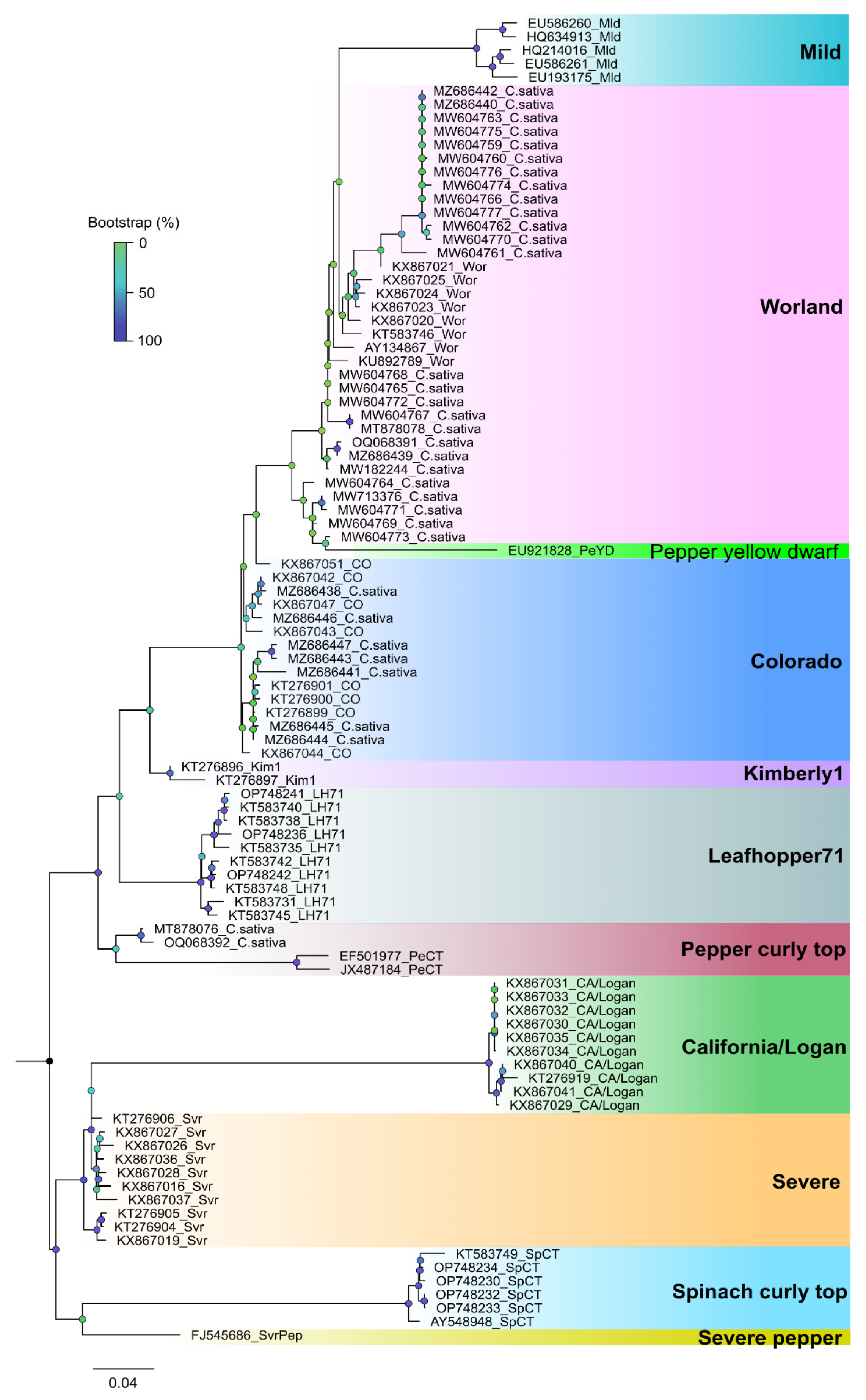
2.4.2. Sequence Diversity and Molecular Phylogeny of Hop Latent Viroid
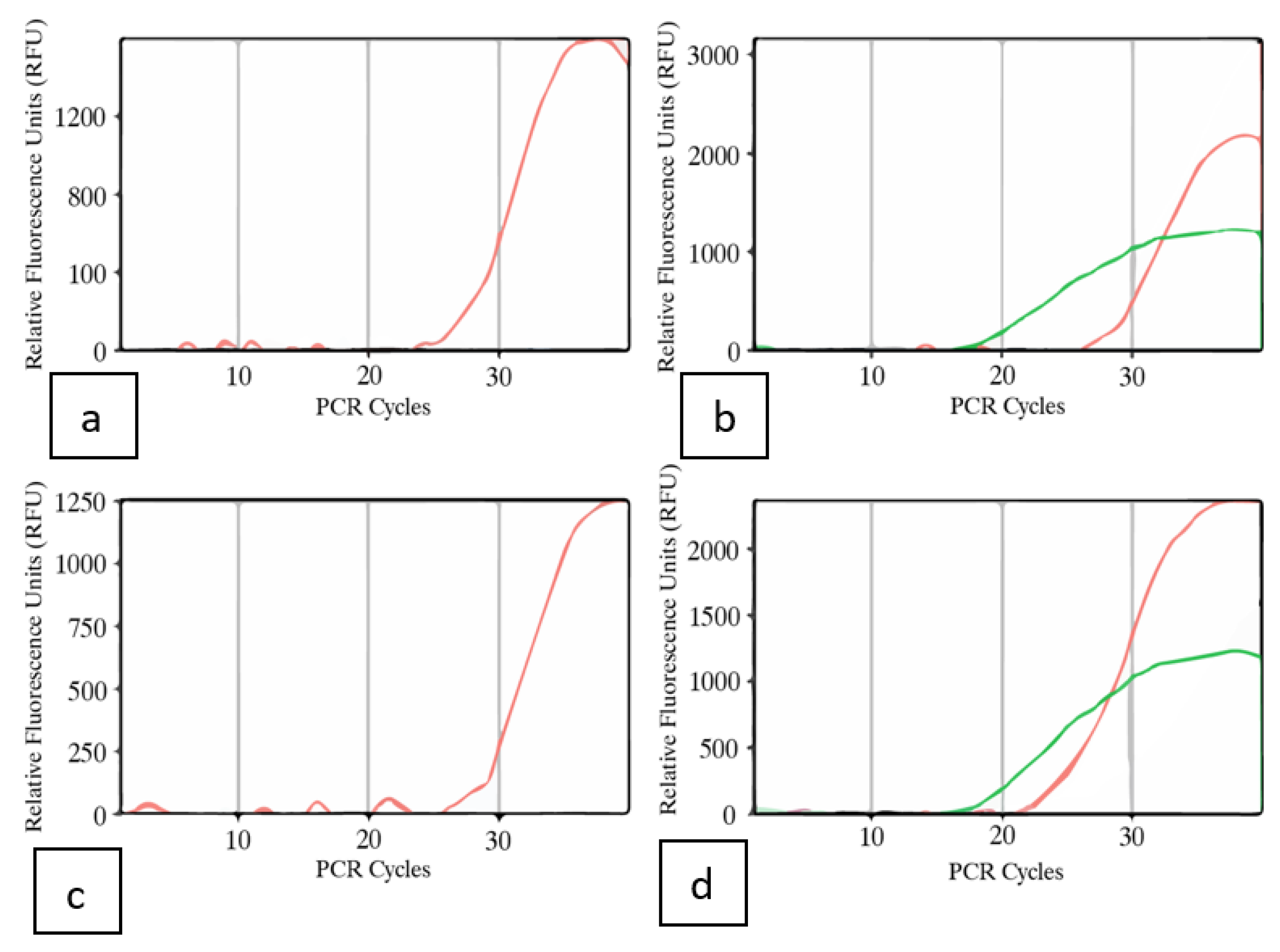
2.4.3. Droplet Digital PCR
2.4.4. Multiplex Taqman RT-qPCR
2.4.5. LAMP Assays
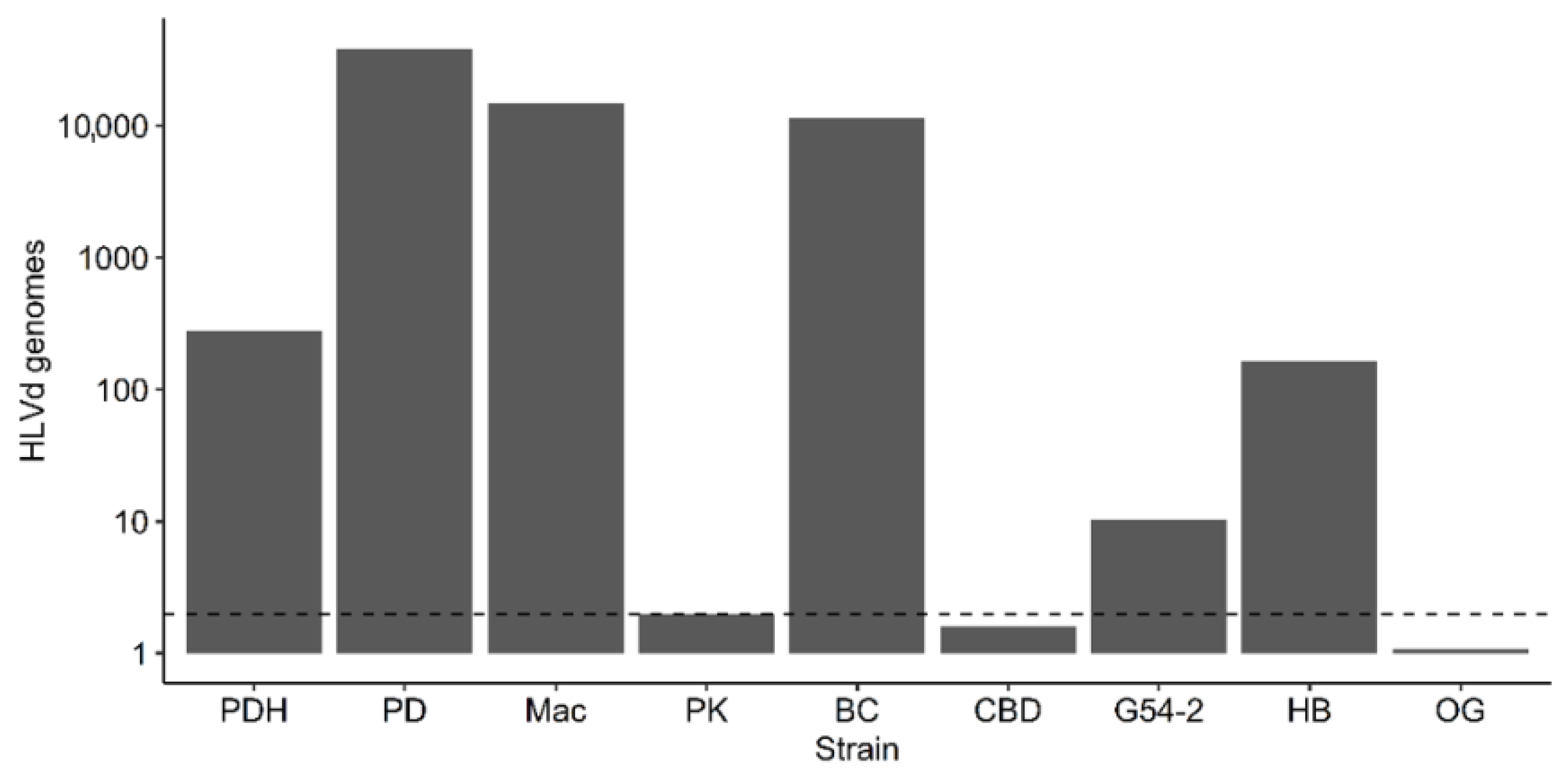
2.5. Monitoring Distribution of HLVd in Various Tissues of Cannabis Plants
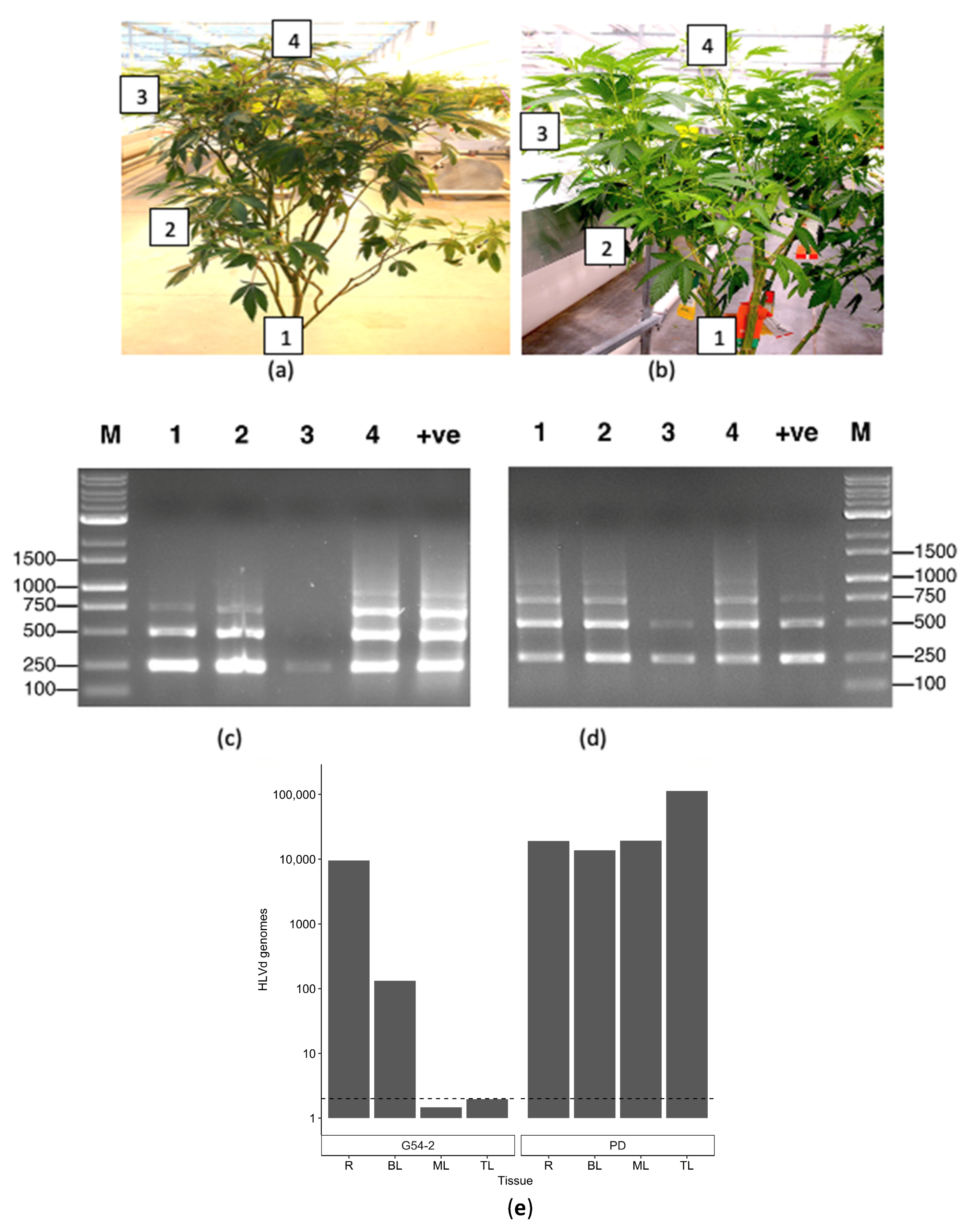
2.5.1. Distribution within Stock Plants
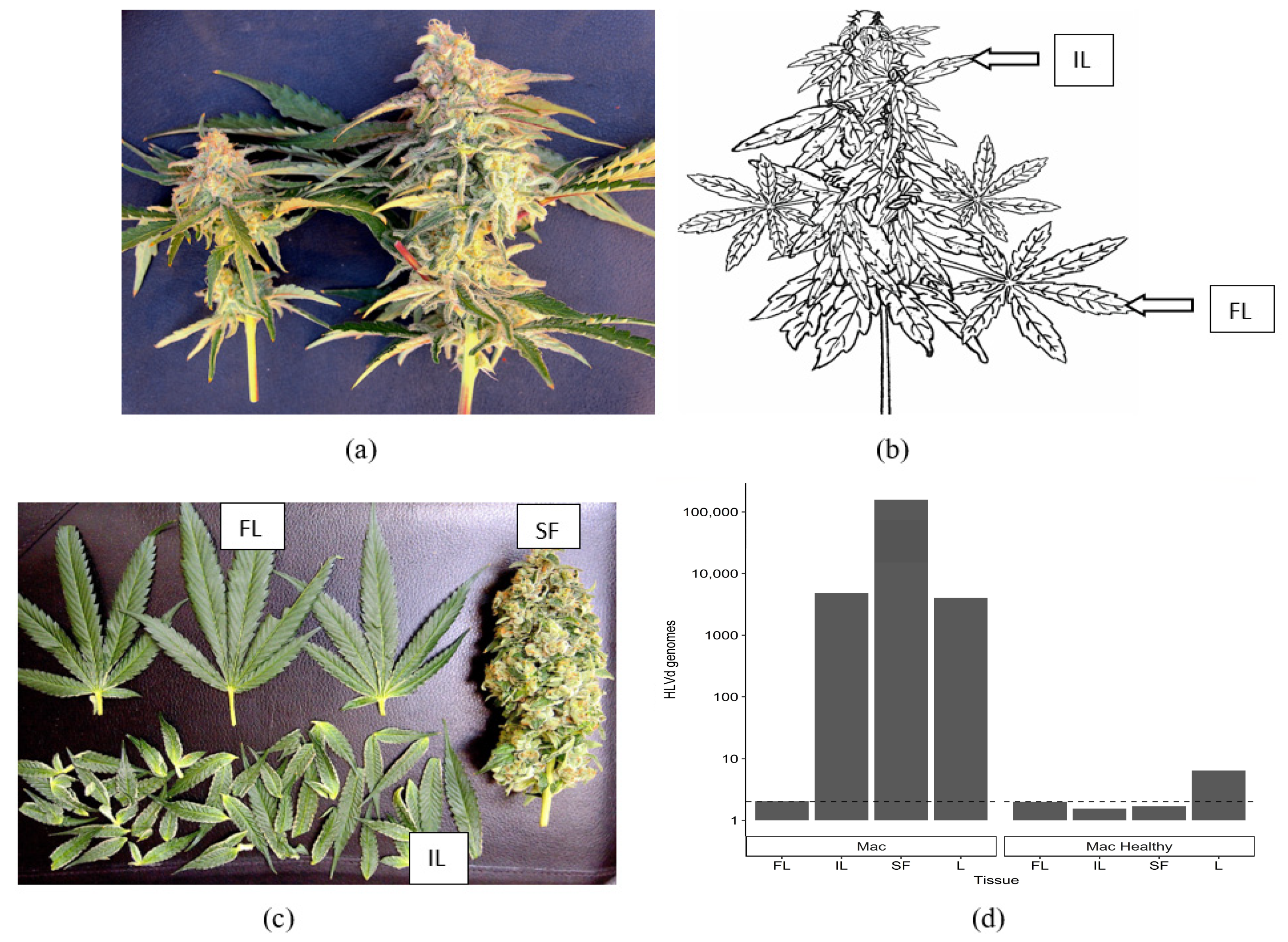
2.5.2. Distribution of HLVd within Inflorescence Tissues
2.5.3. Distribution of HLVd within Cuttings from Infected Stock Plants
2.5.4. Detection of Pathogens in Fresh and Dried Cannabis Inflorescences
2.5.5. Detection of HLVd and CasaMV1 in Cannabis Seed
2.6. Detection of Beet Curly Top Virus in Cannabis Plants
2.7. Diagnostic Approaches to Detect Viral and Viroid Pathogens Affecting Low-THC-Containing Cannabis sativa L. (hemp)
2.7.1. Next-Generation Sequencing (NGS)
2.7.2. RT-PCR with Specific Primer Sets
2.7.3. Sequence Diversity and Molecular Phylogeny of Beet Curly Top Virus
2.7.4. Detection of HLVd in Hemp Seeds and on Thrips
3. Discussion
4. Materials and Methods
4.1. Detection of Fungal and Oomycete Pathogens on Cannabis
4.1.1. Symptoms and Pathogen Isolation
4.1.2. PCR Analysis
4.2. Detection of Viral Pathogens on Cannabis
4.2.1. Symptoms
4.2.2. PCR with Broad-Spectrum and Specific Primer Sets
4.2.3. Transmission Electron Microscopy
4.2.4. Host Range Studies
4.2.5. High-throughput Sequencing (HTS)
4.3. Molecular Assays for Viroid and Other Viral Pathogens
4.3.1. RNA Extractions and cDNA Preparation
4.3.2. Primer Design for RT-PCR

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Primer Name | Sequence (5′–3′) | Source |
|---|---|---|---|
| HLVd | HLVd seq Forward1 | ATACAACTCTTGAGCGCCGA | Eastwell and Nelson [69] |
| HLVd seq Reverse1 | CCACCGGGTAGTTCCCAACT | Eastwell and Nelson [69] | |
| HLVd seq Reverse2 | AGGACGCGAACAAGAAGAAG | This work | |
| HLVd quant Forward1 | GTTGCTTCGGCTTCTTCTTG | This work | |
| HLVd quant Reverse1 | AGTTGTATCCACCGGGTAGT | This work | |
| Cannabis EF1α | Cannabis EF1α Forward | TGTTTTGCACGGATCAGTTTG | Guo [70] |
| Cannabis EF1α Reverse | AATGCCGACCGCTACAGTTC | Guo [70] |
4.4. Sequence Diversity and Molecular Phylogeny of Hop Latent Viroid
4.5. Droplet Digital PCR
4.6. Multiplex Taqman RT-qPCR
4.7. LAMP Assays
4.8. Monitoring Distribution of HLVd in Various Plant Tissues
4.8.1. Distribution within Stock Plants
4.8.2. Distribution within Inflorescence Tissues
4.8.3. Distribution of HLVd within Cuttings from Infected Stock Plants
4.8.4. Presence of Pathogens in Fresh and Dried Cannabis Inflorescences
4.8.5. Detection of HLVd and Mitovirus in Cannabis Seed
4.9. Detection of Beet Curly Top Virus
4.10. Diagnostic Approaches to Detect Viral and Viroid Pathogens Affecting Low THC-Containing Cannabis sativa L. (hemp)
4.10.1. Next-Generation Sequencing (NGS)
4.10.2. RT-PCR with Specific Primer Sets
4.10.3. Sequence Diversity and Molecular Phylogeny of Beet Curly Top Virus
4.10.4. Detection of HLVd on Hemp Seeds and on Thrips
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Punja, Z.K. Emerging diseases of Cannabis sativa and sustainable management. Pest. Manag. Sci. 2021, 77, 3857–3870. [Google Scholar] [CrossRef]
