
Leveraging SARS-CoV-2 Main Protease (Mpro) for COVID-19 Mitigation with Selenium-Based Inhibitors
 , ,
, ,  ,
,  , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
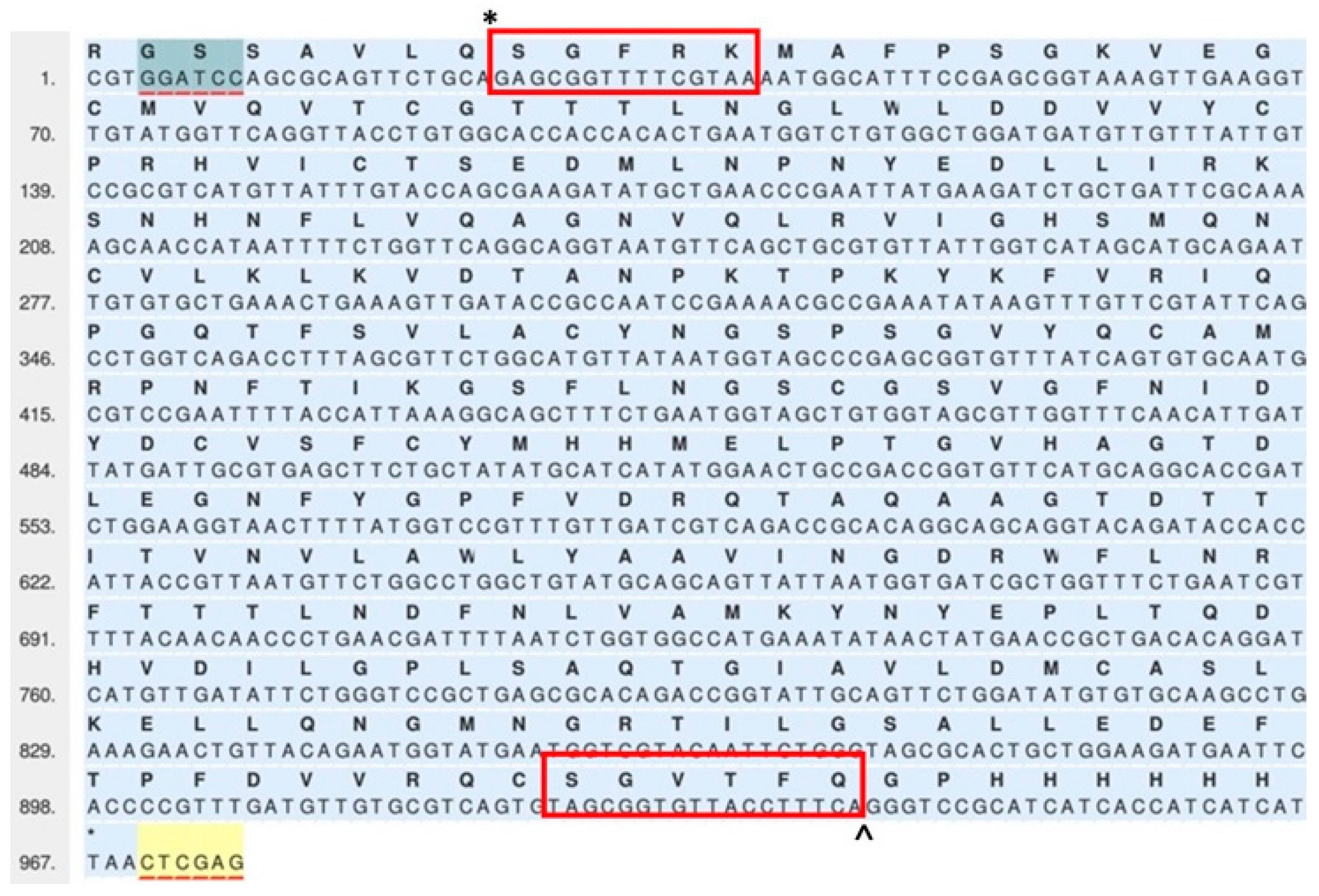
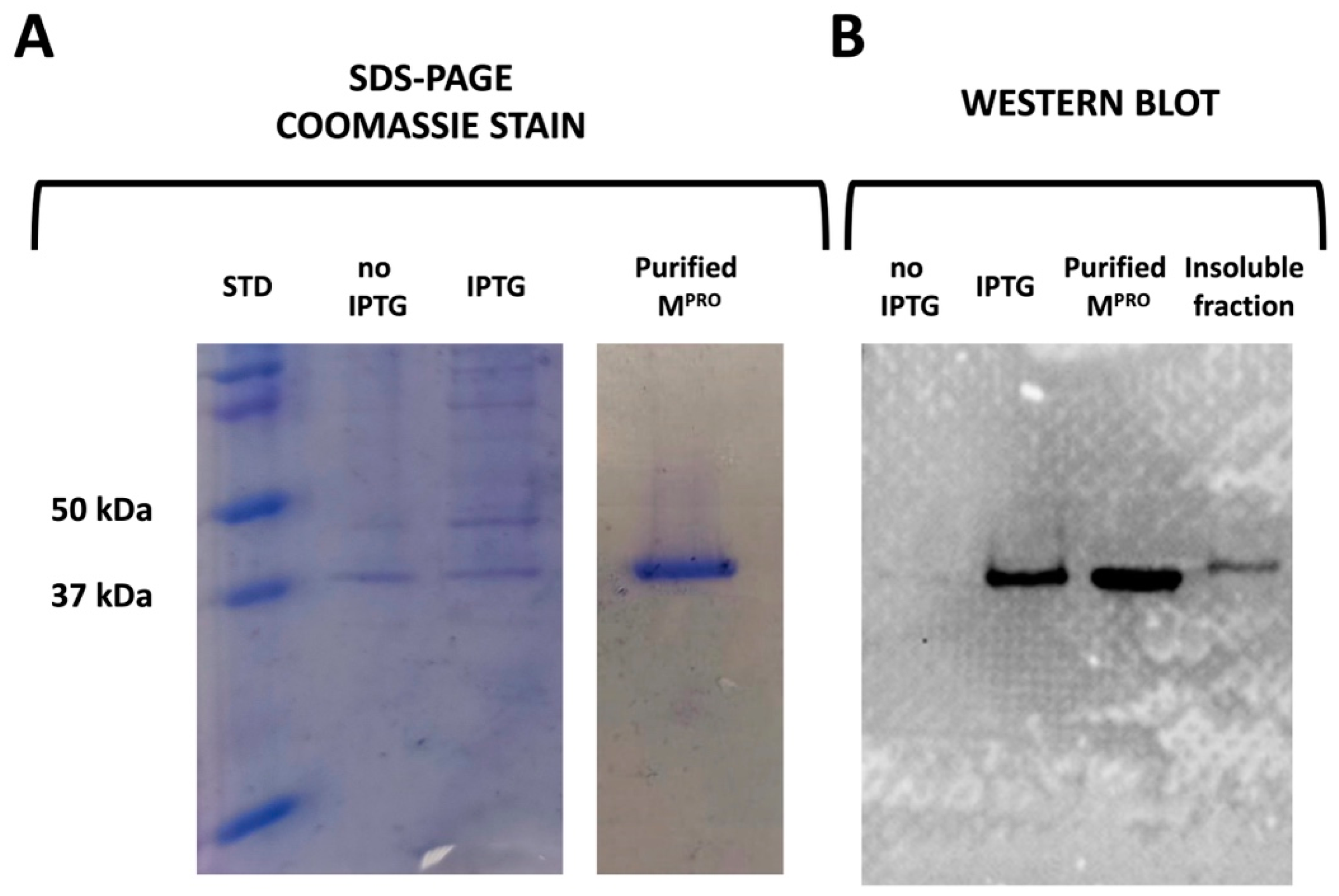
2.1. Cloning, Expression, and Purification
2.2. Determination of the Mpro Activity Using the Spectrophotometric Method
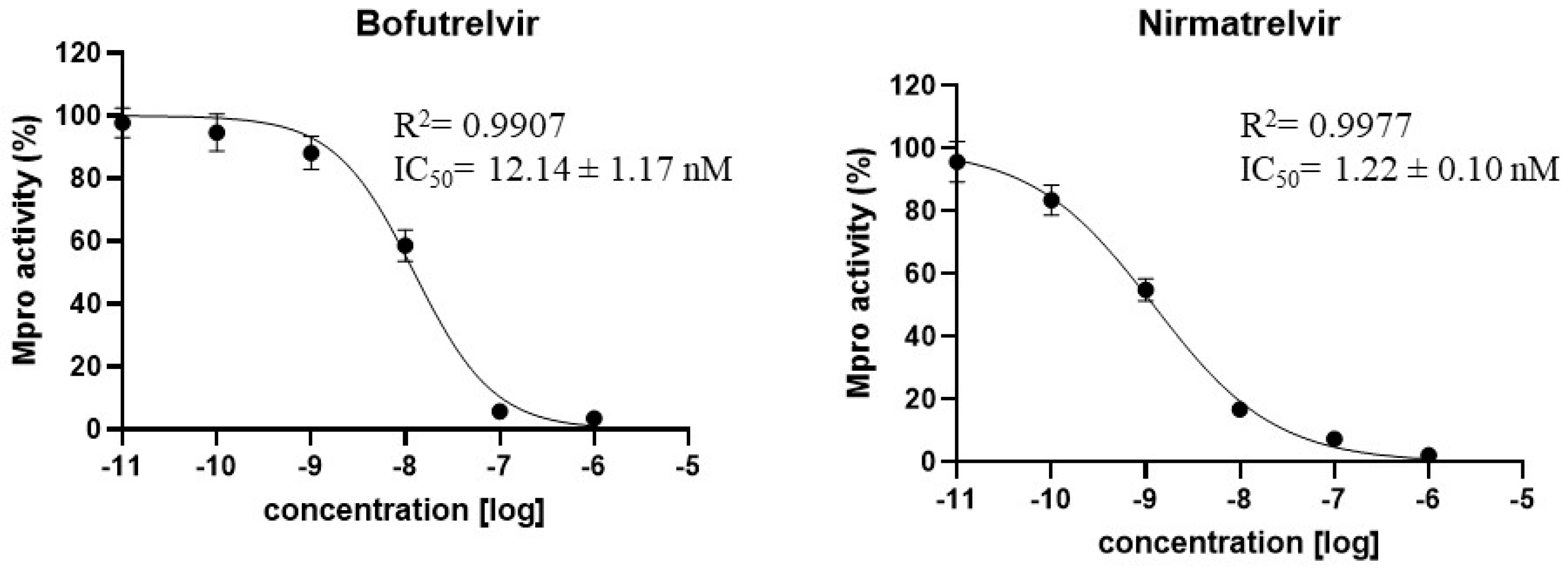
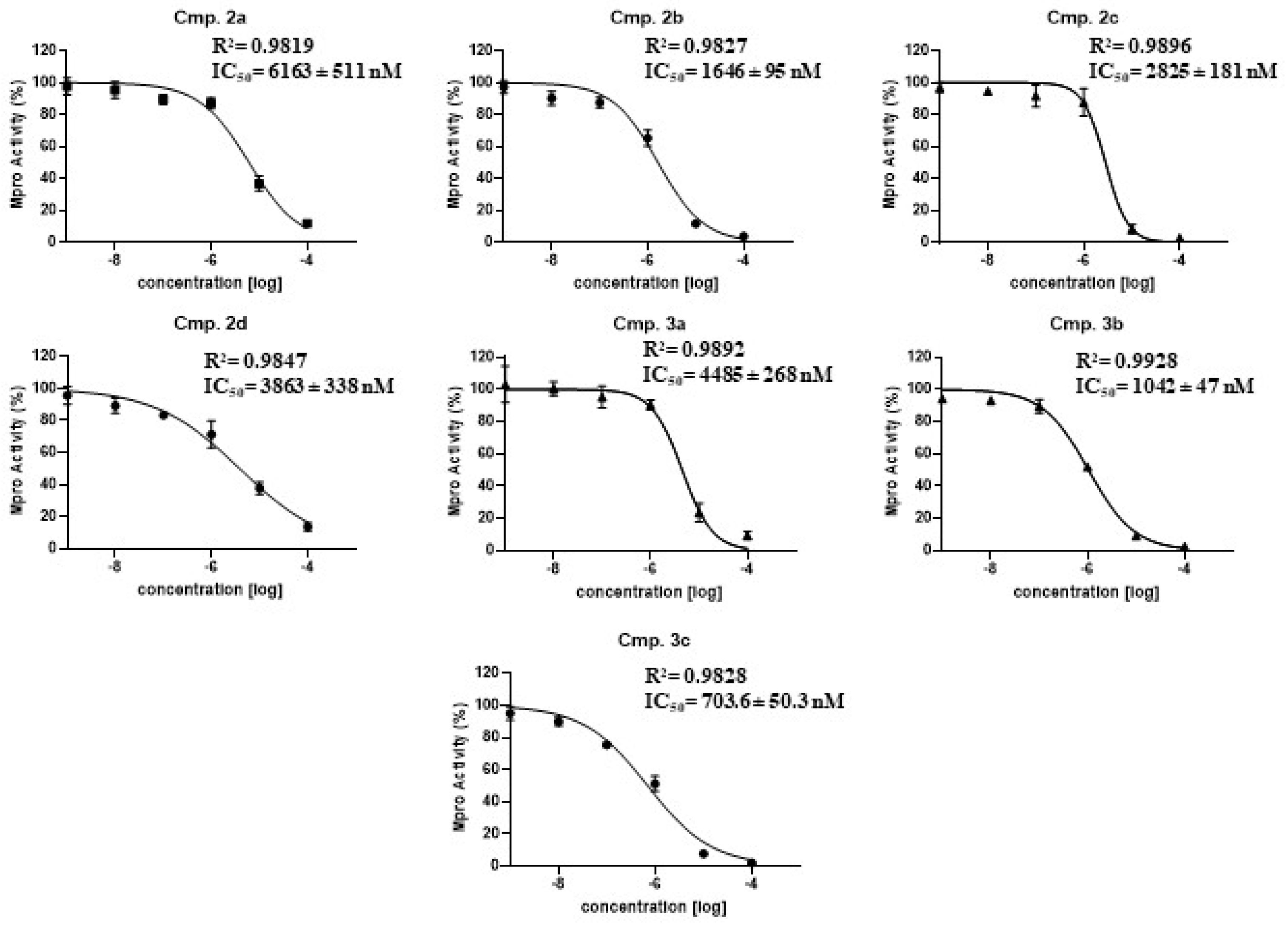
2.3. Determination of the Mpro Half Maximal Inhibitory Concentrations (IC50)
2.4. Computational Study
3. Materials and Methods
3.1. Construction of Mpro Expression Vector
3.2. Mpro Expression and Purification
3.3. SDS-Page and Western Blot
3.4. Enzymatic Protease Assay
3.5. Chemistry
3.5.1. General
3.5.2. General Procedure for the Synthesis of Selenolesters (2)
3.5.3. Synthesis of Se-Phenethyl Benzoselenoate (2a)
3.5.4. Synthesis of Se-(2-Hydroxycyclohexyl) Benzoselenoate (2c)
3.5.5. Synthesis of Se-(4-Sulfamoylbenzyl) (3,5-Dimethylphenyl) Carbamoselenoate (3c)
3.6. Computational Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Markov, P.V.; Ghafari, M.; Beer, M.; Lythgoe, K.; Simmonds, P.; Stilianakis, N.I.; Katzourakis, A. The evolution of SARS-CoV-2. Nat. Rev. Microbiol. 2023, 21, 361–379. [Google Scholar] [CrossRef]
- Li, Y.; Niu, Y. A commentary on “The socio-economic implications of the coronavirus pandemic (COVID-19): A review”. Int. J. Surg. 2021, 95, 106048. [Google Scholar] [CrossRef] [PubMed]
- Sardar, S.; Abdul-Khaliq, I.; Ingar, A.; Amaidia, H.; Mansour, N. ‘COVID-19 lockdown: A protective measure or exacerbator of health inequalities? A comparison between the United Kingdom and India.’ a commentary on “the socio-economic implications of the coronavirus and COVID-19 pandemic: A review”. Int. J. Surg. 2020, 83, 189–191. [Google Scholar] [CrossRef]
- Scherf, M.; Matschke, X.; Rieger, M.O. Stock market reactions to COVID-19 lockdown: A global analysis. Financ. Res. Lett. 2022, 45, 102245. [Google Scholar] [CrossRef]
- Badiani, A.A.; Patel, J.A.; Ziolkowski, K.; Nielsen, F.B.H. Pfizer: The miracle vaccine for COVID-19? Public Health Pract. 2020, 1, 100061. [Google Scholar] [CrossRef] [PubMed]
- Carabelli, A.M.; Peacock, T.P.; Thorne, L.G.; Harvey, W.T.; Hughes, J.; Consortium, C.-G.U.; Peacock, S.J.; Barclay, W.S.; de Silva, T.I.; Towers, G.J.; et al. SARS-CoV-2 variant biology: Immune escape, transmission and fitness. Nat. Rev. Microbiol. 2023, 21, 162–177. [Google Scholar] [CrossRef]
