
Cortistatin as a Novel Multimodal Therapy for the Treatment of Parkinson’s Disease
 , , , , , , and
, , , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Treatment with Cortistatin Reduces MPTP-Induced Locomotor Dysfunction
2.2. Cortistatin Attenuates MPTP-Induced Dopaminergic Neurodegeneration
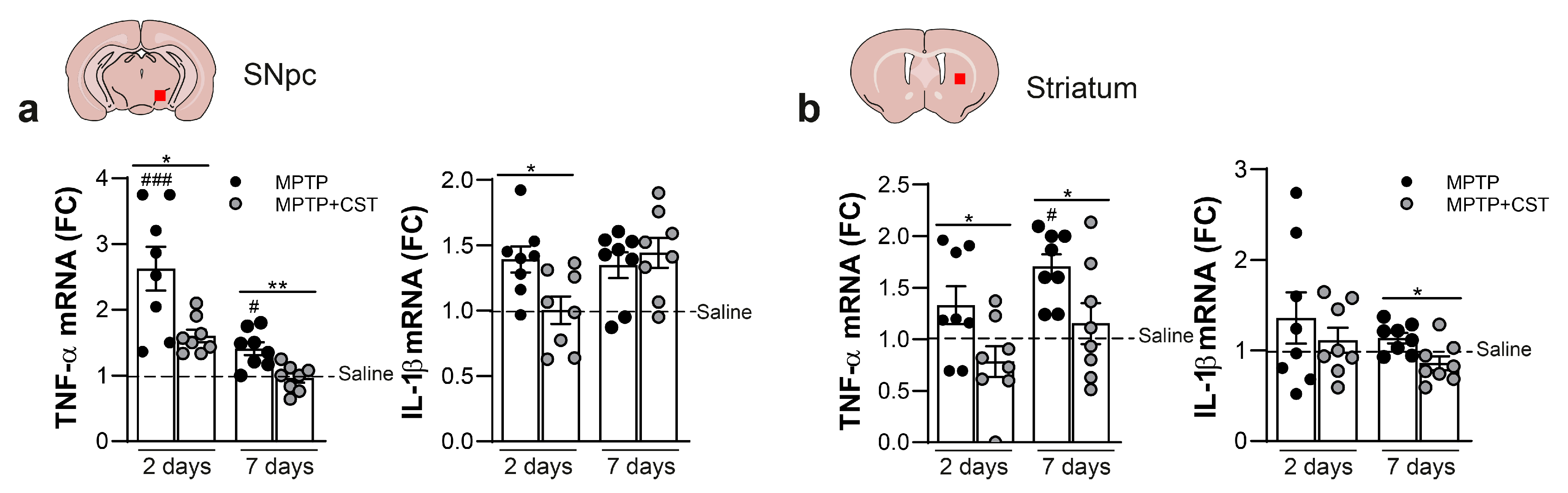
2.3. Cortistatin Modulates the Neuroinflammatory Response Induced by Acute MPTP Administration
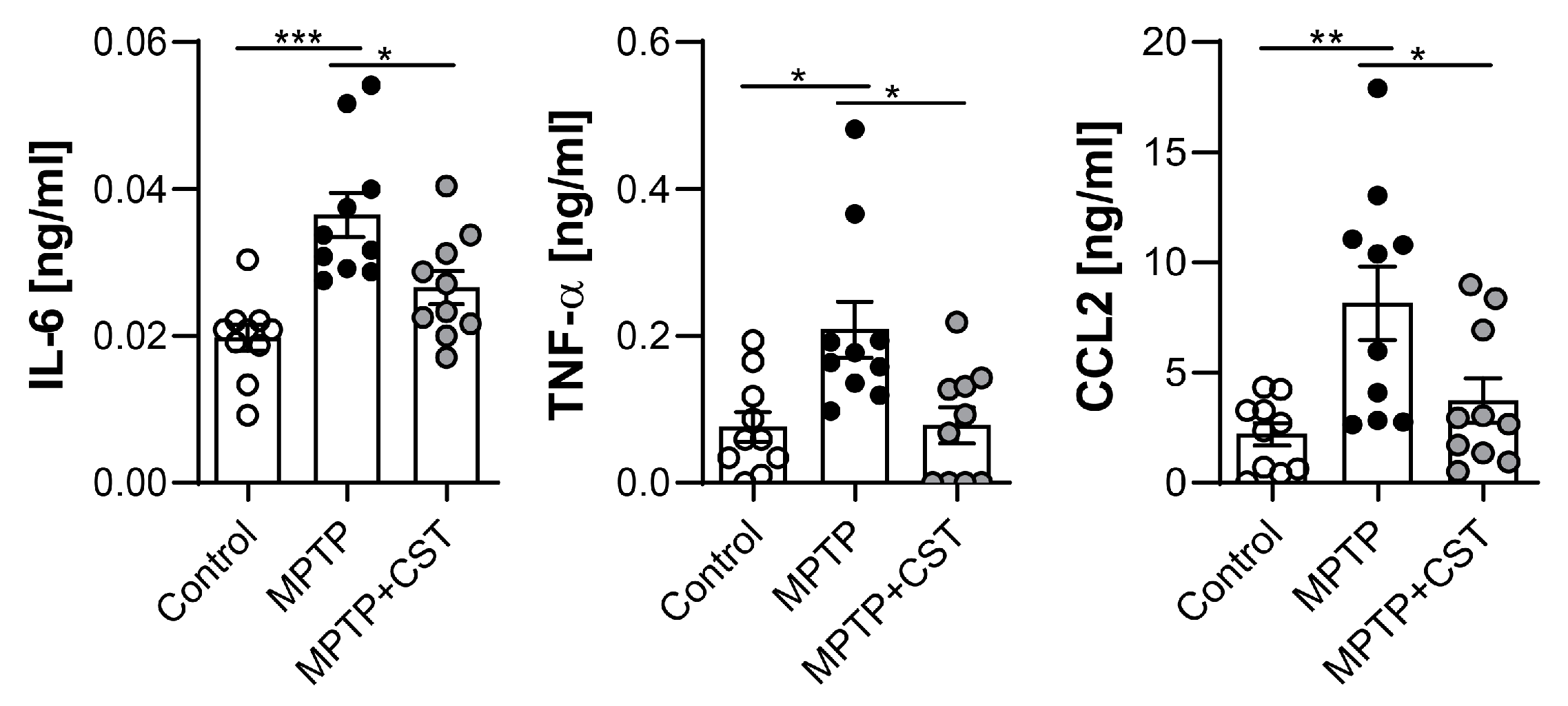
2.4. Cortistatin Alleviates the Peripheral Immune Response Induced by MPTP Intoxication
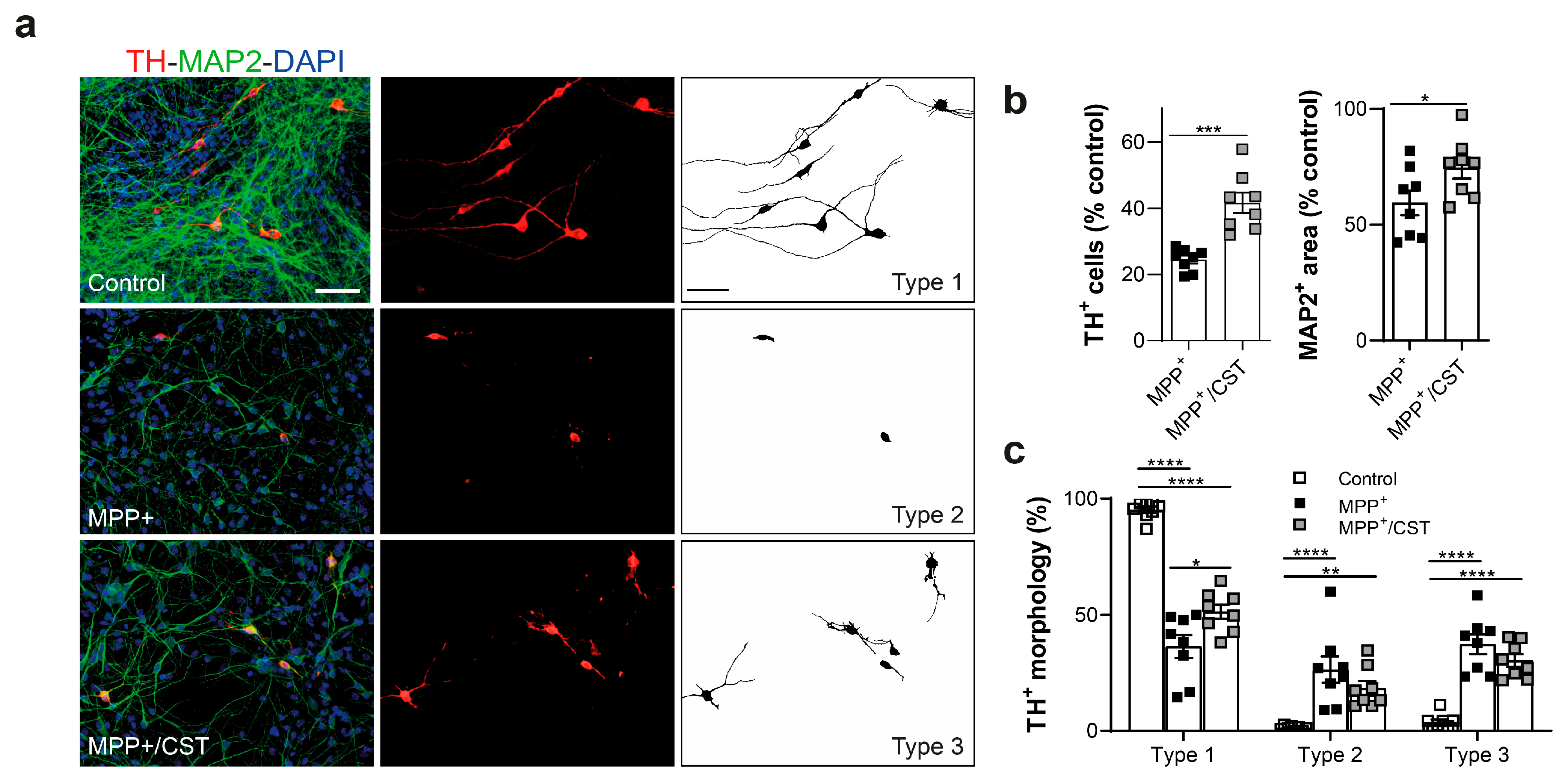
2.5. Protective Effect of Cortistatin on Dopaminergic Neurons Exposed to MPP+
3. Discussion
4. Materials and Methods
4.1. Animals and Ethics Statement
4.2. Reagents
4.3. Induction and Treatment of the PD Model
4.4. Evaluation of Motor Activity
4.5. Histological Analyses: Immunohistochemistry and Immunofluorescence
4.6. Stereology and Densitometry Evaluation
4.7. Microglia Phenotype Characterization
4.8. Determination of Dopamine Levels
4.9. Determination of Gene Expression
4.10. Primary Mesencephalic Cell Culture
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ray Dorsey, E.; Elbaz, A.; Nichols, E.; Abd-Allah, F.; Abdelalim, A.; Adsuar, J.C.; Ansha, M.G.; Brayne, C.; Choi, J.Y.J.; Collado-Mateo, D.; et al. Global, Regional, and National Burden of Parkinson’s Disease, 1990–2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef] [PubMed]
- Marino, B.L.B.; de Souza, L.R.; Sousa, K.P.A.; Ferreira, J.V.; Padilha, E.C.; da Silva, C.H.T.P.; Taft, C.A.; Hage-Melim, L.I.S. Parkinson’s Disease: A Review from Pathophysiology to Treatment. Mini Rev. Med. Chem. 2020, 20, 754–767. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s Disease: A Target for Neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef] [PubMed]
- Grotemeyer, A.; McFleder, R.L.; Wu, J.; Wischhusen, J.; Ip, C.W. Neuroinflammation in Parkinson’s Disease—Putative Pathomechanisms and Targets for Disease-Modification. Front. Immunol. 2022, 13, 878771. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, Y.; Zhou, J. Neuroinflammation in Parkinson’s Disease and Its Potential as Therapeutic Target. Transl. Neurodegener. 2015, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Pajares, M.; Rojo, A.I.; Manda, G.; Boscá, L.; Cuadrado, A. Inflammation in Parkinson’s Disease: Mechanisms and Therapeutic Implications. Cells 2020, 9, 1687. [Google Scholar] [CrossRef]
- Tansey, M.G.; Wallings, R.L.; Houser, M.C.; Herrick, M.K.; Keating, C.E.; Joers, V. Inflammation and Immune Dysfunction in Parkinson Disease. Nat. Rev. Immunol. 2022, 22, 657–673. [Google Scholar] [CrossRef]
- Arena, G.; Sharma, K.; Agyeah, G.; Krüger, R.; Grünewald, A.; Fitzgerald, J.C. Neurodegeneration and Neuroinflammation in Parkinson’s Disease: A Self-Sustained Loop. Curr. Neurol. Neurosci. Rep. 2022, 22, 427–440. [Google Scholar] [CrossRef]
- Kouli, A.; Camacho, M.; Allinson, K.; Williams-Gray, C.H. Neuroinflammation and Protein Pathology in Parkinson’s Disease Dementia. Acta Neuropathol. Commun. 2020, 8, 1–19. [Google Scholar] [CrossRef]
