
Genetic Alterations of NF-κB and Its Regulators: A Rich Platform to Advance Colorectal Cancer Diagnosis and Treatment
 ,
,  ,
, 
Abstract
:1. Introduction
1.1. Overview of Colorectal Cancer (CRC)
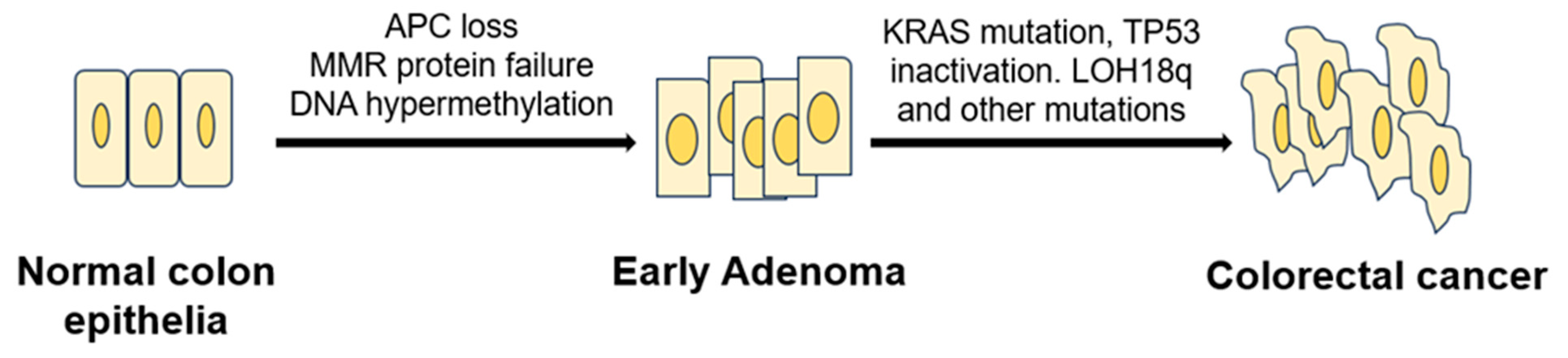
1.2. Model of CRC Initiation and Progression
1.3. Known Genetic Changes in CRC
2. NF-κB Signaling in CRC
3. Genetic Changes of NF-κB Signaling in CRC
3.1. Known NF-κB Genetic Mutations in CRC
3.1.1. Genetic Alterations
3.1.2. Polymorphism in the NF-κB1 Gene in CRC
3.2. Known Genetic Changes of NF-κB Signaling Regulators
3.2.1. Overall Genetic Alterations
3.2.2. Genetic Alterations of NF-κB Positive Regulator, PRMT5 in CRC
3.2.3. Genetic Alterations of NF-κB Negative Regulator, ODAD2 in CRC
4. Research Models CRC
4.1. PDX CRC Models
4.2. Organoid CRC Models
4.3. Applications and Impact of PDX and Organoid CRC Models
5. Conclusions and Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Han, C.J.; Yang, G.S.; Syrjala, K. Symptom Experiences in Colorectal Cancer Survivors after Cancer Treatments: A Systematic Review and Meta-Analysis. Cancer Nurs. 2020, 43, E132–E158. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.; Larsen, P.V.; Søndergaard, J.; Elnegaard, S.; Svendsen, R.P.; Jarbøl, D.E. Specific and Non-Specific Symptoms of Colorectal Cancer and Contact to General Practice. Fam. Pract. 2015, 32, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.S.; Karuniawati, H.; Jairoun, A.A.; Urbi, Z.; Ooi, D.J.; John, A.; Lim, Y.C.; Kibria, K.M.K.; Mohiuddin, A.K.M.; Ming, L.C.; et al. Colorectal Cancer: A Review of Carcinogenesis, Global Epidemiology, Current Challenges, Risk Factors, Preventive and Treatment Strategies. Cancers 2022, 14, 1732. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Z.; Yu, X.; Yang, B.; Zhang, Y.; Zhang, L.; Li, X.; Sun, H. Colorectal Cancer Heterogeneity and Targeted Therapy: Clinical Implications, Challenges and Solutions for Treatment Resistance. Semin. Cell Dev. Biol. 2017, 64, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal Cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef] [PubMed]
- Malki, A.; ElRuz, R.A.; Gupta, I.; Allouch, A.; Vranic, S.; Al Moustafa, A.-E. Molecular Mechanisms of Colon Cancer Progression and Metastasis: Recent Insights and Advancements. Int. J. Mol. Sci. 2021, 22, 130. [Google Scholar] [CrossRef] [PubMed]
- Arvelo, F.; Sojo, F.; Cotte, C. Biology of Colorectal Cancer. Ecancermedicalscience 2015, 9, 520. [Google Scholar] [CrossRef]
- Li, J.; Ma, X.; Chakravarti, D.; Shalapour, S.; DePinho, R.A. Genetic and Biological Hallmarks of Colorectal Cancer. Genes Dev. 2021, 35, 787–820. [Google Scholar] [CrossRef]
- Boland, C.R.; Goel, A. Microsatellite Instability in Colorectal Cancer. Gastroenterology 2010, 138, 2073–2087.e3. [Google Scholar] [CrossRef]
- Mojarad, E.N.; Kuppen, P.J.; Aghdaei, H.A.; Zali, M.R. The CpG Island Methylator Phenotype (CIMP) in Colorectal Cancer. Gastroenterol. Hepatol. Bed Bench 2013, 6, 120–128. [Google Scholar]
- Alzahrani, S.M.; Al Doghaither, H.A.; Al-Ghafari, A.B. General Insight into Cancer: An Overview of Colorectal Cancer. Mol. Clin. Oncol. 2021, 15, 271. [Google Scholar] [CrossRef]
- Tanaka, T. Colorectal Carcinogenesis: Review of Human and Experimental Animal Studies. J. Carcinog. 2009, 8, 5. [Google Scholar] [CrossRef]