- Punja, Z.K.; Collyer, D.; Scott, C.; Lung, S.; Holmes, J.; Sutton, D. Pathogens and molds affecting production and quality of Cannabis sativa L. Front. Plant Sci. 2019, 10, 1120. [Google Scholar] [CrossRef]
- Chiginsky, J.; Langemeier, K.; MacWilliams, J.; Albrecht, T.; Cranshaw, W.; Fulladolsa, A.C.; Kapuscinski, M.; Stenglein, M.; Nachappa, P. First insights into the virus and viroid communities in hemp (Cannabis sativa). Front. Agron. 2021, 3, 778433. [Google Scholar] [CrossRef]
- Punja, Z.K. Epidemiology of Fusarium oxysporum causing root and crown rot of cannabis (Cannabis sativa L., marijuana) plants in commercial greenhouse production. Can. J. Plant Pathol. 2021, 43, 216–235. [Google Scholar] [CrossRef]
- Punja, Z.K. First report of Fusarium proliferatum causing crown and stem rot, and pith necrosis, in cannabis (Cannabis sativa L., marijuana) plants. Can. J. Plant Pathol. 2021, 43, 236–255. [Google Scholar] [CrossRef]
- Punja, Z.K.; Li, N.; Roberts, A.J. The Fusarium solani species complex infecting cannabis (Cannabis sativa L., marijuana) plants and a first report of Fusarium (Cylindrocarpon) lichenicola causing root and crown rot. Can. J. Plant Pathol. 2021, 43, 567–581. [Google Scholar] [CrossRef]
- Punja, Z.K.; Scott, C.; Lung, S. Several Pythium species cause crown and root rot on cannabis (Cannabis sativa L., marijuana) plants grown under commercial greenhouse conditions. Can. J. Plant Pathol. 2022, 44, 66–81. [Google Scholar] [CrossRef]
- Scott, C.; Punja, Z.K. Evaluation of disease management approaches for powdery mildew on Cannabis sativa L. (marijuana) plants. Can. J. Plant Pathol. 2021, 43, 394–412. [Google Scholar] [CrossRef]
- Punja, Z.K. First report of the powdery mildew pathogen of hops, Podosphaeria macularis, naturally infecting cannabis (Cannabis sativa L., marijuana) plants under field conditions. Can. J. Plant Pathol. 2021, 44, 235–249. [Google Scholar] [CrossRef]
- Punja, Z.K.; Ni, L. The bud rot pathogens infecting cannabis (Cannabis sativa L., marijuana) inflorescences: Symptomology, species identification, pathogenicity and biological control. Can. J. Plant Pathol. 2022, 43, 827–854. [Google Scholar] [CrossRef]
- Mahmoud, M.; BenRejeb, I.; Punja, Z.K.; Buirs, L.; Jabaji, S. Understanding bud rot development, caused by Botrytis cinerea, on cannabis (Cannabis sativa L.) plants grown under greenhouse conditions. Botany 2023, 101, 200–231. [Google Scholar] [CrossRef]
- Punja, Z.K.; Scott, C. Organically grown cannabis (Cannabis sativa L.) plants contain a diverse range of culturable epiphytic and endophytic fungi in inflorescences and stem tissues. Botany 2023, 101, 255–269. [Google Scholar] [CrossRef]
- Punja, Z.K. The diverse mycoflora present on dried cannabis (Cannabis sativa L., marijuana) inflorescences in commercial production. Can. J. Plant Pathol. 2021, 43, 88–100. [Google Scholar] [CrossRef]
- Punja, Z.K.; Ni, L.; Lung, S.; Buirs, L. Total yeast and mold levels in high-THC-containing cannabis (Cannabis sativa L.) inflorescences are influenced by genotype, environment, and pre- and post-harvest handling practices. Front. Microbiol. 2023, 14, 1192035. [Google Scholar] [CrossRef] [PubMed]
- Gwinn, K.D.; Leung, M.C.K.; Stephens, A.B.; Punja, Z.K. Fungal and mycotoxin contaminants in cannabis and hemp flowers: Implications for consumer health and directions for further research. Front. Microbiol. 2023, 14, 1278189. [Google Scholar] [CrossRef] [PubMed]