- Picca, A.; Guerra, F.; Calvani, R.; Romano, R.; Josécoelho-Júnior, H.; Bucci, C.; Marzetti, E. Mitochondrial Dysfunction, Protein Misfolding and Neuroinflammation in Parkinson’s Disease: Roads to Biomarker Discovery. Biomolecules 2021, 11, 1508. [Google Scholar] [CrossRef]
- Roodveldt, C.; Labrador-Garrido, A.; Gonzalez-Rey, E.; Lachaud, C.C.; Guilliams, T.; Fernandez-Montesinos, R.; Benitez-Rondan, A.; Robledo, G.; Hmadcha, A.; Delgado, M.; et al. Preconditioning of Microglia by α-Synuclein Strongly Affects the Response Induced by Toll-like Receptor (TLR) Stimulation. PLoS ONE 2013, 8, e0079160. [Google Scholar] [CrossRef] [PubMed]
- Racette, B.A.; Gross, A.; Vouri, S.M.; Camacho-Soto, A.; Willis, A.W.; Searles Nielsen, S. Immunosuppressants and Risk of Parkinson Disease. Ann. Clin. Transl. Neurol. 2018, 5, 870–875. [Google Scholar] [CrossRef] [PubMed]
- Samii, A.; Etminan, M.; Wiens, M.O.; Jafari, S. NSAID Use and the Risk of Parkinson’s Disease: Systematic Review and Meta-Analysis of Observational Studies. Drugs Aging 2009, 26, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Alrouji, M.; Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Saad, H.M.; Batiha, G.E.S. A Story of the Potential Effect of Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) in Parkinson’s Disease: Beneficial or Detrimental Effects. Inflammopharmacology 2023, 31, 673–688. [Google Scholar] [CrossRef] [PubMed]
- Peterson, A.L.; Nutt, J.G. Treatment of Parkinson’s Disease with Trophic Factors. Neurotherapeutics 2008, 5, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, T.B.; Savall, A.S.; Gutierrez, M.E.Z.; Pinton, S. Neurotrophic Factors in Alzheimer’s and Parkinson’s Diseases: Implications for Pathogenesis and Therapy. Neural Regen. Res. 2017, 12, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Li, Y.; Jiao, Q.; Du, X.; Jiang, H. Cerebral Dopamine Neurotrophic Factor: A Potential Therapeutic Agent for Parkinson’s Disease. Neurosci. Bull. 2017, 33, 568. [Google Scholar] [CrossRef]
- De Lecea, L.; Criado, J.R.; Prospero-Garcia, Ó.; Gautvik, K.M.; Schweitzer, P.; Danielson, P.E.; Dunlop, C.L.M.; Siggins, G.R.; Henriksen, S.J.; Sutcliffe, J.G. A Cortical Neuropeptide with Neuronal Depressant and Sleep-Modulating Properties. Nature 1996, 381, 242–245. [Google Scholar] [CrossRef]
- Delgado, M.; Gonzalez-Rey, E. Role of Cortistatin in the Stressed Immune System. Front. Horm. Res. 2017, 48, 110–120. [Google Scholar] [CrossRef]
- Rauca, C.; Schäfer, K.; Höllt, V. Effects of Somatostatin, Octreotide and Cortistatin on Ischaemic Neuronal Damage Following Permanent Middle Cerebral Artery Occlusion in the Rat. Naunyn Schmiedebergs Arch. Pharmacol. 1999, 360, 633–638. [Google Scholar] [CrossRef]