- Chu, P.-C.; Lin, P.-C.; Wu, H.-Y.; Lin, K.-T.; Wu, C.; Bekaii-Saab, T.; Lin, Y.-J.; Lee, C.-T.; Lee, J.-C.; Chen, C.-S. Mutant KRAS Promotes Liver Metastasis of Colorectal Cancer, in Part, by Upregulating the MEK-Sp1-DNMT1-miR-137-YB-1-IGF-IR Signaling Pathway. Oncogene 2018, 37, 3440–3455. [Google Scholar] [CrossRef]
- Meng, M.; Zhong, K.; Jiang, T.; Liu, Z.; Kwan, H.Y.; Su, T. The Current Understanding on the Impact of KRAS on Colorectal Cancer. Biomed. Pharmacother. 2021, 140, 111717. [Google Scholar] [CrossRef]
- Midthun, L.; Shaheen, S.; Deisch, J.; Senthil, M.; Tsai, J.; Hsueh, C.-T. Concomitant KRAS and BRAF Mutations in Colorectal Cancer. J. Gastrointest. Oncol. 2019, 10, 577–581. [Google Scholar] [CrossRef]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular Basis of Colorectal Cancer. N. Engl. J. Med. 2009, 361, 2449–2460. [Google Scholar] [CrossRef] [PubMed]
- Maslankova, J.; Vecurkovska, I.; Rabajdova, M.; Katuchova, J.; Kicka, M.; Gayova, M.; Katuch, V. Regulation of Transforming Growth Factor-β Signaling as a Therapeutic Approach to Treating Colorectal Cancer. World J. Gastroenterol. 2022, 28, 4744–4761. [Google Scholar] [CrossRef]
- Armaghany, T.; Wilson, J.D.; Chu, Q.; Mills, G. Genetic Alterations in Colorectal Cancer. Gastrointestinal Cancer Res GCR 2012, 5, 19–27. [Google Scholar]
- Mendelaar, P.A.J.; Smid, M.; van Riet, J.; Angus, L.; Labots, M.; Steeghs, N.; Hendriks, M.P.; Cirkel, G.A.; van Rooijen, J.M.; Ten Tije, A.J.; et al. Whole Genome Sequencing of Metastatic Colorectal Cancer Reveals Prior Treatment Effects and Specific Metastasis Features. Nat. Commun. 2021, 12, 574. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB Signaling in Inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Peng, H.; Huang, Y.; Kong, W.; Cui, Q.; Du, J.; Jin, H. Post-translational Modifications of IκBα: The State of the Art. Front. Cell Dev. Biol. 2020, 8, 574706. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D. Introduction to NF-κB: Players, pathways, perspectives. Oncogene 2006, 25, 6680–6684. [Google Scholar] [CrossRef] [PubMed]
- Shih, V.F.-S.; Tsui, R.; Caldwell, A.; Hoffmann, A. A Single NF-κB System for Both Canonical and Non-Canonical Signaling. Cell Res. 2011, 21, 86–102. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-κB Pathway for the Therapy of Diseases: Mechanism and Clinical Study. Signal Transduct. Target. Ther. 2020, 5, 209. [Google Scholar] [CrossRef] [PubMed]
- Motolani, A.; Martin, M.; Sun, M.; Lu, T. Phosphorylation of the Regulators, a Complex Facet of NF-κB Signaling in Cancer. Biomolecules 2021, 11, 15. [Google Scholar] [CrossRef]
- Sun, S.-C. Non-Canonical NF-κB Signaling Pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef]
- Martin, M.; Sun, M.; Motolani, A.; Lu, T. The Pivotal Player: Components of NF-κB Pathway as Promising Biomarkers in Colorectal Cancer. Int. J. Mol. Sci. 2021, 22, 7429. [Google Scholar] [CrossRef]
- Hai Ping, P.; Feng Bo, T.; Li, L.; Nan Hui, Y.; Hong, Z. IL-1β/NF-κB Signaling Promotes Colorectal Cancer Cell Growth through miR-181a/PTEN Axis. Arch. Biochem. Biophys. 2016, 604, 20–26. [Google Scholar] [CrossRef]
- Soleimani, A.; Rahmani, F.; Ferns, G.A.; Ryzhikov, M.; Avan, A.; Hassanian, S.M. Role of the NF-κB Signaling Pathway in the Pathogenesis of Colorectal Cancer. Gene 2020, 726, 144132. [Google Scholar] [CrossRef]
- CBioPortal for Cancer Genomics. (n.d.). Available online: https://www.cbioportal.org/study/summary?id=coad_silu_2022 (accessed on 18 September 2023).
