
Hydrophilic Reduction-Resistant Spin Labels of Pyrrolidine and Pyrroline Series from 3,4-Bis-hydroxymethyl-2,2,5,5-tetraethylpyrrolidine-1-oxyl
 , , , and
, , , and
Abstract
:1. Introduction
2. Results and Discussion
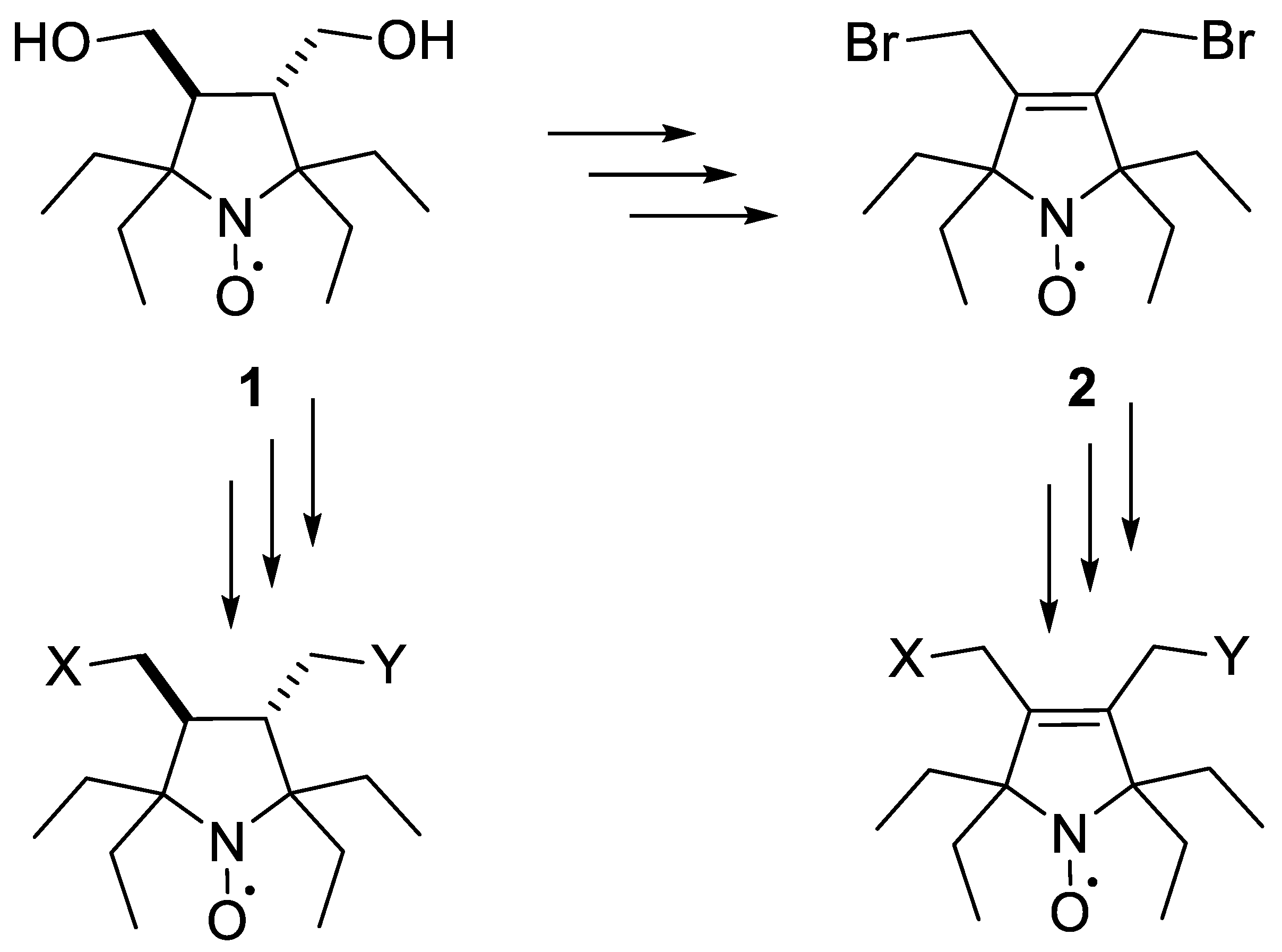