- Vasilaki, A.; Lanneau, C.; Dournand, P.; De Lecea, L.; Gardette, R.; Epelbaum, J. Cortistatin Affects Glutamate Sensitivity in Mouse Hypothalamic Neurons through Activation of Sst2 Somatostatin Receptor Subtype. Neuroscience 1999, 88, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Braun, H.; Schulz, S.; Becker, A.; Schröder, H.; Höllt, V. Protective Effects of Cortistatin (CST-14) against Kainate-Induced Neurotoxicity in Rat Brain. Brain Res. 1998, 803, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.T.; Wen, L.L.; Pao, H.P.; Wang, J.Y. Cortistatin Is Induced in Brain Tissue and Exerts Neuroprotection in a Rat Model of Bacterial Meningoencephalitis. J. Infect. Dis. 2011, 204, 1563–1572. [Google Scholar] [CrossRef] [PubMed]
- Wen, Q.; Ding, Q.; Wang, J.; Yin, Y.; Xu, S.; Ju, Y.; Ji, H.; Liu, B. Cortistatin-14 Exerts Neuroprotective Effect Against Microglial Activation, Blood-Brain Barrier Disruption, and Cognitive Impairment in Sepsis-Associated Encephalopathy. J. Immunol. Res. 2022, 2022, 3334145. [Google Scholar] [CrossRef] [PubMed]
- Falo, C.P.; Benitez, R.; Caro, M.; Morell, M.; Forte-Lago, I.; Hernandez-Cortes, P.; Sanchez-Gonzalez, C.; O’valle, F.; Delgado, M.; Gonzalez-Rey, E. The Neuropeptide Cortistatin Alleviates Neuropathic Pain in Experimental Models of Peripheral Nerve Injury. Pharmaceutics 2021, 13, 947. [Google Scholar] [CrossRef] [PubMed]
- Souza-Moreira, L.; Morell, M.; Delgado-Maroto, V.; Pedreño, M.; Martinez-Escudero, L.; Caro, M.; O’Valle, F.; Luque, R.; Gallo, M.; de Lecea, L.; et al. Paradoxical Effect of Cortistatin Treatment and Its Deficiency on Experimental Autoimmune Encephalomyelitis. J. Immunol. 2013, 191, 2144–2154. [Google Scholar] [CrossRef]
- Carrasco, E.; Hernández, C.; de Torres, I.; Farrés, J.; Simó, R. Lowered Cortistatin Expression Is an Early Event in the Human Diabetic Retina and Is Associated with Apoptosis and Glial Activation. Mol. Vis. 2008, 14, 1496. [Google Scholar]
- Meredith, G.E.; Rademacher, D.J. MPTP Mouse Models of Parkinson’s Disease: An Update. J. Parkinsons Dis. 2011, 1, 19. [Google Scholar] [CrossRef]
- Taylor, T.N.; Greene, J.G.; Miller, G.W. Behavioral Phenotyping of Mouse Models of Parkinson’s Disease. Behav. Brain Res. 2010, 211, 1–10. [Google Scholar] [CrossRef]
- Fernagut, P.O.; Chalon, S.; Diguet, E.; Guilloteau, D.; Tison, F.; Jaber, M. Motor Behaviour Deficits and Their Histopathological and Functional Correlates in the Nigrostriatal System of Dopamine Transporter Knockout Mice. Neuroscience 2003, 116, 1123–1130. [Google Scholar] [CrossRef]
- Leem, Y.H.; Park, J.S.; Park, J.E.; Kim, D.Y.; Kim, H.S. Neurogenic Effects of Rotarod Walking Exercise in Subventricular Zone, Subgranular Zone, and Substantia Nigra in MPTP-Induced Parkinson’s Disease Mice. Sci. Rep. 2022, 12, 10544. [Google Scholar] [CrossRef] [PubMed]
- Daubner, S.C.; Le, T.; Wang, S. Tyrosine Hydroxylase and Regulation of Dopamine Synthesis. Arch. Biochem. Biophys. 2011, 508, 1. [Google Scholar] [CrossRef] [PubMed]
- Badanjak, K.; Fixemer, S.; Smajić, S.; Skupin, A.; Grünewald, A. The Contribution of Microglia to Neuroinflammation in Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 4676. [Google Scholar] [CrossRef] [PubMed]
- Kam, T.I.; Hinkle, J.T.; Dawson, T.M.; Dawson, V.L. Microglia and Astrocyte Dysfunction in Parkinson’s Disease. Neurobiol. Dis. 2020, 144, 105028. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, R.; Nisa Awan, M.U.; Bai, J. The Mechanism and Function of Glia in Parkinson’s Disease. Front. Cell Neurosci. 2022, 16, 903469. [Google Scholar] [CrossRef]