- McKernan, K.; Spangler, J.; Helbert, Y.; Lynch, R.C.; Devitt-Lee, A.; Zhang, L.; Orphe, W.; Warner, J.; Foss, T.; Hudalla, C.J.; et al. Metagenomic analysis of medicinal cannabis samples; pathogenic bacteria, toxigenic fungi, and beneficial microbes grow in culture-based yeast and mold tests. F1000Research 2016, 5, 2471. [Google Scholar] [CrossRef]
- McKernan, K.; Spangler, J.; Zhang, L.; Tadigotla, V.; Helbert, Y.; Foss, T.; Smith, D. Cannabis microbiome sequencing reveals several mycotoxic fungi native to dispensary grade Cannabis flowers. F1000Research 2016, 4, 1422. [Google Scholar] [CrossRef] [PubMed]
- Thompson, G.R., III; Tuscano, J.M.; Dennis, M.; Singapuri, A.; Libertini, S.; Gaudino, R.; Torres, A.; Delisle, J.P.M.; Gillece, J.D.; Schupp, J.M.; et al. A microbiome assessment of medical marijuana. Clin. Microbiol. Infect. 2017, 23, 269–270. [Google Scholar] [CrossRef]
- McKernan, K.; Helbert, Y.; Kane, L.; Houde, N.; Zhang, L.; McLaughlin, S. Whole genome sequencing of colonies derived from cannabis flowers and the impact of media selection on benchmarking total yeast and mold detection tools. F1000Research 2021, 10, 624. [Google Scholar] [CrossRef]
- Nibert, M.L.; Vong, M.; Fugate, K.K.; Debat, H.J. Evidence for contemporary plant mitoviruses. Virology 2018, 518, 14–24. [Google Scholar] [CrossRef]
- Punja, Z.K.; Wang, K.; Lung, S.; Buirs, L. Symptomology, prevalence, and impact of Hop latent viroid on greenhouse-grown cannabis (Cannabis sativa L.) plants in Canada. Can. J. Plant Pathol. 2023; in press. [Google Scholar]
- Righetti, L.; Paris, R.; Ratti, C.; Calassanzio, M.; Onofri, C.; Calzolari, D.; Menzel, W.; Knierim, D.; Magagnini, G.; Pacifico, D.; et al. Not the one, but the only one: About cannabis cryptic virus in plants showing ‘Hemp streak’ disease symptoms. Eur. J. Plant Pathol. 2018, 150, 575–588. [Google Scholar] [CrossRef]
- Ziegler, A.; Matoušek, J.; Steger, G.; Schubert, J. Complete sequence of a cryptic virus from hemp (Cannabis sativa). Arch. Virol. 2012, 157, 383–385. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Chen, X.; Punja, Z.K. Molecular and biological characterization of a mitovirus in Chalara elegans (Thielaviopsis basicola). Phytopathology 2006, 96, 468–479. [Google Scholar] [CrossRef] [PubMed]
- Di Silvestre, D.; Passignani, G.; Rossi, R.; Ciuffo, M.; Turina, M.; Vigani, G.; Mauri, P.L. Presence of a mitovirus is associated with alteration of the mitochondrial proteome, as revealed by protein-protein interaction (PPI) and co-expression network models in Chenopodium quinoa plants. Biology 2022, 11, 95. [Google Scholar] [CrossRef] [PubMed]
- Brunstein, J. Cannabis mitovirus—What you need to know. An introduction and state of knowledge. Segra Int. 2022. Available online: https://www.segra-intl.com/mitoviruses (accessed on 15 October 2023).
- Kegler, H.; Spaar, D. On the virus susceptibility of varieties of Cannabis sativa L. Arch. Phytopathol. Plant Prot. 1997, 30, 457–464. [Google Scholar] [CrossRef]
- Miotti, N.; Passera, A.; Ratti, C.; Dall’Ara, M.; Casati, P. A guide to cannabis virology: From the virome investigation to the development of viral biotechnological tools. Viruses 2023, 15, 1532. [Google Scholar] [CrossRef]
- D’Amato, F. Role of somatic mutations in the evolution of higher plants. Caryologia 1997, 50, 1–15. [Google Scholar] [CrossRef]