- Roelands, J.; Kuppen, P.J.K.; Ahmed, E.I.; Mall, R.; Masoodi, T.; Singh, P.; Monaco, G.; Raynaud, C.; de Miranda, N.F.C.C.; Ferraro, L.; et al. An Integrated Tumor, Immune and Microbiome Atlas of Colon Cancer. Nat. Med. 2023, 29, 1273–1286. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Castranova, V.; Shi, X.; Demers, L.M. New Insights into the Role of Nuclear Factor-KappaB, a Ubiquitous Transcription Factor in the Initiation of Diseases. Clin. Chem. 1999, 45, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Wang, B.; Miyagi, M.; She, Y.; Gopalan, B.; Huang, D.B.; Ghosh, G.; Stark, G.R.; Lu, T. PRMT5 Dimethylates R30 of the p65 Subunit to Activate NF-κB. Proc. Natl. Acad. Sci. USA 2013, 110, 13516–13521. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Jackson, M.W.; Wang, B.; Yang, M.; Chance, M.R.; Miyagi, M.; Gudkov, A.V.; Stark, G.R. Regulation of NF-kappaB by NSD1/FBXL11-Dependent Reversible Lysine Methylation of p65. Proc. Natl. Acad. Sci. USA 2010, 107, 46–51. [Google Scholar] [CrossRef]
- Alidoust, M.; Shamshiri, A.K.; Tajbakhsh, A.; Gheibihayat, S.M.; Mazloom, S.M.; Alizadeh, F.; Pasdar, A. The Significant Role of a Functional Polymorphism in the NF-κB1 Gene in Breast Cancer: Evidence from an Iranian Cohort. Future Oncol. 2021, 17, 4895–4905. [Google Scholar] [CrossRef] [PubMed]
- Weichert, W.; Boehm, M.; Gekeler, V.; Bahra, M.; Langrehr, J.; Neuhaus, P.; Denkert, C.; Imre, G.; Weller, C.; Hofmann, H.-P.; et al. High Expression of RelA/p65 is Associated with Activation of Nuclear Factor-κB-Dependent Signaling in Pancreatic Cancer and Marks a Patient Population with Poor Prognosis. Br. J. Cancer 2007, 97, 523–530. [Google Scholar] [CrossRef]
- Lu, T.; Sathe, S.S.; Swiatkowski, S.M.; Hampole, C.V.; Stark, G.R. Secretion of Cytokines and Growth Factors as a General Cause of Constitutive NFkappaB Activation in Cancer. Oncogene 2004, 23, 2138–2145. [Google Scholar] [CrossRef]
- Nagel, D.; Vincendeau, M.; Eitelhuber, A.C.; Krappmann, D. Mechanisms and consequences of constitutive NF-κB activation in B-cell lymphoid malignancies. Oncogene 2014, 33, 5655–5665. [Google Scholar] [CrossRef]
- Lewander, A.; Butchi, A.K.R.; Gao, J.; He, L.-J.; Lindblom, A.; Arbman, G.; Carstensen, J.; Zhang, Z.-Y.; The Swedish Low-Risk Colorectal Cancer Study Group; Sun, X.-F. Polymorphism in the Promoter Region of the NFKB1 Gene Increases the Risk of Sporadic Colorectal Cancer in Swedish but Not in Chinese Populations. Scand. J. Gastroenterol. 2007, 42, 1332–1338. [Google Scholar] [CrossRef]
- Lu, T.; Stark, G.R. NF-κB: Regulation by Methylation. Cancer Res. 2015, 75, 3692–3695. [Google Scholar] [CrossRef]
- Prabhu, L.; Martin, M.; Chen, L.; Demir, Ö.; Jin, J.; Huang, X.; Motolani, A.; Sun, M.; Jiang, G.; Nakshatri, H.; et al. Inhibition of PRMT5 by Market Drugs as a Novel Cancer Therapeutic Avenue. Genes Dis. 2022, 10, 267–283. [Google Scholar] [CrossRef]
- Prabhu, L.; Wei, H.; Chen, L.; Demir, Ö.; Sandusky, G.; Sun, E.; Wang, J.; Mo, J.; Zeng, L.; Fishel, M.; et al. Adapting AlphaLISA High Throughput Screen to Discover a Novel Small-Molecule Inhibitor Targeting Protein Arginine Methyltransferase 5 in Pancreatic and Colorectal Cancers. Oncotarget 2017, 8, 39963–39977. [Google Scholar] [CrossRef]
- Martin, M.; Mundade, R.; Hartley, A.V.; Jiang, G.; Jin, J.; Sun, S.; Safa, A.; Sandusky, G.; Liu, Y.; Lu, T. Using VBIM Technique to Discover ARMC4/ODAD2 as a Novel Negative Regulator of NF-κB and a New Tumor Suppressor in Colorectal Cancer. Int. J. Mol. Sci. 2022, 23, 2732. [Google Scholar] [CrossRef] [PubMed]
- Abumustafa, W.; Zamer, B.A.; Khalil, B.A.; Hamad, M.; Maghazachi, A.A.; Muhammad, J.S. Protein Arginine N-Methyltransferase 5 in Colorectal Carcinoma: Insights into Mechanisms of Pathogenesis and Therapeutic Strategies. Biomed. Pharmacother. 2022, 145, 112368. [Google Scholar] [CrossRef] [PubMed]