- Morrison, H.; Young, K.; Qureshi, M.; Rowe, R.K.; Lifshitz, J. Quantitative Microglia Analyses Reveal Diverse Morphologic Responses in the Rat Cortex after Diffuse Brain Injury. Sci. Rep. 2017, 7, 13211. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Sierra, A.; Stevens, B.; Tremblay, M.E.; Aguzzi, A.; Ajami, B.; Amit, I.; Audinat, E.; Bechmann, I.; Bennett, M.; et al. Microglia States and Nomenclature: A Field at Its Crossroads. Neuron 2022, 110, 3458–3483. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Arjona, M.d.M.; Grondona, J.M.; Granados-Durán, P.; Fernández-Llebrez, P.; López-Ávalos, M.D. Microglia Morphological Categorization in a Rat Model of Neuroinflammation by Hierarchical Cluster and Principal Components Analysis. Front. Cell Neurosci. 2017, 11, 235. [Google Scholar] [CrossRef]
- Fletcher, E.J.R.; Finlay, C.J.; Amor Lopez, A.; Crum, W.R.; Vernon, A.C.; Duty, S. Neuroanatomical and Microglial Alterations in the Striatum of Levodopa-Treated, Dyskinetic Hemi-Parkinsonian Rats. Front. Neurosci. 2020, 14, 567222. [Google Scholar] [CrossRef]
- Cappelletti, C.; Henriksen, S.P.; Geut, H.; Rozemuller, A.J.M.; van de Berg, W.D.J.; Pihlstrøm, L.; Toft, M. Transcriptomic Profiling of Parkinson’s Disease Brains Reveals Disease Stage Specific Gene Expression Changes. Acta Neuropathol. 2023, 146, 227–244. [Google Scholar] [CrossRef]
- Tipton, K.F.; Singer, T.P. Advances in Our Understanding of the Mechanisms of the Neurotoxicity of MPTP and Related Compounds. J. Neurochem. 1993, 61, 1191–1206. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.M.; Callahan, L.M.; Casaceli, C.; Chen, L.; Kiser, G.L.; Chui, B.; Kaysser-Kranich, T.M.; Sendera, T.J.; Palaniappan, C.; Federoff, H.J. Dysregulation of Gene Expression in the 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine-Lesioned Mouse Substantia Nigra. J. Neurosci. 2004, 24, 7445–7454. [Google Scholar] [CrossRef] [PubMed]
- Howells, D.W.; Porritt, M.J.; Wong, J.Y.F.; Batchelor, P.E.; Kalnins, R.; Hughes, A.J.; Donnan, G.A. Reduced BDNF MRNA Expression in the Parkinson’s Disease Substantia Nigra. Exp. Neurol. 2000, 166, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Kordower, J.H.; Emborg, M.E.; Bloch, J.; Ma, S.Y.; Chu, Y.; Leventhal, L.; McBride, J.; Chen, E.Y.; Palfi, S.; Roitberg, B.Z.; et al. Neurodegeneration Prevented by Lentiviral Vector Delivery of GDNF in Primate Models of Parkinson’s Disease. Science 2000, 290, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Palasz, E.; Wysocka, A.; Gasiorowska, A.; Chalimoniuk, M.; Niewiadomski, W.; Niewiadomska, G. BDNF as a Promising Therapeutic Agent in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 1170. [Google Scholar] [CrossRef] [PubMed]
- Whone, A.; Luz, M.; Boca, M.; Woolley, M.; Mooney, L.; Dharia, S.; Broadfoot, J.; Cronin, D.; Schroers, C.; Barua, N.U.; et al. Randomized Trial of Intermittent Intraputamenal Glial Cell Line-Derived Neurotrophic Factor in Parkinson’s Disease. Brain 2019, 142, 512–525. [Google Scholar] [CrossRef]