- Li, M.; Wang, H.; Tian, L.; Pang, Z.; Yang, Q.; Huang, T.; Fan, J.; Song, L.; Tong, Y.; Fan, H. COVID-19 vaccine development: Milestones, lessons and prospects. Signal Transduct. Target. Ther. 2022, 7, 146–177. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Mishevich, J.L.; Mesecar, A.; Mitsuya, H. Recent Drug Development and Medicinal Chemistry Approaches for the Treatment of SARS-CoV-2 Infection and COVID-19. ChemMedChem 2022, 17, e202200440. [Google Scholar] [CrossRef]
- Lui, G.; Guaraldi, G. Drug treatment of COVID-19 infection. Curr. Opin. Pulm. Med. 2023, 29, 174–183. [Google Scholar] [CrossRef]
- Ravaghi, H.; Naidoo, V.; Mataria, A.; Khalil, M. Hospitals early challenges and interventions combatting COVID-19 in the Eastern Mediterranean Region. PLoS ONE 2022, 17, e0268386. [Google Scholar] [CrossRef]
- Sharun, K.; Tiwari, R.; Dhama, J.; Dhama, K. Dexamethasone to combat cytokine storm in COVID-19: Clinical trials and preliminary evidence. Int. J. Surg. 2020, 82, 179–181. [Google Scholar] [CrossRef]
- da Silva, S.J.R.; do Nascimento, J.C.F.; Mendes, R.P.G.; Guarines, K.M.; da Silva, C.T.A.; da Silva, P.G.; de Magalhaes, J.J.F.; Vigar, J.R.J.; Silva, A.; Kohl, A.; et al. Two Years into the COVID-19 Pandemic: Lessons Learned. ACS Infect. Dis. 2020, 8, 1758–1814. [Google Scholar] [CrossRef] [PubMed]
- Debnath, S.; Agarwal, A.; Kumar, N.R.; Bedi, A. Selenium-Based Drug Development for Antioxidant and Anticancer Activity. Future Pharmacol. 2022, 2, 595–607. [Google Scholar] [CrossRef]
- Ali, W.; Benedetti, R.; Handzlik, J.; Zwergel, C.; Battistelli, C. The innovative potential of seleniumcontaining agents for fighting cancer and viral infections. Drug Discov. Today 2021, 26, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Dong, H.; Zhang, X.; Wang, Y.; Su, L.; Xu, H. Selenium as an emerging versatile player in heterocycles and natural products modification. Drug Discov. Today 2022, 27, 2268–2277. [Google Scholar] [CrossRef]
- Angeli, A.; Ferraroni, M.; Capperucci, A.; Tanini, D.; Costantino, G.; Supuran, C.T. Selenocarbamates As a Prodrug-Based Approach to Carbonic Anhydrase Inhibition. ChemMedChem 2022, 17, e202200085. [Google Scholar] [CrossRef]