- Shifteh, D.; Sapir, T.; Pahmer, M.; Haimowitz, A.; Goel, S.; Maitra, R. Protein Arginine Methyltransferase 5 as a Therapeutic Target for KRAS Mutated Colorectal Cancer. Cancers 2020, 12, 2091. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Hartley, A.V.; Motolani, A.; Jiang, G.; Safa, A.; Prabhu, L.; Liu, Y.; Lu, T. A Complex Signature Network That Controls the Upregulation of PRMT5 in Colorectal Cancer. Genes Dis. 2021, 9, 285–287. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, S.; Bouley, R.A.; Yoder, R.J.; Petreaca, R.C. Protein Arginine Methyltransferase 5 (PRMT5) Mutations in Cancer Cells. Int. J. Mol. Sci. 2023, 24, 6042. [Google Scholar] [CrossRef] [PubMed]
- Hartley, A.V.; Wang, B.; Jiang, G.; Wei, H.; Sun, M.; Prabhu, L.; Safa, A.; Liu, Y.; Lu, T. Regulation of a PRMT5/NF-κB Axis by Phosphorylation of PRMT5 at Serine 15 in Colorectal Cancer. Int. J. Mol. Sci. 2020, 21, 3684. [Google Scholar] [CrossRef] [PubMed]
- Hartley, A.V.; Wang, B.; Mundade, R.; Jiang, G.; Sun, M.; Wei, H.; Sun, S.; Liu, Y.; Lu, T. PRMT5-Mediated Methylation of YBX1 Regulates NF-κB Activity in Colorectal Cancer. Sci. Rep. 2020, 10, 15934. [Google Scholar] [CrossRef] [PubMed]
- Burtin, F.; Mullins, C.S.; Linnebacher, M. Mouse models of colorectal cancer: Past, present and future perspectives. World J. Gastroenterol. 2020, 26, 1394–1426. [Google Scholar] [CrossRef]
- Liu, X.; Xin, Z.; Wang, K. Patient-derived xenograft model in colorectal cancer basic and translational research. Animal Model. Exp. Med. 2023, 6, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Abdolahi, S.; Ghazvinian, Z.; Muhammadnejad, S.; Saleh, M.; Asadzadeh Aghdaei, H.; Baghaei, K. Patient-derived xenograft (PDX) models, applications and challenges in cancer research. J. Transl. Med. 2022, 20, 206. [Google Scholar] [CrossRef] [PubMed]
- Puig, I.; Chicote, I.; Tenbaum, S.P.; Arques, O.; Herance, J.R.; Gispert, J.D.; Jimenez, J.; Landolfi, S.; Caci, K.; Allende, H.; et al. A personalized preclinical model to evaluate the metastatic potential of patient-derived colon cancer initiating cells. Clin. Cancer Res. 2013, 19, 6787–6801. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, G.J. Applications of patient-derived tumor xenograft models and tumor organoids. J. Hematol. Oncol. 2020, 13, 4. [Google Scholar] [CrossRef] [PubMed]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernandez-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef] [PubMed]
- van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef] [PubMed]
- Porru, M.; Zizza, P.; Panera, N.; Alisi, A.; Biroccio, A.; Leonetti, C. Harnessing Omics Approaches on Advanced Preclinical Models to Discovery Novel Therapeutic Targets for the Treatment of Metastatic Colorectal Cancer. Cancers 2020, 12, 1830. [Google Scholar] [CrossRef] [PubMed]
- Noto, F.K.; Yeshi, T. Humanized Mouse and Rat PDX Cancer Models. In Patient-Derived Xenograft Models of Human Cancer; Wang, Y., Lin, D., Gout, P.W., Eds.; Springer: Cham, Switzerland, 2017; pp. 43–57. [Google Scholar]
- Fumagalli, A.; Suijkerbuijk, S.J.E.; Begthel, H.; Beerling, E.; Oost, K.C.; Snippert, H.J.; van Rheenen, J.; Drost, J. A surgical orthotopic organoid transplantation approach in mice to visualize and study colorectal cancer progression. Nat. Protoc. 2018, 13, 235–247. [Google Scholar] [CrossRef]
- Georges, L.M.C.; De Wever, O.; Galvan, J.A.; Dawson, H.; Lugli, A.; Demetter, P.; Zlobec, I. Cell Line Derived Xenograft Mouse Models Are a Suitable in vivo Model for Studying Tumor Budding in Colorectal Cancer. Front. Med. 2019, 6, 139. [Google Scholar] [CrossRef]
- Stastna, M.; Janeckova, L.; Hrckulak, D.; Kriz, V.; Korinek, V. Human Colorectal Cancer from the Perspective of Mouse Models. Genes 2019, 10, 788. [Google Scholar] [CrossRef]