- Kordower, J.H.; Palfi, S.; Chen, E.-Y.; Ma, S.Y.; Sendera, T.; Cochran, E.J.; Mufson, E.J.; Penn, R.; Goetz, C.G.; Comella, C.D. Clinicopathological Findings Following Intraventricular Glial-derived Neurotrophic Factor Treatment in a Patient with Parkinson’s Disease. Ann. Neurol. 1999, 46, 219–224. [Google Scholar] [CrossRef]
- Maynard, K.R.; Kardian, A.; Hill, J.L.; Mai, Y.; Barry, B.; Hallock, H.L.; Jaffe, A.E.; Martinowich, K. TrkB Signaling Influences Gene Expression in Cortistatin-Expressing Interneurons. eNeuro 2020, 7, 1–14. [Google Scholar] [CrossRef]
- Jiang, J.H.; Peng, Y.L.; Liang, X.Y.; Li, S.; Chang, X.; Li, L.F.; Chang, M. Centrally Administered Cortistatin-14 Induces Antidepressant-like Effects in Mice via Mediating Ghrelin and GABAA Receptor Signaling Pathway. Front. Pharmacol. 2018, 9, 767. [Google Scholar] [CrossRef]
- Yasuda, Y.; Shimoda, T.; Uno, K.; Tateishi, N.; Furuya, S.; Yagi, K.; Suzuki, K.; Fujita, S. The Effects of MPTP on the Activation of Microglia/Astrocytes and Cytokine/Chemokine Levels in Different Mice Strains. J. Neuroimmunol. 2008, 204, 43–51. [Google Scholar] [CrossRef]
- Streit, W.J.; Conde, J.R.; Fendrick, S.E.; Flanary, B.E.; Mariani, C.L. Role of Microglia in the Central Nervous System’s Immune Response. Neurol. Res. 2005, 27, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Fujita, A.; Yamaguchi, H.; Yamasaki, R.; Cui, Y.; Matsuoka, Y.; Yamada, K.; Kira, J. Connexin 30 Deficiency Attenuates A2 Astrocyte Responses and Induces Severe Neurodegeneration in a 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Hydrochloride Parkinson’s Disease Animal Model. J. Neuroinflamm. 2018, 15, 227. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, J.M.; Schardien, K.; Wigdahl, B.; Nonnemacher, M.R. Roles of Neuropathology-Associated Reactive Astrocytes: A Systematic Review. Acta Neuropathol. Commun. 2023, 11, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Machado, V.; Zöller, T.; Attaai, A.; Spittau, B. Microglia-Mediated Neuroinflammation and Neurotrophic Factor-Induced Protection in the MPTP Mouse Model of Parkinson’s Disease-Lessons from Transgenic Mice. Int. J. Mol. Sci. 2016, 17, 151. [Google Scholar] [CrossRef]
- Liberatore, G.T.; Jackson-Lewis, V.; Vukosavic, S.; Mandir, A.S.; Vila, M.; Mcauliffe, W.G.; Dawson, V.L.; Dawson, T.M.; Przedborski, S. Inducible Nitric Oxide Synthase Stimulates Dopaminergic Neurodegeneration in the MPTP Model of Parkinson Disease. Nat. Med. 1999, 5, 1403–1409. [Google Scholar] [CrossRef]
- Wu, D.C.; Jackson-Lewis, V.; Vila, M.; Tieu, K.; Teismann, P.; Vadseth, C.; Choi, D.K.; Ischiropoulos, H.; Przedborski, S. Blockade of Microglial Activation Is Neuroprotective in the 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Mouse Model of Parkinson Disease. J. Neurosci. 2002, 22, 1763–1771. [Google Scholar] [CrossRef]
- Brochard, V.; Combadière, B.; Prigent, A.; Laouar, Y.; Perrin, A.; Beray-Berthat, V.; Bonduelle, O.; Alvarez-Fischer, D.; Callebert, J.; Launay, J.M.; et al. Infiltration of CD4+ Lymphocytes into the Brain Contributes to Neurodegeneration in a Mouse Model of Parkinson Disease. J. Clin. Investig. 2009, 119, 182–192. [Google Scholar] [CrossRef]