- Tanini, D.; Carradori, S.; Capperucci, A.; Lupori, L.; Zara, S.; Ferraroni, M.; Ghelardini, C.; Di Cesare Mannelli, L.; Micheli, L.; Lucarini, E.; et al. Chalcogenides-incorporating carbonic anhydrase inhibitors concomitantly reverted oxaliplatin-induced neuropathy and enhanced antiproliferative action. Eur. J. Med. Chem. 2021, 225, 113793. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Ma, C.; Hu, Y.; Townsend, J.A.; Lagarias, P.I.; Marty, M.T.; Kolocouris, A.; Wang, J. Ebselen, Disulfiram, Carmofur, PX-12, Tideglusib, and Shikonin Are Nonspecific Promiscuous SARS-CoV-2 Main Protease Inhibitors. ACS Pharmacol. Transl. Sci. 2020, 3, 1265–1277. [Google Scholar] [CrossRef]
- Amporndanai, K.; Meng, X.; Shang, W.; Jin, Z.; Rogers, M.; Zhao, Y.; Rao, Z.; Liu, Z.J.; Yang, H.; Zhang, L.; et al. Inhibition mechanism of SARS-CoV-2 main protease by ebselen and its derivatives. Nat. Commun. 2021, 12, 3061. [Google Scholar] [CrossRef]
- Kovacs, T.; Kurtan, K.; Varga, Z.; Nagy, P.; Panyi, G.; Zakany, F. Veklury(R) (remdesivir) formulations inhibit initial membrane-coupled events of SARS-CoV-2 infection due to their sulfobutylether-beta-cyclodextrin content. Br. J. Pharmacol. 2023, 180, 2064–2084. [Google Scholar] [CrossRef] [PubMed]
- Pilote, S.; Simard, C.; Drolet, B. Remdesivir (VEKLURY) for Treating COVID-19: Guinea Pig Ex Vivo and In Vivo Cardiac Electrophysiological Effects. J. Cardiovasc. Pharmacol. 2022, 80, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Kelley, B.; De Moor, P.; Douglas, K.; Renshaw, T.; Traviglia, S. Monoclonal antibody therapies for COVID-19: Lessons learned and implications for the development of future products. Curr. Opin. Biotechnol. 2022, 78, 102798–102805. [Google Scholar] [CrossRef] [PubMed]
- Ravi, G.; Eerike, M.; Konda, V.R.; Bisoi, D.; Raj, G.M.; Priyadarshini, R.; Mali, K.R.; Chaliserry, L.F. Efficacy and Safety of Anti-SARS-CoV-2 Monoclonal Antibodies: An Updated Review. Monoclon. Antibodies Immunodiagn. Immunother. 2023, 42, 77–94. [Google Scholar] [CrossRef]
- Harris, E. Nasally Administered Monoclonal Antibody for COVID-19. JAMA-J. Am. Med. Assoc. 2023, 329, 1142–1152. [Google Scholar] [CrossRef] [PubMed]
- Kip, K.E.; McCreary, E.K.; Collins, K.; Minnier, T.E.; Snyder, G.M.; Garrard, W.; McKibben, J.C.; Yealy, D.M.; Seymour, C.W.; Huang, D.; et al. Evolving Real-World Effectiveness of Monoclonal Antibodies for Treatment of COVID-19 A Cohort Study. Ann. Intern. Med. 2023, 176, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Wynter-Adams, D.; Thomas-Brown, P. COVID-19 vaccine hesitancy in a developing country: Prevalence, explanatory factors and implications for the future. Public Health 2023, 217, 146–154. [Google Scholar] [CrossRef]