- Yang, C.; Xiao, W.; Wang, R.; Hu, Y.; Yi, K.; Sun, X.; Wang, G.; Xu, X. Tumor organoid model of colorectal cancer. Oncol. Lett. 2023, 26, 328. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.S.; Liu, C.Y.; Wen, D.; Gao, D.Z.; Lin, S.; He, H.F.; Zhao, X.F. Recent advances in the development of transplanted colorectal cancer mouse models. Transl. Res. 2022, 249, 128–143. [Google Scholar] [CrossRef] [PubMed]
- Shultz, L.D.; Brehm, M.A.; Garcia-Martinez, J.V.; Greiner, D.L. Humanized mice for immune system investigation: Progress, promise and challenges. Nat. Rev. Immunol. 2012, 12, 786–798. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.M.; Xue, A.; Mittal, A.; Samra, J.S.; Smith, R.; Hugh, T.J. Patient-derived xenograft models of colorectal cancer in pre-clinical research: A systematic review. Oncotarget 2016, 7, 66212–66225. [Google Scholar] [CrossRef] [PubMed]
- Wimsatt, J.H.; Montgomery, C.; Thomas, L.S.; Savard, C.; Tallman, R.; Innes, K.; Jrebi, N. Assessment of a mouse xenograft model of primary colorectal cancer with special reference to perfluorooctane sulfonate. PeerJ 2018, 6, e5602. [Google Scholar] [CrossRef]
- Sheng, Y.; Qiang, W.; Guo, S. Impact of orthotopic versus subcutaneous implantation on patient-derived xenograft transcriptomic profile. Eur. J. Cancer 2022, 174, S9. [Google Scholar] [CrossRef]
- Hoffman, R.M. Patient-derived orthotopic xenografts: Better mimic of metastasis than subcutaneous xenografts. Nat. Rev. Cancer 2015, 15, 451–452. [Google Scholar] [CrossRef]
- Day, C.P.; Merlino, G.; Van Dyke, T. Preclinical mouse cancer models: A maze of opportunities and challenges. Cell 2015, 163, 39–53. [Google Scholar] [CrossRef]
- Sveen, A.; Bruun, J.; Eide, P.W.; Eilertsen, I.A.; Ramirez, L.; Murumagi, A.; Arjama, M.; Danielsen, S.A.; Kryeziu, K.; Elez, E.; et al. Colorectal Cancer Consensus Molecular Subtypes Translated to Preclinical Models Uncover Potentially Targetable Cancer Cell Dependencies. Clin. Cancer Res. 2018, 24, 794–806. [Google Scholar] [CrossRef]
- Julien, S.; Merino-Trigo, A.; Lacroix, L.; Pocard, M.; Goere, D.; Mariani, P.; Landron, S.; Bigot, L.; Nemati, F.; Dartigues, P.; et al. Characterization of a large panel of patient-derived tumor xenografts representing the clinical heterogeneity of human colorectal cancer. Clin. Cancer Res. 2012, 18, 5314–5328. [Google Scholar] [CrossRef]
- Lu, Y.; Huang, R.; Ying, J.; Li, X.; Jiao, T.; Guo, L.; Zhou, H.; Wang, H.; Tuersuntuoheti, A.; Liu, J.; et al. RING finger 138 deregulation distorts NF-small ka, CyrillicB signaling and facilities colitis switch to aggressive malignancy. Signal Transduct. Target. Ther. 2022, 7, 185. [Google Scholar] [CrossRef] [PubMed]
- Lindner, A.U.; Carberry, S.; Monsefi, N.; Barat, A.; Salvucci, M.; O’Byrne, R.; Zanella, E.R.; Cremona, M.; Hennessy, B.T.; Bertotti, A.; et al. Systems analysis of protein signatures predicting cetuximab responses in KRAS, NRAS, BRAF and PIK3CA wild-type patient-derived xenograft models of metastatic colorectal cancer. Int. J. Cancer 2020, 147, 2891–2901. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Wang, G.; Chen, W.; Zhu, Z.; Liu, Y.; Huang, Z.; Huang, Y.; Du, P.; Yang, Y.; Liu, C.Y.; et al. Co-inhibition of BET proteins and NF-kappaB as a potential therapy for colorectal cancer through synergistic inhibiting MYC and FOXM1 expressions. Cell Death Dis. 2018, 9, 315. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Korn, J.M.; Ferretti, S.; Monahan, J.E.; Wang, Y.; Singh, M.; Zhang, C.; Schnell, C.; Yang, G.; Zhang, Y.; et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat. Med. 2015, 21, 1318–1325. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.; Van Es, J.H.; Van den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef] [PubMed]
- Tuveson, D.; Clevers, H. Cancer modeling meets human organoid technology. Science 2019, 364, 952–955. [Google Scholar] [CrossRef] [PubMed]