- Gosselin, D.; Skola, D.; Coufal, N.G.; Holtman, I.R.; Schlachetzki, J.C.M.; Sajti, E.; Jaeger, B.N.; O’Connor, C.; Fitzpatrick, C.; Pasillas, M.P.; et al. An Environment-Dependent Transcriptional Network Specifies Human Microglia Identity. Science 2017, 356, 1248–1259. [Google Scholar] [CrossRef]
- Surmeier, D.J.; Obeso, J.A.; Halliday, G.M. Selective Neuronal Vulnerability in Parkinson Disease. Nat. Rev. Neurosci. 2017, 18, 101–113. [Google Scholar] [CrossRef]
- Fricker, M.; Neher, J.J.; Zhao, J.W.; Théry, C.; Tolkovsky, A.M.; Brown, G.C. MFG-E8 Mediates Primary Phagocytosis of Viable Neurons during Neuroinflammation. J. Neurosci. 2012, 32, 2657–2666. [Google Scholar] [CrossRef]
- Neher, J.J.; Neniskyte, U.; Zhao, J.-W.; Bal-Price, A.; Tolkovsky, A.M.; Brown, G.C. Inhibition of Microglial Phagocytosis Is Sufficient to Prevent Inflammatory Neuronal Death. J. Immunol. 2011, 186, 4973–4983. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, Y.; Yoshikawa, E.; Sekine, Y.; Futatsubashi, M.; Kanno, T.; Ogusu, T.; Torizuka, T. Microglial Activation and Dopamine Terminal Loss in Early Parkinson’s Disease. Ann. Neurol. 2005, 57, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cao, L.; Song, Y.; Kang, Z.; Liu, T.; Ding, J.; Hu, G.; Lu, M. Mitochondrial Glutamine Transporter SLC1A5_var, a Potential Target to Suppress Astrocyte Reactivity in Parkinson’s Disease. Cell Death Dis. 2022, 13, 946. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ding, L.; Chen, H.; Zhang, M.; Ma, R.; Zheng, S.; Gong, J.; Zhang, Z.; Xu, H.; Xu, P.; et al. Cntnap4 Partial Deficiency Exacerbates α-Synuclein Pathology through Astrocyte–Microglia C3-C3aR Pathway. Cell Death Dis. 2023, 14, 285. [Google Scholar] [CrossRef] [PubMed]
- Khakh, B.S.; Deneen, B. The Emerging Nature of Astrocyte Diversity. Annu. Rev. Neurosci. 2019, 42, 187–207. [Google Scholar] [CrossRef] [PubMed]
- Chai, H.; Diaz-Castro, B.; Shigetomi, E.; Monte, E.; Octeau, J.C.; Yu, X.; Cohn, W.; Rajendran, P.S.; Vondriska, T.M.; Whitelegge, J.P.; et al. Neural Circuit-Specialized Astrocytes: Transcriptomic, Proteomic, Morphological, and Functional Evidence. Neuron 2017, 95, 531–549.e9. [Google Scholar] [CrossRef] [PubMed]
- Nagatsu, T.; Mogi, M.; Ichinose, H.; Togari, A. Changes in cytokines and neurotrophins in Parkinson’s Disease. Adv. Res. Neurodeger. 2000, 277–290. [Google Scholar] [CrossRef]
- Ferger, B.; Leng, A.; Mura, A.; Hengerer, B.; Feldon, J. Genetic Ablation of Tumor Necrosis Factor-Alpha (TNF-Alpha) and Pharmacological Inhibition of TNF-Synthesis Attenuates MPTP Toxicity in Mouse Striatum. J. Neurochem. 2004, 89, 822–833. [Google Scholar] [CrossRef]
- Sriram, K.; Matheson, J.M.; Benkovic, S.A.; Miller, D.B.; Luster, M.I.; O’Callaghan, J.P. Deficiency of TNF Receptors Suppresses Microglial Activation and Alters the Susceptibility of Brain Regions to MPTP-Induced Neurotoxicity: Role of TNF-Alpha. FASEB J. 2006, 20, 670–682. [Google Scholar] [CrossRef]