- Iacobucci, G. COVID-19: Evusheld protects the most vulnerable patients, analysis shows. BMJ-Br. Med. J. 2022, 379, o2690. [Google Scholar] [CrossRef]
- Niknam, Z.; Jafari, A.; Golchin, A.; Danesh Pouya, F.; Nemati, M.; Rezaei-Tavirani, M.; Rasmi, Y. Potential therapeutic options for COVID-19: An update on current evidence. Eur. J. Med. Res. 2022, 27, 6–20. [Google Scholar] [CrossRef]
- Ng, T.I.; Correia, I.; Seagal, J.; DeGoey, D.A.; Schrimpf, M.R.; Hardee, D.J.; Noey, E.L.; Kati, W.M. Antiviral Drug Discovery for the Treatment of COVID-19 Infections. Viruses 2022, 14, 961. [Google Scholar] [CrossRef]
- Kabinger, F.; Stiller, C.; Schmitzova, J.; Dienemann, C.; Kokic, G.; Hillen, H.S.; Hobartner, C.; Cramer, P. Mechanism of molnupiravir-induced SARS-CoV-2 mutagenesis. Nat. Struct. Mol. Biol. 2021, 28, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Kamal, L.; Ramadan, A.; Farraj, S.; Bahig, L.; Ezzat, S. The pill of recovery; Molnupiravir for treatment of COVID-19 patients; a systematic review. Saudi Pharm. J. 2022, 30, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Buxeraud, J.; Faure, S.; Fougere, E. Nirmatrelvir/ritonavir (Paxlovid(R)), a treatment for COVID-19. Actual. Pharm. 2022, 61, 10–12. [Google Scholar] [PubMed]
- Yang, D.W.; Ju, M.J.; Wang, H.; Jia, Y.C.; Wang, X.D.; Fang, H.; Fan, J. Proxalutamide for the treatment of COVID-19 rebound following Paxlovid treatment: Report of four cases and review of the literature. J. Clin. Lab. Anal. 2023, 37, e24880. [Google Scholar] [CrossRef]
- Chavda, V.P.; Vuppu, S.; Mishra, T.; Kamaraj, S.; Patel, A.B.; Sharma, N.; Chen, Z.S. Recent review of COVID-19 management: Diagnosis, treatment and vaccination. Pharmacol. Rep. 2022, 74, 1120–1148. [Google Scholar] [CrossRef]
- Hashemian, S.M.R.; Sheida, A.; Taghizadieh, M.; Memar, M.Y.; Hamblin, M.R.; Bannazadeh Baghi, H.; Sadri Nahand, J.; Asemi, Z.; Mirzaei, H. Paxlovid (Nirmatrelvir/Ritonavir): A new approach to COVID-19 therapy? Biomed. Pharmacother. 2023, 162, 114367. [Google Scholar] [CrossRef]
- Eltayb, W.A.; Abdalla, M.; Rabie, A.M. Novel Investigational Anti-SARS-CoV-2 Agent Ensitrelvir “S-217622”: A Very Promising Potential Universal Broad-Spectrum Antiviral at the Therapeutic Frontline of Coronavirus Species. ACS Omega 2023, 8, 5234–5246. [Google Scholar] [CrossRef]