- Matano, M.; Date, S.; Shimokawa, M.; Takano, A.; Fujii, M.; Ohta, Y.; Watanabe, T.; Kanai, T.; Sato, T. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat. Med. 2015, 21, 256–262. [Google Scholar] [CrossRef]
- Hofer, M.; Lutolf, M.P. Engineering organoids. Nat. Rev. Mater. 2021, 6, 402–420. [Google Scholar] [CrossRef]
- Kim, S.C.; Park, J.W.; Seo, H.Y.; Kim, M.; Park, J.H.; Kim, G.H.; Lee, J.O.; Shin, Y.K.; Bae, J.M.; Koo, B.K.; et al. Multifocal Organoid Capturing of Colon Cancer Reveals Pervasive Intratumoral Heterogenous Drug Responses. Adv. Sci. 2022, 9, e2103360. [Google Scholar] [CrossRef]
- Okazawa, Y.; Mizukoshi, K.; Koyama, Y.; Okubo, S.; Komiyama, H.; Kojima, Y.; Goto, M.; Habu, S.; Hino, O.; Sakamoto, K.; et al. High-sensitivity Detection of Micrometastases Generated by GFP Lentivirus-transduced Organoids Cultured from a Patient-derived Colon Tumor. J. Vis. Exp. 2018, 136, e57374. [Google Scholar]
- Shimokawa, M.; Ohta, Y.; Nishikori, S.; Matano, M.; Takano, A.; Fujii, M.; Date, S.; Sugimoto, S.; Kanai, T.; Sato, T. Visualization and targeting of LGR5(+) human colon cancer stem cells. Nature 2017, 545, 187–192. [Google Scholar] [CrossRef] [PubMed]
- LeSavage, B.L.; Suhar, R.A.; Broguiere, N.; Lutolf, M.P.; Heilshorn, S.C. Next-generation cancer organoids. Nat. Mater. 2022, 21, 143–159. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Shostak, K.; Wathieu, C.; Tielens, S.; Chariot, A. NF-kappaB Signaling in Ex-Vivo Mouse Intestinal Organoids. Methods Mol. Biol. 2021, 2366, 283–292. [Google Scholar] [PubMed]
- Daghero, H.; Doffe, F.; Varela, B.; Yozzi, V.; Verdes, J.M.; Crispo, M.; Bollati-Fogolin, M.; Pagotto, R. Jejunum-derived NF-kappaB reporter organoids as 3D models for the study of TNF-alpha-induced inflammation. Sci. Rep. 2022, 12, 14425. [Google Scholar] [CrossRef] [PubMed]
- Ooft, S.N.; Weeber, F.; Dijkstra, K.K.; McLean, C.M.; Kaing, S.; van Werkhoven, E.; Schipper, L.; Hoes, L.; Vis, D.J.; van de Haar, J.; et al. Patient-derived organoids can predict response to chemotherapy in metastatic colorectal cancer patients. Sci. Transl. Med. 2019, 11, eaay2574. [Google Scholar] [CrossRef] [PubMed]
- Dahlmann, M.; Gambara, G.; Brzezicha, B.; Popp, O.; Pachmayr, E.; Wedeken, L.; Pflaume, A.; Mokritzkij, M.; Gul-Klein, S.; Brandl, A.; et al. Peritoneal metastasis of colorectal cancer (pmCRC): Identification of predictive molecular signatures by a novel preclinical platform of matching pmCRC PDX/PD3D models. Mol. Cancer 2021, 20, 129. [Google Scholar] [CrossRef]
- Huang, L.; Bockorny, B.; Paul, I.; Akshinthala, D.; Frappart, P.O.; Gandarilla, O.; Bose, A.; Sanchez-Gonzalez, V.; Rouse, E.E.; Lehoux, S.D.; et al. PDX-derived organoids model in vivo drug response and secrete biomarkers. JCI Insight 2020, 5, e135544. [Google Scholar] [CrossRef]
- Liu, H.; Liang, Z.; Cheng, S.; Huang, L.; Li, W.; Zhou, C.; Zheng, X.; Li, S.; Zeng, Z.; Kang, L. Mutant KRAS Drives Immune Evasion by Sensitizing Cytotoxic T-Cells to Activation-Induced Cell Death in Colorectal Cancer. Adv. Sci. 2023, 10, e2203757. [Google Scholar] [CrossRef]
- Braoudaki, M.; Ahmad, M.S.; Mustafov, D.; Seriah, S.; Siddiqui, M.N.; Siddiqui, S.S. Chemokines and chemokine receptors in colorectal cancer; multifarious roles and clinical impact. Semin. Cancer Biol. 2022, 86, 436–449. [Google Scholar] [CrossRef]
- De La Rochere, P.; Guil-Luna, S.; Decaudin, D.; Azar, G.; Sidhu, S.S.; Piaggio, E. Humanized mice for the study of immuno-oncology. Trends Immunol. 2018, 39, 748–763. [Google Scholar] [CrossRef] [PubMed]
- Bouffi, C.; Wikenheiser-Brokamp, K.A.; Chaturvedi, P.; Sundaram, N.; Goddard, G.R.; Wunderlich, M.; Brown, N.E.; Staab, J.F.; Latanich, R.; Zachos, N.C.; et al. In vivo development of immune tissue in human intestinal organoids transplanted into humanized mice. Nat. Biotechnol. 2023, 41, 824–831. [Google Scholar] [CrossRef] [PubMed]