- McKey, D.; Elias, M.; Pujol, B.; Duputié, A. The evolutionary ecology of clonally propagated domesticated plants: Tansley review. New Phytol. 2010, 186, 318–332. [Google Scholar] [CrossRef]
- Adamek, K.; Jones, A.M.P.; Torkamaneh, D. Accumulation of somatic mutations leads to genetic mosaicism in cannabis. Plant Genome 2022, 15, 184–196. [Google Scholar] [CrossRef]
- Fitz-James, M.; Cavalli, G. Molecular mechanisms of transgenerational epigenetic inheritance. Nat. Rev. Genet. 2022, 23, 325–341. [Google Scholar] [CrossRef] [PubMed]
- Hadad, L.; Luria, N.; Smith, E.; Sela, N.; Lachman, O.; Dombrovsky, A. Lettuce chlorosis virus disease: A new threat to cannabis production. Viruses 2019, 11, 802. [Google Scholar] [CrossRef] [PubMed]
- Giladi, Y.; Hadad, L.; Luria, N.; Cranshaw, W.; Lachman, O.; Dombrovsky, A. First report of Beet curly top virus infecting Cannabis sativa in western Colorado. Plant Dis. 2020, 104, 999. [Google Scholar] [CrossRef]
- Hu, J.; Masson, R.; Dickey, L. First report of Beet curly top virus infecting industrial hemp (Cannabis sativa) in Arizona. Plant Dis. 2021, 105, 1233. [Google Scholar] [CrossRef] [PubMed]
- Melgarejo, T.A.; Chen, L.-F.; Rojas, M.R.; Schilder, A.; Gilbertson, R.L. Curly top disease of hemp (Cannabis sativa) in California is caused by mild-type strains of Beet curly top virus often in mixed infection. Plant Dis. 2022, 106, 3022–3026. [Google Scholar] [CrossRef] [PubMed]
- Jarugula, S.; Wagstaff, C.; Mitra, A.; Crowder, D.W.; Gang, D.R.; Naidu, R.A. First reports of Beet curly top virus, Citrus yellow vein-associated virus, and Hop latent viroid in industrial hemp (Cannabis sativa) in Washington State. Plant Dis. 2023; in press. [Google Scholar]
- Kwon, S.-J.; Bodaghi, S.; Dang, T.; Gadhave, K.R.; Ho, T.; Osman, F.; Al Rwahnih, M.; Tzanetakis, I.E.; Simon, A.E.; Vidalakis, G. Complete nucleotide sequence, genome organization, and comparative genomic analyses of Citrus yellow-vein associated virus (CYVaV). Front. Microbiol. 2021, 12, 1371. [Google Scholar] [CrossRef] [PubMed]
- Schoener, L.; Wang, S. Hemp abnormal growth is attributed to mono-, di-, or tri-infections of Spiroplasma citri, Candidatus Phytoplasma trifolii, and Beet curly top virus. PhytoFrontiers, 2023; in press. [Google Scholar] [CrossRef]
- Rivedal, H.M.; Temple, T.; Thomas, W.J.; Ocamb, C.M.; Funke, C.; Skillman, V.; Jones, G.; Shrestha, G.; Achala, K.C.; Dung, J.K.S.; et al. First report of Spiroplasma citri associated with disease in field-grown hemp (Cannabis sativa L.) in the United States. Plant Dis. 2023; in press. [Google Scholar]
- Strausbaugh, C.A.; Eujayl, I.A.; Wintermantel, W.M. Beet curly top virus strains associated with sugar beet in Idaho, Oregon, and a western U.S. Collection. Plant Dis. 2017, 101, 1373–1382. [Google Scholar] [CrossRef] [PubMed]
- Ng, J.C.; Falk, B.W. Virus-vector interactions mediating nonpersistent and semipersistent transmission of plant viruses. Annu. Rev. Phytopathol. 2006, 44, 183–212. [Google Scholar] [CrossRef]
- McKernan, K.; Palmer, C. Hop latent viroid detection in leafhoppers (Nephotettix cincticeps) feeding on Cannabis sativa sap. Nepetalactone Newsl. 2023. Available online: https://anandamide.substack.com/p/hop-latent-viroid-detection-in-leafhoppers (accessed on 1 October 2023).