- Yadav, R.; Chaudhary, J.K.; Jain, N.; Chaudhary, P.K.; Khanra, S.; Dhamija, P.; Sharma, A.; Kumar, A.; Handu, S. Role of Structural and Non-Structural Proteins and Therapeutic Targets of SARS-CoV-2 for COVID-19. Cells 2021, 10, 821. [Google Scholar] [CrossRef]
- Mori, M.; Capasso, C.; Carta, F.; Donald, W.A.; Supuran, C.T. A deadly spillover: SARS-CoV-2 outbreak. Expert Opin. Ther. Pat. 2020, 30, 481–485. [Google Scholar] [CrossRef]
- Capasso, C.; Nocentini, A.; Supuran, C.T. Protease inhibitors targeting the main protease and papain-like protease of coronaviruses. Expert Opin. Ther. Pat. 2021, 31, 309–324. [Google Scholar] [CrossRef]
- Deniz, S.; Uysal, T.K.; Capasso, C.; Supuran, C.T.; Ozensoy Guler, O. Is carbonic anhydrase inhibition useful as a complementary therapy of COVID-19 infection? J. Enzym. Inhib. Med. Chem. 2021, 36, 1230–1235. [Google Scholar] [CrossRef] [PubMed]
- Citarella, A.; Scala, A.; Piperno, A.; Micale, N. SARS-CoV-2 Mpro: A Potential Target for Peptidomimetics and Small-Molecule Inhibitors. Biomolecules 2021, 11, 607. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.M.; Samant, N.; Schneider-Nachum, G.; Barkan, D.T.; Yilmaz, N.K.; Schiffer, C.A.; Moquin, S.A.; Dovala, D.; Bolon, D.N.A. Comprehensive fitness landscape of SARS-CoV-2 Mpro reveals insights into viral resistance mechanisms. eLife 2022, 11, e77433. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Xiong, Y.; Zhu, G.H.; Zhang, Y.N.; Zhang, Y.W.; Huang, P.; Ge, G.B. The SARS-CoV-2 main protease (Mpro): Structure, function, and emerging therapies for COVID-19. MedComm (2020) 2022, 3, e151. [Google Scholar] [CrossRef] [PubMed]
- Mengist, H.M.; Dilnessa, T.; Jin, T. Structural Basis of Potential Inhibitors Targeting SARS-CoV-2 Main Protease. Front. Chem. 2021, 9, 622898–622916. [Google Scholar] [CrossRef] [PubMed]
- Waugh, D.S. An overview of enzymatic reagents for the removal of affinity tags. Protein Expr. Purif. 2011, 80, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.L.; Lin, D.Z.; Sun, X.Y.Y.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Razali, R.; Subbiah, V.K.; Budiman, C. Technical Data of Heterologous Expression and Purification of SARS-CoV-2 Proteases Using Escherichia coli System. Data 2021, 6, 99. [Google Scholar] [CrossRef]