- Ramadass, V.; Vaiyapuri, T.; Tergaonkar, V. Small Molecule NF-κB Pathway Inhibitors in Clinic. Int. J. Mol. Sci. 2020, 21, 5164. [Google Scholar] [CrossRef] [PubMed]
- Motolani, A.; Martin, M.; Sun, M.; Lu, T. NF-κB and cancer therapy drugs. In Reference Module in Biomedical Sciences; Book Chapter; Elsevier Press: Amsterdam, The Netherlands, 2021. [Google Scholar] [CrossRef]
- Crespo, M.; Vilar, E.; Tsai, S.; Chang, K.; Amin, S.; Srinivasan, T.; Zhang, T.; Pipalia, N.H.; Chen, H.J.; Witherspoon, M.; et al. Colonic Organoids Derived from Human Pluripotent Stem Cells for Modeling Colorectal Cancer and Drug Testing. Nat. Med. 2017, 23, 878–884. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Gene Description | Protein | Protein Mutation | Mutation Type | Copy Number |
|---|---|---|---|---|---|
| Rel | C-Rel proto-oncogene, NF-κB subunit | Rel | R22C | Missense | Diploid |
| G288S | Missense | Diploid | |||
| R108Q | Missense | Diploid | |||
| G229D | Missense | Diploid | |||
| X285_splice | Splice | Diploid | |||
| RelA | RelA proto-oncogene, NF-κB subunit | RelA (p65) | R166W | Missense | Diploid |
| D446H | Missense | Diploid | |||
| N139del | IF del | Diploid | |||
| H487Pfs*4 | FS ins | Diploid | |||
| P521L | Missense | Diploid | |||
| T357A | Missense | Diploid | |||
| Q287* | Nonsense | ShallowDel | |||
| RelB | RELB Proto-Oncogene, NF-κB Subunit | RelB | A29V | Missense | Diploid |
| P314L | Missense | Gain | |||
| R434W | Missense | Diploid | |||
| T494M | Missense | Diploid | |||
| G530Afs*5 | FS del | Diploid | |||
| E53* | Nonsense | Diploid | |||
| P482L | Missense | Diploid | |||
| P482L | Missense | ShallowDel | |||
| V379I | Missense | Diploid | |||
| G522R | Missense | Diploid | |||
| Y539H | Missense | Diploid | |||
| V353M | Missense | Diploid | |||
| C306Y | Missense | Diploid | |||
| NF-κB1 | NF-κB subunit 1 | p105/p50 | R613C | Missense | Diploid |
| A901T | Missense | Diploid | |||
| G477V | Missense | Diploid | |||
| D436G | Missense | Diploid | |||
| NF-κB2 | NF-κB subunit 2 | p100/p52 | Y294Ifs*4 | FS del | Diploid |
| A514T | Missense | Diploid | |||
| A867V | Missense | Diploid | |||
| K252N | Missense | Diploid | |||
| T806M | Missense | Diploid | |||
| L474M | Missense | Diploid | |||
| A121T | Missense | Diploid | |||
| K252M | Missense | Diploid | |||
| M1? | Nonstart | Diploid |
| Gene Symbol | Gene Description | Protein | Protein Mutation | Mutation Type | Copy Number |
|---|---|---|---|---|---|
| Chuk (IKBKA) | Inhibitor of nuclear factor κB kinase subunit α | IKKα | E82* | Nonsense | Diploid |
| P700del | IF del | Diploid | |||
| X577_splice | Splice | Diploid | |||
| E513* | Nonsense | Diploid | |||
| L50P | Missense | ShallowDel | |||
| IKBKB | Inhibitor of nuclear factor κB kinase subunit β | IKKβ | R582Q | Missense | Diploid |
| P551L | Missense | Diploid | |||
| A454T | Missense | Diploid | |||
| N225Tfs*25 | FS del | Gain | |||
| Q438H | Missense | Gain | |||
| A481V | Missense | Diploid | |||
| X159_splice | Splice | Gain | |||
| IRAK1 | Interleukin 1 receptor associated kinase 1 | IRAK1 | R51C | Missense | Diploid |
| T383A | Missense | Diploid | |||
| T234M | Missense | Diploid | |||
| A78T | Missense | Diploid | |||
| R61C | Missense | Diploid | |||
| E259D | Missense | Diploid | |||
| C43R | Missense | Diploid | |||
| KDM2A | Lysine demethylase 2A | KDM2A | P597Afs*34 | FS ins | Diploid |
| T162M | Missense | Diploid | |||
| N1083S | Missense | Diploid | |||
| H452R | Missense | Diploid | |||
| A576S | Missense | Diploid | |||
| P729L | Missense | Diploid | |||
| R733G | Missense | Diploid | |||
| S416G | Missense | Diploid | |||
| MAP3K7 | Mitogen-activated protein kinase kinase kinase 7 | MAP3K7 (TAK1) | R226W | Missense | Diploid |