- Nachappa, P.; Challacombe, J.; Margolies, D.C.; Nechols, J.R.; Whitfield, A.E.; Rotenberg, D. Tomato spotted wilt virus benefits its thrips vector by modulating metabolic and plant defense pathways in tomato. Front. Plant Sci. 2020, 11, 575564. [Google Scholar] [CrossRef]
- Gezovitch, O.; Luria, N.; Lachman, O.; Sela, N.; Smith, E.; Dombrovsky, A. Aviv Dombrovsky Cucurbit chlorotic yellows virus, a crinivirus infecting Cannabis sativa plants. Plant Pathol. 2023. [Google Scholar] [CrossRef]
- Adkar-Purushothama, C.R.; Sano, T.; Perreault, J.-P. Hop latent viroid: A hidden threat to the cannabis industry. Viruses 2023, 15, 681. [Google Scholar] [CrossRef] [PubMed]
- Notomi, T.; Okayama, H.; Masubuchi, H.; Yonekawa, T.; Watanabe, K.; Amino, N.; Hase, T. Loop-mediated isothermal amplification of DNA. Nucl. Acids Res. 2000, 28, e63. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Gutierrez, S.V.; Goodwin, S.B. Loop-mediated isothermal amplification for detection of plant pathogens in wheat (Triticum aestivum). Front. Plant Sci. 2022, 13, 857673. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Vu, N.T. Progress of loop-mediated isothermal amplification technique in molecular diagnosis of plant diseases. Appl. Biol. Chem. 2017, 60, 169–180. [Google Scholar] [CrossRef]
- Hsieh, K.; Mage, P.L.; Csordas, A.T.; Eisenstein, M.; Soh, H.T. Simultaneous elimination of carryover contamination and detection of DNA with uracil-DNA-glycosylase-supplemented loop-mediated isothermal amplification (UDG-LAMP). Chem. Commun. 2014, 50, 3747–3749. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.-M.; Jeon, H.-S.; Kim, J.-J.; Shin, Y.-K.; Lee, Y.-J.; Yeo, S.-G.; Park, C.-K. Uracil-DNA glycosylase-treated reverse transcription loop-mediated isothermal amplification for rapid detection of avian influenza virus preventing carry-over contamination. J. Vet. Sci. 2016, 17, 421–425. [Google Scholar] [CrossRef]
- Baek, Y.H.; Um, J.; Antigua, K.J.C.; Park, J.H.; Kim, Y.; Oh, S.; Kim, Y.I.; Choi, W.S.; Kim, S.G.; Jeong, J.H.; et al. Development of a reverse transcription-loop-mediated isothermal amplification as a rapid early-detection method for novel SARS-CoV-2. Emerg. Microbes Infect. 2020, 9, 998–1007. [Google Scholar] [CrossRef]
- Ti, V.L.D.; Herbst, K.; Boerner, K.; Meurer, M.; Kremer, L.P.; Kirrmaier, D.; Freistaedter, A.; Papagiannidis, D.; Galmozzi, C.; Stanifer, M.L.; et al. A colorimetric RT-LAMP assay and LAMP-sequencing for detecting SARS-CoV-2 RNA in clinical samples. Sci. Transl. Med. 2020, 12, eabc7075. [Google Scholar]
- Fernandez, A.M.; Marti, A.; Parungao, M.; Hollin, J.; Selimotic, B.; Farrar, G.; Seyler, T.; Anand, A.; Ahmad, R. A novel, precise and high-throughput technology for viroid detection in Cannabis (MFDetectTM). Viruses 2023, 15, 1487. [Google Scholar] [CrossRef]
- Gutiérrez-Aguirre, I.; Rački, N.; Dreo, T.; Ravnikar, M. Droplet digital PCR for absolute quantification of pathogens. In Plant Pathology. Techniques and Protocols. Methods in Molecular Biology; Lacomme, C., Ed.; Humana Press: New York, NY, USA, 2015; Volume 1302. [Google Scholar] [CrossRef]
- Zhao, Y.; Xia, Q.; Yin, Y.; Wang, Z. Comparison of droplet digital PCR and quantitative PCR assays for quantitative detection of Xanthomonas citri subsp. citri. PLoS ONE 2016, 11, e0159004. [Google Scholar] [CrossRef]