- Khan, M.M.; Zaheer, S.; Nehman, J.; Zaheer, A. Suppression of Glia Maturation Factor Expression Prevents 1-Methyl-4-Phenylpyridinium (MPP+)-Induced Loss of Mesencephalic Dopaminergic Neurons. Neuroscience 2014, 277, 196–205. [Google Scholar] [CrossRef]
- Parillaud, V.R.; Lornet, G.; Monnet, Y.; Privat, A.L.; Haddad, A.T.; Brochard, V.; Bekaert, A.; de Chanville, C.B.; Hirsch, E.C.; Combadière, C.; et al. Analysis of Monocyte Infiltration in MPTP Mice Reveals That Microglial CX3CR1 Protects against Neurotoxic Over-Induction of Monocyte-Attracting CCL2 by Astrocytes. J. Neuroinflamm. 2017, 14, 60. [Google Scholar] [CrossRef] [PubMed]
- Bolin, L.M.; Strycharska-Orczyk, I.; Murray, R.; Langston, J.W.; Di Monte, D. Increased Vulnerability of Dopaminergic Neurons in MPTP-Lesioned Interleukin-6 Deficient Mice. J. Neurochem. 2002, 83, 167–175. [Google Scholar] [CrossRef]
- Wang, L.; Wu, X.; Yang, G.; Hu, N.; Zhao, Z.; Zhao, L.; Li, S. Cannabidiol Alleviates the Damage to Dopaminergic Neurons in 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine-Induced Parkinson’s Disease Mice Via Regulating Neuronal Apoptosis and Neuroinflammation. Neuroscience 2022, 498, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Delgado, M.; Ganea, D. Neuroprotective Effect of Vasoactive Intestinal Peptide (VIP) in a Mouse Model of Parkinson’s Disease by Blocking Microglial Activation. FASEB J. 2003, 17, 944–946. [Google Scholar] [CrossRef] [PubMed]
- Liu, B. Modulation of Microglial Pro-Inflammatory and Neurotoxic Activity for the Treatment of Parkinson’s Disease. AAPS J. 2006, 8, E606. [Google Scholar] [CrossRef] [PubMed]
- Moon, M.; Kim, H.G.; Hwang, L.; Seo, J.H.; Kim, S.; Hwang, S.; Kim, S.; Lee, D.; Chung, H.; Oh, M.S.; et al. Neuroprotective Effect of Ghrelin in the 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Mouse Model of Parkinson’s Disease by Blocking Microglial Activation. Neurotox. Res. 2009, 15, 332–347. [Google Scholar] [CrossRef] [PubMed]
- Bayliss, J.A.; Lemus, M.; Santos, V.V.; Deo, M.; Elsworth, J.D.; Andrews, Z.B. Acylated but Not Des-Acyl Ghrelin Is Neuroprotective in an MPTP Mouse Model of Parkinson’s Disease. J. Neurochem. 2016, 137, 460–471. [Google Scholar] [CrossRef]
- Gerhard, A.; Pavese, N.; Hotton, G.; Turkheimer, F.; Es, M.; Hammers, A.; Eggert, K.; Oertel, W.; Banati, R.B.; Brooks, D.J. In Vivo Imaging of Microglial Activation with [11C](R)-PK11195 PET in Idiopathic Parkinson’s Disease. Neurobiol. Dis. 2006, 21, 404–412. [Google Scholar] [CrossRef]
- Dias, V.; Junn, E.; Mouradian, M.M. The Role of Oxidative Stress in Parkinson’s Disease. J. Parkinsons Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef]
- Sjöstedt, E.; Zhong, W.; Fagerberg, L.; Karlsson, M.; Mitsios, N.; Adori, C.; Oksvold, P.; Edfors, F.; Limiszewska, A.; Hikmet, F.; et al. An Atlas of the Protein-Coding Genes in the Human, Pig, and Mouse Brain. Science 2020, 367, eaay5947. [Google Scholar] [CrossRef]
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ (accessed on 14 October 2023).
- Brain Map—Brain-Map.Org. Available online: https://portal.brain-map.org/ (accessed on 14 October 2023).