- Sacco, M.D.; Ma, C.; Lagarias, P.; Gao, A.; Townsend, J.A.; Meng, X.; Dube, P.; Zhang, X.; Hu, Y.; Kitamura, N.; et al. Structure and inhibition of the SARS-CoV-2 main protease reveal strategy for developing dual inhibitors against Mpro and cathepsin L. Sci. Adv. 2020, 6, eabe0751. [Google Scholar] [CrossRef]
- Bhardwaj, V.K.; Singh, R.; Das, P.; Purohit, R. Evaluation of acridinedione analogs as potential SARS-CoV-2 main protease inhibitors and their comparison with repurposed anti-viral drugs. Comput. Biol. Med. 2021, 128, 104117–104128. [Google Scholar] [CrossRef]
- Ullrich, S.; Nitsche, C. The SARS-CoV-2 main protease as drug target. Bioorganic Med. Chem. Lett. 2020, 30, 127377–127384. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Fang, C.; Zhang, Q.; Zhang, R.; Zhao, X.; Duan, Y.; Wang, H.; Zhu, Y.; Feng, L.; Zhao, J.; et al. Crystal structure of SARS-CoV-2 main protease in complex with protease inhibitor PF-07321332. Protein Cell 2022, 13, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Shao, Y.; Peng, X.; Liang, B.; Xu, J.; Xing, D. Review of preclinical data of PF-07304814 and its active metabolite derivatives against SARS-CoV-2 infection. Front. Pharmacol. 2022, 13, 1035969–1035978. [Google Scholar] [CrossRef]
- Jiang, H.; Zou, X.; Zeng, P.; Zeng, X.; Zhou, X.; Wang, J.; Zhang, J.; Li, J. Crystal structures of main protease (Mpro) mutants of SARS-CoV-2 variants bound to PF-07304814. Mol. Biomed. 2023, 4, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lin, C.; Zhou, X.; Zhong, F.; Zeng, P.; McCormick, P.J.; Jiang, H.; Zhang, J. Structural Basis of Main Proteases of Coronavirus Bound to Drug Candidate PF-07304814. J. Mol. Biol. 2022, 434, 167706–167716. [Google Scholar] [CrossRef] [PubMed]
- Karniadakis, I.; Mazonakis, N.; Tsioutis, C.; Papadakis, M.; Markaki, I.; Spernovasilis, N. Oral Molnupiravir and Nirmatrelvir/Ritonavir for the Treatment of COVID-19: A Literature Review with a Focus on Real-World Evidence. Infect. Dis. Rep. 2023, 15, 662–678. [Google Scholar] [CrossRef]
- Liu, T.H.; Wu, J.Y.; Huang, P.Y.; Hsu, W.H.; Chuang, M.H.; Tsai, Y.W.; Lai, C.C.; Huang, C.Y. Clinical efficacy of nirmatrelvir plus ritonavir in patients with COVID-19 and preexisting cardiovascular diseases. Expert Rev. Anti-Infect. Ther. 2023, 1–8. [Google Scholar] [CrossRef]
- Tanini, D.; Capperucci, A. Synthesis and Applications of Organic Selenols. Adv. Synth. Catal. 2021, 363, 5360–5385. [Google Scholar] [CrossRef]