- Dupas, E.; Legendre, B.; Olivier, V.; Poliakoff, F.; Manceau, C.; Cunty, A. Comparison of real-time PCR and droplet digital PCR for the detection of Xylella fastidiosa in plants. J. Microbiol. Meth. 2019, 162, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Chen, R.; Muhae-Ud-Din, G.; Fang, M.; Li, T.; Yang, Y.; Chen, W.; Gao, L. Development of real-time PCR and droplet digital PCR based marker for the detection of Tilletia caries inciting common bunt of wheat. Front. Plant Sci. 2022, 13, 1031611. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Liu, E.; Liu, H.; Jin, X.; Niu, C.; Gao, Y.; Su, X. A droplet digital PCR assay for detection and quantification of Verticillium nonalfalfae and V. albo-atrum. Front. Cell. Infect. Microbiol. 2022, 12, 1972. [Google Scholar] [CrossRef] [PubMed]
- Amoia, S.S.; Minafra, A.; Ligorio, A.; Cavalieri, V.; Boscia, D.; Saponari, M.; Loconsole, G. Detection of Xylella fastidiosa in host plants and insect vectors by droplet digital PCR. Agriculture 2023, 13, 716. [Google Scholar] [CrossRef]
- Flores, R.; Hernandez, C.; Martinez de Alba, A.E.; Daros, J.-A.; Di Serio, F. Viroids and viroid-host interactions. Annu. Rev. Phytopathol. 2005, 43, 117–139. [Google Scholar] [CrossRef] [PubMed]
- Navarro, B.; Flores, R.; Di Serio, F. Advances in viroid-host interactions. Annu. Rev. Virol. 2021, 8, 305–325. [Google Scholar] [CrossRef] [PubMed]
- Pitt, W.J.; Kairy, L.; Villa, E.; Nalam, V.J.; Nachappa, P. Virus infection and host plant suitability affect feeding behaviors of cannabis aphid (Hemiptera: Aphididae), a newly described vector of potato virus Y. Environ. Entomol. 2022, 51, 322–331. [Google Scholar] [CrossRef]
- Rivedale, H.M.; Funke, C.N.; Frost, K.E. An overview of pathogens associated with biotic stresses in hemp crops in Oregon, 2019 to 2020. Plant Dis. 2022, 106, 1334–1340. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Hitchborn, J.H.; Hills, G.J. The use of negative staining in the electron microscopic examination of plant viruses in crude extracts. Virology 1965, 27, 528–540. [Google Scholar] [CrossRef]
- Su, L.; Li, Z.N.; Bernardy, M.G.; Wiersma, P.A.; Cheng, Z.H.; Xiang, Y. The complete nucleotide sequence and genome organization of Pea streak virus (genus Carlavirus). Arch. Virol. 2015, 160, 2651–2654. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- Eastwell, K.C.; Nelson, M.E. Occurrence of viroids in commercial hop (Humulus lupulus L.) production areas of Washington State. Plant Health Prog. 2007, 8, 1–9. [Google Scholar] [CrossRef]
- Guo, R.; Guo, H.; Zhang, Q.; Guo, M.; Xu, Y.; Zeng, M.; Yang, M. Evaluation of reference genes for RT-qPCR analysis in wild and cultivated Cannabis. Biosci. Biotech. Biochem. 2018, 82, 1902–1910. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA 11: Molecular Evolutionary Genetics Analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Mark, D.; Sikazwe, G.; Saggaf, M.; Tairo, F.; Ngunguru, J.; Chiunga, E.; Lusana, J.; Kweka, E.; Maghembe, R.; Bachwenkizi, H. Assessing the effect of sample storage time on viral detection using a rapid and cost-effective CTAB-based extraction method. Res. Sq. 2022, preprint. [Google Scholar] [CrossRef]
- Hataya, T.; Hikage, K.; Suda, N.; Nagata, T.; Li, S.; Itoga, Y.; Tanikoshi, T.; Shikata, E. Detection of hop latent viroid using reverse transcription and polymerase chain reaction (RT-PCR). Ann. Phytopathol. Soc. Jpn. 1992, 58, 677–684. [Google Scholar] [CrossRef]