- Dong, J.; Song, N.; Xie, J.; Jiang, H. Ghrelin Antagonized 1-Methyl-4-Phenylpyridinium (MPP(+))-Induced Apoptosis in MES23.5 Cells. J. Mol. Neurosci. 2009, 37, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Li, L.J.; Wang, J.; Xie, J.X. Ghrelin Antagonizes MPTP-Induced Neurotoxicity to the Dopaminergic Neurons in Mouse Substantia Nigra. Exp. Neurol. 2008, 212, 532–537. [Google Scholar] [CrossRef] [PubMed]
- Dalm, V.A.; Van Hagen, P.M.; Van Koetsveld, P.M.; Langerak, A.W.; Van Der Lely, A.J.; Lamberts, S.W.; Hofland, L.J. Cortistatin Rather than Somatostatin as a Potential Endogenous Ligand for Somatostatin Receptors in the Human Immune System. J. Clin. Endocrinol. Metab. 2003, 88, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Gahete, M.D.; Rubio, A.; Durán-Prado, M.; Avila, J.; Luque, R.M.; Castaño, J.P. Expression of Somatostatin, cortistatin, and their receptors, as well as dopamine receptors, but not of neprilysin, are reduced in the temporal lobe of Alzheimer’s disease patients. J. Alzheimers Dis. 2010, 20, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Castillo-González, J.; Ruiz, J.L.; Serrano-Martínez, I.; Forte-Lago, I.; Ubago-Rodriguez, A.; Caro, M.; Pérez-Gómez, J.M.; Benítez-Troncoso, A.; Andrés-León, E.; Sánchez-Navarro, M.; et al. Cortistatin Deficiency Reveals a Dysfunctional Brain Endothelium with Impaired Gene Pathways, Exacerbated Immune Activation, and Disrupted Barrier Integrity. J. Neuroinflamm. 2023, 20, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Morell, M.; Camprubí-Robles, M.; Culler, M.D.; de Lecea, L.; Delgado, M. Cortistatin Attenuates Inflammatory Pain via Spinal and Peripheral Actions. Neurobiol. Dis. 2014, 63, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Percie du Sert, N.; Hurst, V.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; et al. The ARRIVE Guidelines 2.0: Updated Guidelines for Reporting Animal Research. Br. J. Pharmacol. 2020, 177, 3617. [Google Scholar] [CrossRef] [PubMed]
- Paxinos, G.; Franklin, K.B.J. Paxinos and Franklin’s the Mouse Brain in Stereotaxic Coordinates, 2nd ed.; Academic Press: San Diego, CA, USA, 2001; ISBN 0125476361. [Google Scholar]
- West, M.J. New Stereological Methods for Counting Neurons. Neurobiol. Aging 1993, 14, 275–285. [Google Scholar] [CrossRef]
- Muñoz-Manchado, A.B.; Villadiego, J.; Romo-Madero, S.; Suárez-Luna, N.; Bermejo-Navas, A.; Rodríguez-Gõmez, J.A.; Garrido-Gil, P.; Labandeira-García, J.L.; Echevarría, M.; Lõpez-Barneo, J.; et al. Chronic and Progressive Parkinson’s Disease MPTP Model in Adult and Aged Mice. J. Neurochem. 2016, 136, 373–387. [Google Scholar] [CrossRef]
- Villadiego, J.; Muñoz-Manchado, A.B.; Sobrino, V.; Bonilla-Henao, V.; Suárez-Luna, N.; Ortega-Sáenz, P.; Pardal, R.; López-Barneo, J.; Toledo-Aral, J.J. Protection and Repair of the Nigrostriatal Pathway with Stem-Cell-Derived Carotid Body Glomus Cell Transplants in Chronic MPTP Parkinsonian Model. Int. J. Mol. Sci. 2023, 24, 5575. [Google Scholar] [CrossRef]
- Young, K.; Morrison, H. Quantifying Microglia Morphology from Photomicrographs of Immunohistochemistry Prepared Tissue Using ImageJ. J. Vis. Exp. 2018, 136, e57648. [Google Scholar] [CrossRef]
- Doube, M.; Klosowski, M.M.; Arganda-Carreras, I.; Cordelières, F.P.; Dougherty, R.P.; Jackson, J.S.; Schmid, B.; Hutchinson, J.R.; Shefelbine, S.J. BoneJ: Free and Extensible Bone Image Analysis in ImageJ. Bone 2010, 47, 1076–1079. [Google Scholar] [CrossRef] [PubMed]
- Arganda-Carreras, I.; Fernández-González, R.; Muñoz-Barrutia, A.; Ortiz-De-Solorzano, C. 3D Reconstruction of Histological Sections: Application to Mammary Gland Tissue. Microsc. Res. Tech. 2010, 73, 1019–1029. [Google Scholar] [CrossRef] [PubMed]
- Karperien, A.; Ahammer, H.; Jelinek, H.F. Quantitating the Subtleties of Microglial Morphology with Fractal Analysis. Front. Cell Neurosci. 2013, 7, 1–34. [Google Scholar] [CrossRef]
- Gaven, F.; Marin, P.; Claeysen, S. Primary Culture of Mouse Dopaminergic Neurons. J. Vis. Exp. 2014, 91, e51751. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serrano-Martínez, I.; Pedreño, M.; Castillo-González, J.; Ferraz-de-Paula, V.; Vargas-Rodríguez, P.; Forte-Lago, I.; Caro, M.; Campos-Salinas, J.; Villadiego, J.; Peñalver, P.; et al. Cortistatin as a Novel Multimodal Therapy for the Treatment of Parkinson’s Disease. Int. J. Mol. Sci. 2024, 25, 694. https://doi.org/10.3390/ijms25020694
Serrano-Martínez I, Pedreño M, Castillo-González J, Ferraz-de-Paula V, Vargas-Rodríguez P, Forte-Lago I, Caro M, Campos-Salinas J, Villadiego J, Peñalver P, et al. Cortistatin as a Novel Multimodal Therapy for the Treatment of Parkinson’s Disease. International Journal of Molecular Sciences. 2024; 25(2):694. https://doi.org/10.3390/ijms25020694
Chicago/Turabian StyleSerrano-Martínez, Ignacio, Marta Pedreño, Julia Castillo-González, Viviane Ferraz-de-Paula, Pablo Vargas-Rodríguez, Irene Forte-Lago, Marta Caro, Jenny Campos-Salinas, Javier Villadiego, Pablo Peñalver, and et al. 2024. "Cortistatin as a Novel Multimodal Therapy for the Treatment of Parkinson’s Disease" International Journal of Molecular Sciences 25, no. 2: 694. https://doi.org/10.3390/ijms25020694