- Tanini, D.; Scarpelli, S.; Ermini, E.; Capperucci, A. Seleno-Michael Reaction of Stable Functionalised Alkyl Selenols: A Versatile Tool for the Synthesis of Acyclic and Cyclic Unsymmetrical Alkyl and Vinyl Selenides. Adv. Synth. Catal. 2019, 361, 2337–2346. [Google Scholar] [CrossRef]
- Tanini, D.; Tiberi, C.; Gellini, C.; Salvi, P.R.; Capperucci, A. A Straightforward Access to Stable β-Functionalized Alkyl Selenols. Adv. Synth. Catal. 2018, 360, 3367–3375. [Google Scholar] [CrossRef]
- Angeli, A.; Tanini, D.; Nocentini, A.; Capperucci, A.; Ferraroni, M.; Gratteri, P.; Supuran, C.T. Selenols: A new class of carbonic anhydrase inhibitors. Chem. Commun. 2019, 55, 648–651. [Google Scholar] [CrossRef]
- Angeli, A.; Carta, F.; Donnini, S.; Capperucci, A.; Ferraroni, M.; Tanini, D.; Supuran, C.T. Selenolesterase enzyme activity of carbonic anhydrases. Chem. Commun. 2020, 56, 4444–4447. [Google Scholar] [CrossRef]
- Capperucci, A.; Petrucci, A.; Faggi, C.; Tanini, D. Click Reaction of Selenols with Isocyanates: Rapid Access to Selenocarbamates as Peroxide-Switchable Reservoir of Thiol-peroxidase-Like Catalysts. Adv. Synth. Catal. 2021, 363, 4256–4263. [Google Scholar] [CrossRef]
- Ishihara, H.; Koketsu, M.; Fukuta, Y.; Nada, F. Reaction of lithium aluminum hydride with elemental selenium: Its application as a selenating reagent into organic molecules. J. Am. Chem. Soc. 2001, 123, 8408–8409. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.X.; Goreshnik, I.; Coventry, B.; Case, J.B.; Miller, L.; Kozodoy, L.; Chen, R.E.; Carter, L.; Walls, A.C.; Park, Y.J.; et al. De novo design of picomolar SARS-CoV-2 miniprotein inhibitors. Science 2020, 370, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, R.; Agarwal, D.K.; Ranaweera, C.B.; Ishiguro, S.; Conda-Sheridan, M.; Gaudreault, N.N.; Richt, J.A.; Tamura, M.; Comer, J. De novo design of a stapled peptide targeting SARS-CoV-2 spike protein receptor-binding domain. RSC Med. Chem. 2023, 14, 1722–1733. [Google Scholar] [CrossRef]
- Hoffman, R.L.; Kania, R.S.; Brothers, M.A.; Davies, J.F.; Ferre, R.A.; Gajiwala, K.S.; He, M.; Hogan, R.J.; Kozminski, K.; Li, L.Y.; et al. Discovery of Ketone-Based Covalent Inhibitors of Coronavirus 3CL Proteases for the Potential Therapeutic Treatment of COVID-19. J. Med. Chem. 2020, 63, 12725–12747. [Google Scholar] [CrossRef]