- Lu, W.; Zhang, Z.; Xu, P.; Liu, S.; Wang, H.; Jiang, D.; Li, S. Simultaneous detection of three viroid species infecting hops in China by multiplex RT-PCR. J. Phytopathol. 2012, 160, 308–310. [Google Scholar] [CrossRef]
- Strausbaugh, C.A.; Wintermantel, W.M.; Gillen, A.M.; Eujayl, I.A. Curly top survey in the Western United States. Phytopathology 2008, 98, 1212–1217. [Google Scholar] [CrossRef]
- Chen, L.-F.; Brannigan, K.; Clark, R.; Gilbertson, R.L. Characterization of curtoviruses associated with curly top disease of tomato in California and monitoring for these viruses in beet leafhoppers. Plant. Dis. 2010, 94, 99–108. [Google Scholar] [CrossRef]
- Mangeot-Peter, L.; Legay, S.; Hausman, J.-F.; Esposito, S.; Guerriero, G. Identification of reference genes for RT-qPCR data normalization in Cannabis sativa stem tissues. Int. J. Mol. Sci. 2016, 17, 1556. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. TrimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]














| Cannabis | Genotype HB | Genotype HB | Genotype OG | Genotype OG |
|---|---|---|---|---|
| Pathogen | HLVd | CasaMV1 | HLVd | CasaMV1 |
| Genome size (kb) | 256 | 2752 | 256 | 2748 |
| Total reads in sample | 10,586 | 402,487 | 24,120 | 101,117 |
| Reads per million | 4248 | 161,500 | 8507 | 35,664 |
| Reads per million per kb | 16,593 | 58,685 | 33,231 | 12,978 |
| Average sequencing depth | 4549 | 19,775 | 10,241 | 4912 |
| Tissue Type Sampled | Tissue Type Sampled | ||||||
|---|---|---|---|---|---|---|---|
| Cannabis Genotype | Leaf | Petiole | Root | Cannabis Genotype | Leaf | Petiole | Root |
| PD #1 | + | NT | + | BM #1 | − | − | − |
| PD #2 | − | NT | + | BM #2 | + | + | + |
| PD #3 | + | NT | + | BM #3 | − | − | + |
| PD #4 | − | NT | − | BM #4 | − | − | + |
| PK | − | NT | + | BM #5 | + | + | + |
| SC | − | NT | − | BM #6 | − | − | + |
| LM1 | + | + | + | 111 | − | − | − |
| GP #1 | + | + | + | PNW #1 | + | + | + |
| GP #2 | − | − | − | PNW #2 | − | − | + |
| GP #3 | − | − | + | PNW #3 | − | − | + |
| GP #4 | + | − | + | BCP #1 | − | NT | + |
| GP #5 | − | NT | + | BCP #2 | − | NT | + |
| GP #6 | − | NT | − | BCP #3 | + | NT | + |
| GP #7 | − | NT | + | CTQ #1 | − | NT | + |
| G55 | − | NT | + | CTQ #2 | − | NT | + |
| Total positive/total | 5/15 | 2/15 | 11/15 | 4/15 | 3/10 | 13/15 | |
| Seed Sample | CT Cycle Threshold | HLVd Present (+)/Absent (−) |
|---|---|---|
| Seed #1 | − | − |
| Seed #2 | 18.94 | + |
| Seed #3 | 18.56 | + |
| Seed #4 | 18.36 | + |
| Seed #5 | 17.10 | + |
| Seed #6 | − | − |
| Seed #7 | − | − |
| Seed #8 | 28.67 | + |
| Seed #9 | 33.20 | + |
| Seed #10 | 33.66 | + |
| 2019 | 2021 | 2022 |
|---|---|---|
| Cannabis sativa mitovirus 1 | Cannabis sativa mitovirus 1 | Cannabis sativa mitovirus 1 |
| Beet curly top virus, strains CO, BCTV-Wor | Beet curly top virus, strains CO, BCTV-Wor | Beet curly top virus, strains CO, BCTV-Wor |
| Hop latent viroid | Alfalfa mosaic virus | Tomato bushy stunt virus |
| Tobacco streak virus | Cannabis cryptic virus | Cannabis cryptic virus |
| Citrus yellow-vein-associated virus |
| Molecular Techniques | Pathogen(s) Detected | Detection in Various Tissues |
|---|---|---|
| Universal fungal primers | Fusarium, Pythium, Alternaria, Penicillium, Golovinomyces, Botrytis | Leaves, stem, root, flower (cannabis) |
| Virus-group primers | None | |
| NGS, HLVd-specific primers | Hop latent viroid | Leaves, roots, flower, seeds (cannabis, hemp) |