| T169Dfs*7 | FS ins | Diploid | |||
| R238Q | Missense | Diploid | |||
| L255V | Missense | Diploid | |||
| D488V | Missense | Diploid | |||
| R248Q | Missense | Diploid | |||
| P256S | Missense | Diploid | |||
| D343Y | Missense | Diploid | |||
| R463K | Missense | Diploid | |||
| NFKBIA | NF-κB inhibitor α | IκBα | E41A | Missense | Diploid |
| P147H | Missense | Diploid | |||
| ODAD2 | Outer dynein arm docking complex subunit 2 | ODAD2 | T9M | Missense | Diploid |
| R841C | Missense | Diploid | |||
| R1032C | Missense | Diploid | |||
| K304T | Missense | Diploid | |||
| I517T | Missense | Diploid | |||
| L813I | Missense | Diploid | |||
| A29V | Missense | Diploid | |||
| A556V | Missense | Diploid | |||
| L298* | FS del | Gain | |||
| E626* | Nonsense | Diploid | |||
| I543T | Missense | Diploid | |||
| A445E | Missense | Diploid | |||
| L154I | Missense | Diploid | |||
| G536D | Missense | Diploid | |||
| A820S | Missense | ShallowDel | |||
| S661A | Missense | Diploid | |||
| S743Y | Missense | Diploid | |||
| PRMT5 | Protein arginine methyltransferase 5 | PRMT5 | H47Y | Missense | Diploid |
| V413L | Missense | Diploid | |||
| R256Q | Missense | Diploid | |||
| Y535S | Missense | Diploid | |||
| L287V | Missense | Diploid | |||
| E57K | Missense | Gain | |||
| TAB1 | TGFβ activated kinase 1 (MAP3K7) binding protein 1 | TAB1 | Y293C | Missense | Diploid |
| E96D | Missense | Diploid | |||
| A310G | Missense | Diploid | |||
| L361Q | Missense | Diploid | |||
| TAB2 | TGF-β activated kinase 1 (MAP3K7) binding protein 2 | TAB2 | A672T | Missense | Diploid |
| R579I | Missense | Diploid | |||
| N211K | Missense | Diploid | |||
| R72C | Missense | Diploid | |||
| A182V | Missense | Diploid | |||
| TRAF2 | TNF receptor associated factor 2 | TRAF2 | P9Lfs*77 | FS del | Diploid |
| P9Lfs*77 | FS del | Amp | |||
| E122K | Missense | Diploid | |||
| A3T | Missense | Diploid | |||
| A494V | Missense | Diploid |
| Classification | Protein | Protein Mutation Types | Alteration Frequency, % | Total Protein Mutation Types | Total Alteration Frequency, % | ||
|---|---|---|---|---|---|---|---|
| NF-κB family members | Rel | 5 | 2.5 | 38 | 113 | 14.8 | 53.7 |
| RelA (p65) | 7 | 2.5 | |||||
| RelB | 13 | 5.0 | |||||
| P105/p50 | 4 | 1.8 | |||||
| P100/p52 | 9 | 3.0 | |||||
| NF-κB pathway regulators | IKKα | 5 | 1.4 | 75 | 38.9 | ||
| IKKβ | 7 | 6.0 | |||||
| IRAK1 | 7 | 4.0 | |||||
| KDM2A | 8 | 2.8 | |||||
| MAP3K7 | 9 | 4.0 | |||||
| NFKBIA | 2 | 1.1 | |||||
| ODAD2 | 17 | 7.0 | |||||
| PRMT5 | 6 | 5.0 | |||||
| TAB1 | 4 | 1.8 | |||||
| TAB2 | 5 | 3.0 | |||||
| TRAF2 | 5 | 2.8 | |||||
| Model | Advantages | Disadvantages | Reference |
|---|---|---|---|
| Immortalized cancer cell lines |
|
| [58,59] |
| GEMMs |
|
| [51,52,58] |
| CDXs |
|
| [53,59,60] |
| PDXs |
|
| [54,55,61] |
| Organoids |
|
| [56,57,62,63] |
| Humanized mice |
|
| [53,64,65] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alipourgivi, F.; Motolani, A.; Qiu, A.Y.; Qiang, W.; Yang, G.-Y.; Chen, S.; Lu, T. Genetic Alterations of NF-κB and Its Regulators: A Rich Platform to Advance Colorectal Cancer Diagnosis and Treatment. Int. J. Mol. Sci. 2024, 25, 154. https://doi.org/10.3390/ijms25010154
Alipourgivi F, Motolani A, Qiu AY, Qiang W, Yang G-Y, Chen S, Lu T. Genetic Alterations of NF-κB and Its Regulators: A Rich Platform to Advance Colorectal Cancer Diagnosis and Treatment. International Journal of Molecular Sciences. 2024; 25(1):154. https://doi.org/10.3390/ijms25010154
Chicago/Turabian StyleAlipourgivi, Faranak, Aishat Motolani, Alice Y. Qiu, Wenan Qiang, Guang-Yu Yang, Shuibing Chen, and Tao Lu. 2024. "Genetic Alterations of NF-κB and Its Regulators: A Rich Platform to Advance Colorectal Cancer Diagnosis and Treatment" International Journal of Molecular Sciences 25, no. 1: 154. https://doi.org/10.3390/ijms25010154