- Narayanan, A.; Narwal, M.; Majowicz, S.A.; Varricchio, C.; Toner, S.A.; Ballatore, C.; Brancale, A.; Murakami, K.S.; Jose, J. Identification of SARS-CoV-2 inhibitors targeting Mpro and PLpro using in-cell-protease assay. Commun. Biol. 2022, 5, 169–185. [Google Scholar] [CrossRef]
- Wang, Y.C.; Yang, W.H.; Yang, C.S.; Hou, M.H.; Tsai, C.L.; Chou, Y.Z.; Hung, M.C.; Chen, Y. Structural basis of SARS-CoV-2 main protease inhibition by a broad-spectrum anti-coronaviral drug. Am. J. Cancer Res. 2020, 10, 2535–2545. [Google Scholar]
- Schrödinger Suite Release 2022-4. (a) Maestro v.13.2; (b) Prime, v.5.5; (c) Epik, v.6.0; (d) Impact, v.9.5; (e) Macromodel v.13.6. (f) Glide, v.9.5; Schrödinger, LLC: New York, NY, USA, 2022. [Google Scholar]
- Nocentini, A.; Capasso, C.; Supuran, C.T. Perspectives on the design and discovery of alpha-ketoamide inhibitors for the treatment of novel coronavirus: Where do we stand and where do we go? Expert Opin. Drug Discov. 2022, 17, 547–557. [Google Scholar] [CrossRef]
- ElNaggar, M.H.; Elgazar, A.A.; Gamal, G.; Hamed, S.M.; Elsayed, Z.M.; El-Ashrey, M.K.; Abood, A.; El Hassab, M.A.; Soliman, A.M.; El-Domany, R.A.; et al. Identification of sulphonamide-tethered N-((triazol-4-yl)methyl)isatin derivatives as inhibitors of SARS-CoV-2 main protease. J. Enzym. Inhib. Med. Chem. 2023, 38, 2234665–2234675. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Luca, V.; Angeli, A.; Nocentini, A.; Gratteri, P.; Pratesi, S.; Tanini, D.; Carginale, V.; Capperucci, A.; Supuran, C.T.; Capasso, C. Leveraging SARS-CoV-2 Main Protease (Mpro) for COVID-19 Mitigation with Selenium-Based Inhibitors. Int. J. Mol. Sci. 2024, 25, 971. https://doi.org/10.3390/ijms25020971
De Luca V, Angeli A, Nocentini A, Gratteri P, Pratesi S, Tanini D, Carginale V, Capperucci A, Supuran CT, Capasso C. Leveraging SARS-CoV-2 Main Protease (Mpro) for COVID-19 Mitigation with Selenium-Based Inhibitors. International Journal of Molecular Sciences. 2024; 25(2):971. https://doi.org/10.3390/ijms25020971
Chicago/Turabian StyleDe Luca, Viviana, Andrea Angeli, Alessio Nocentini, Paola Gratteri, Silvia Pratesi, Damiano Tanini, Vincenzo Carginale, Antonella Capperucci, Claudiu T. Supuran, and Clemente Capasso. 2024. "Leveraging SARS-CoV-2 Main Protease (Mpro) for COVID-19 Mitigation with Selenium-Based Inhibitors" International Journal of Molecular Sciences 25, no. 2: 971. https://doi.org/10.3390/ijms25020971