| NGS, Mitovirus-specific primers | Mitovirus | Leaves (cannabis, hemp) |
| NGS, BCTV-specific primers | Beet curly top virus | Leaves (cannabis, hemp) |
| NGS, CYVaV-specific primers | Citrus yellow-vein-associated virus | Leaves (hemp) |
| NGS, TSV-specific primers | Tobacco streak virus | Leaves (hemp) |
| NGS | Alfalfa mosaic virus | Leaves (hemp) |
| NGS | Tomato bushy stunt virus | Leaves (hemp) |
| Target | Primer Name | Sequence (5′–3′) | Source |
|---|---|---|---|
| HLVd | HLVd Forward1 | ATACAACTCTTGAGCGCCGA | Hataya et al. [75] |
| HLVd Reverse1 | CCACCGGGTAGTTCCCAACT | Hataya et al. [75] | |
| HLVd Probe 1 | TCTTCGAGCCCTTGCCACCA | This work | |
| HLVd Forward2 | AGTTGCTTCGGCTTCTT | Lu et al. [76] | |
| HLVd Reverse2 | CCATCATACAGGTAAGTCAC | Lu et al. [76] | |
| HLVd Probe 2 | TGCGTGGAACGGCTCCTTCT | This work | |
| Cannabis UBQ | Cannabis UBQ Forward | TACTGCGCCAGCTAACAAAC | Guo [70] |
| Cannabis UBQ Reverse | GCACCCGTCTGACCTGAATC | Guo [70] | |
| Cannabis UBQ Probe | ACAATGCAGCAAATGCTCACTCTACAGCAGTCA | This work |
| Primer | Sequence (5′–3′) |
|---|---|
| HLVd_LAMP_F3 | CGAGCTTTACCTGCAGAAGT |
| HLVd_LAMP_B3 | TGAAGAAGGAGCCGTTCCA |
| HLVd_LAMP_LF | CCCTTGCCACCATACAGG |
| HLVd_LAMP_LB | CGCGGCGACCTGAAGTT |
| HLVd_LAMP_FIP | TAGGTTTCCCCGGGGATCCCCCCCTCTGGGGAATACACT |
| HLVd_LAMP_BIP | CGGAGATCGAGCGCCAGTTCGCAGGACGCGAACAAGAA |
| Target | Primer | Name | Sequence (5′–3′) | Source |
|---|---|---|---|---|
| BCTV-Universal | Forward | BCTV2-F | GTGGATCAATTTCCAGACAATTATC | Strausbaugh et al. [77] |
| Reverse | BCTV2-R | CCCATAAGAGCCATATCAAACTTC | ||
| BCTV-Worland | Forward | BMCTVv2825 | TGATCGAGGCATGGTT | Chen et al. [78] |
| Reverse | BGc396 | CAACTGGTCGATACTGCTAG | ||
| BCTV-Severe | Forward | BSCTVv2688 | GCTGGTACTTCGATGTTG | Chen et al. [78] |
| Reverse | BGc396 | CAACTGGTCGATACTGCTAG | ||
| BCTV-Colorado | Forward | BCTVCO-F | TGCGAGGACGCTTCTTGATT | Chiginsky et al. [3] |
| Reverse | BCTVCO-R | GGGCCGACTCTTATTTTCGG |
| Target | Sequence (5′–3′) | Reference |
|---|---|---|
| Actin | TTGCTGGTCGTGATCTTACTG GTCTCCATCTCCTGCTCAAAG | Mangeot-Peter et al. [79] |
| BCTV universal | GCTTGGTCAAGAGAAGT/ CAACTGGTCGATACTGCTAG | Strausbaugh et al. [41] |
| CasaMV1 | GACGTCTTCTTGTTGTGGCTAGTA GTTCATAGGCAACTGAGGTTCTTT | Chiginsky et al. [3] |
| CYVaV | CCAGACAGGTGTTTCGAGCAT CAATCACTGCAAATCGCG | Kwon et al. [38] |
| TSV | TGGTGTTGACGAGTAATCGTAGTT GAAGCATTCATCAAACAATAGTCG | Chiginsky et al. [3] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Punja, Z.K.; Kahl, D.; Reade, R.; Xiang, Y.; Munz, J.; Nachappa, P. Challenges to Cannabis sativa Production from Pathogens and Microbes—The Role of Molecular Diagnostics and Bioinformatics. Int. J. Mol. Sci. 2024, 25, 14. https://doi.org/10.3390/ijms25010014
Punja ZK, Kahl D, Reade R, Xiang Y, Munz J, Nachappa P. Challenges to Cannabis sativa Production from Pathogens and Microbes—The Role of Molecular Diagnostics and Bioinformatics. International Journal of Molecular Sciences. 2024; 25(1):14. https://doi.org/10.3390/ijms25010014
Chicago/Turabian StylePunja, Zamir K., Dieter Kahl, Ron Reade, Yu Xiang, Jack Munz, and Punya Nachappa. 2024. "Challenges to Cannabis sativa Production from Pathogens and Microbes—The Role of Molecular Diagnostics and Bioinformatics" International Journal of Molecular Sciences 25, no. 1: 14. https://doi.org/10.3390/ijms25010014