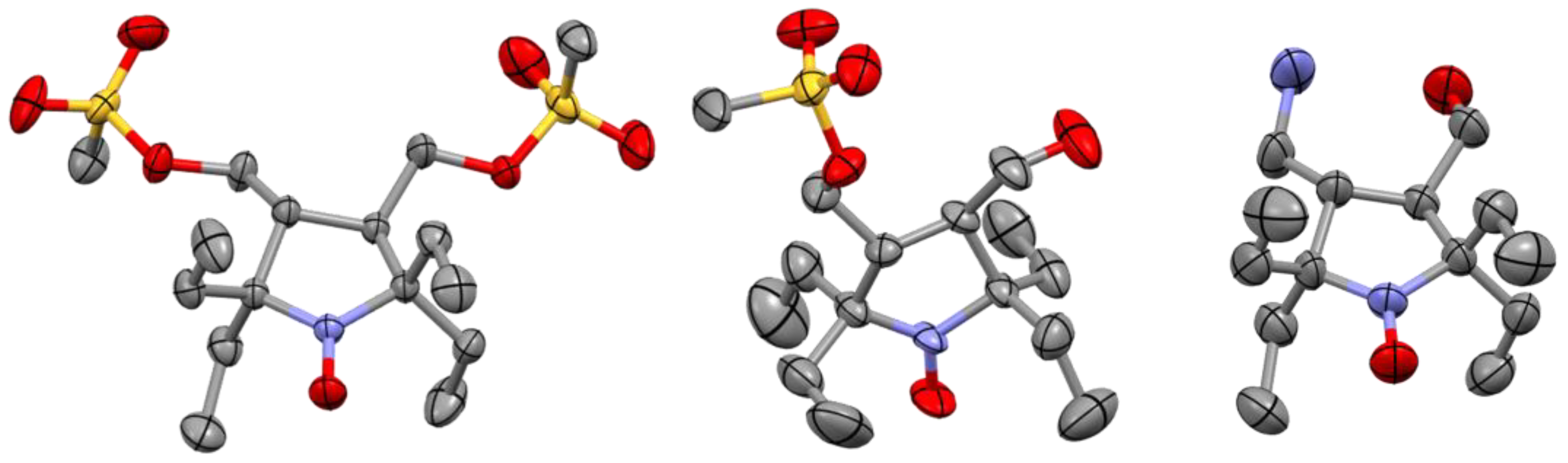
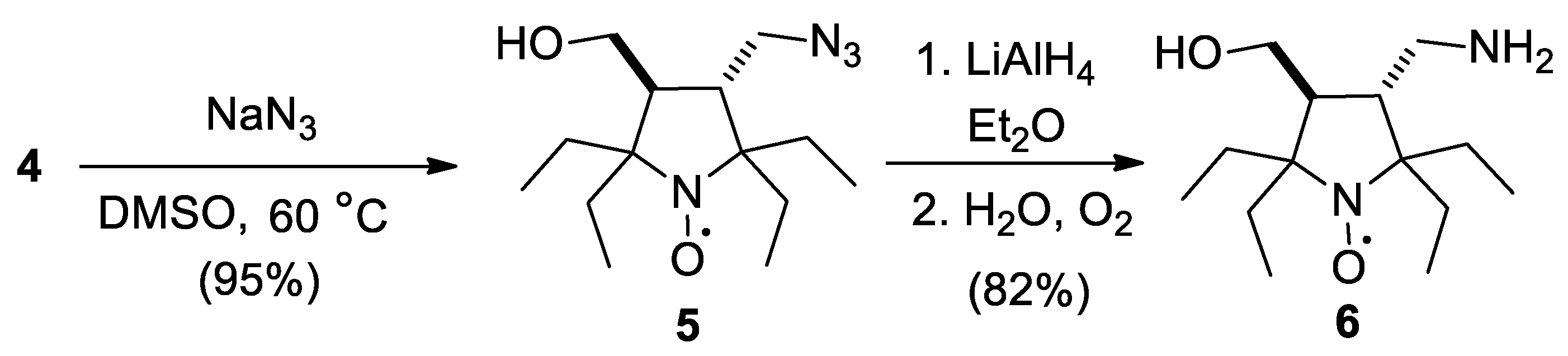
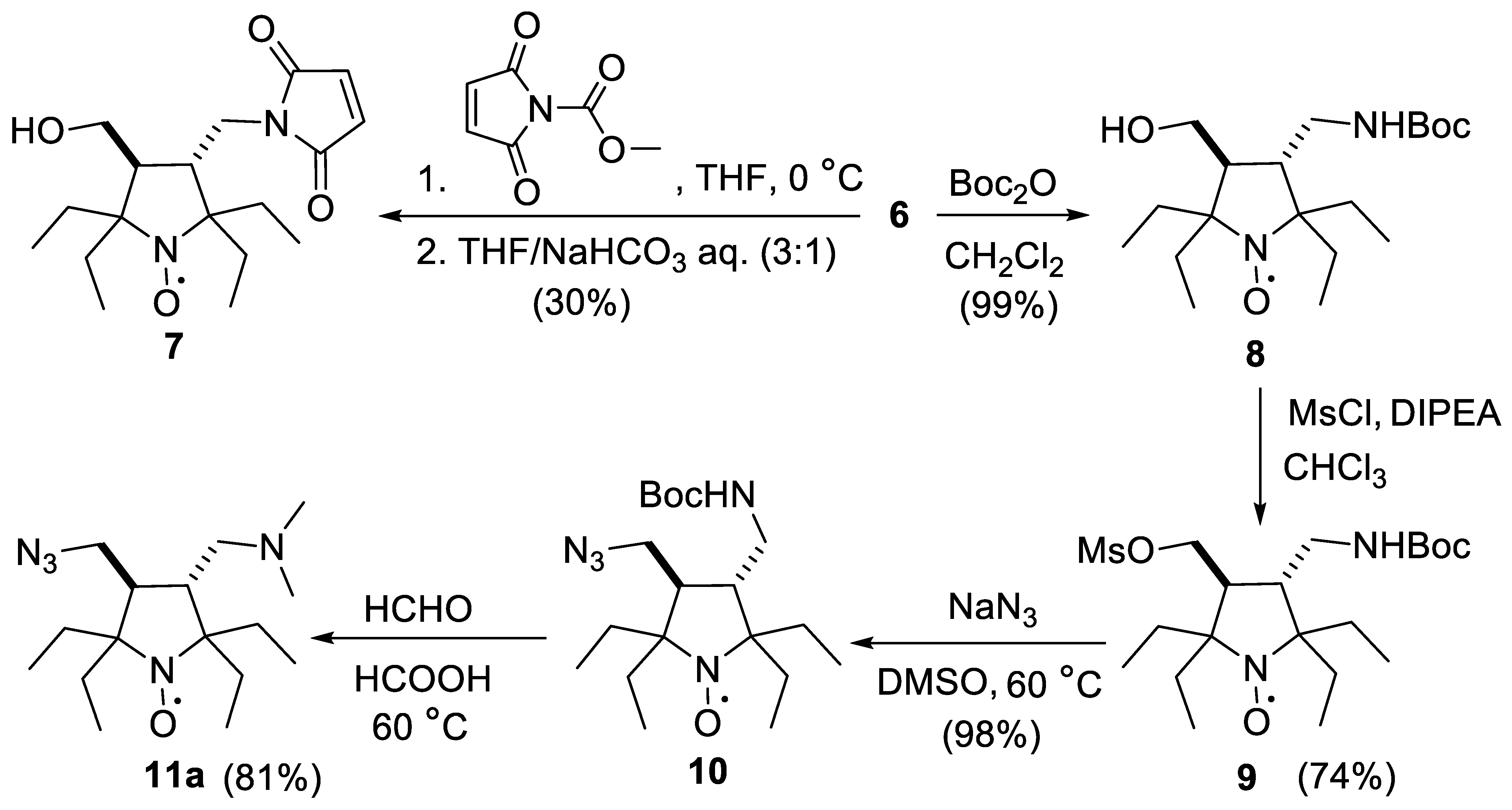
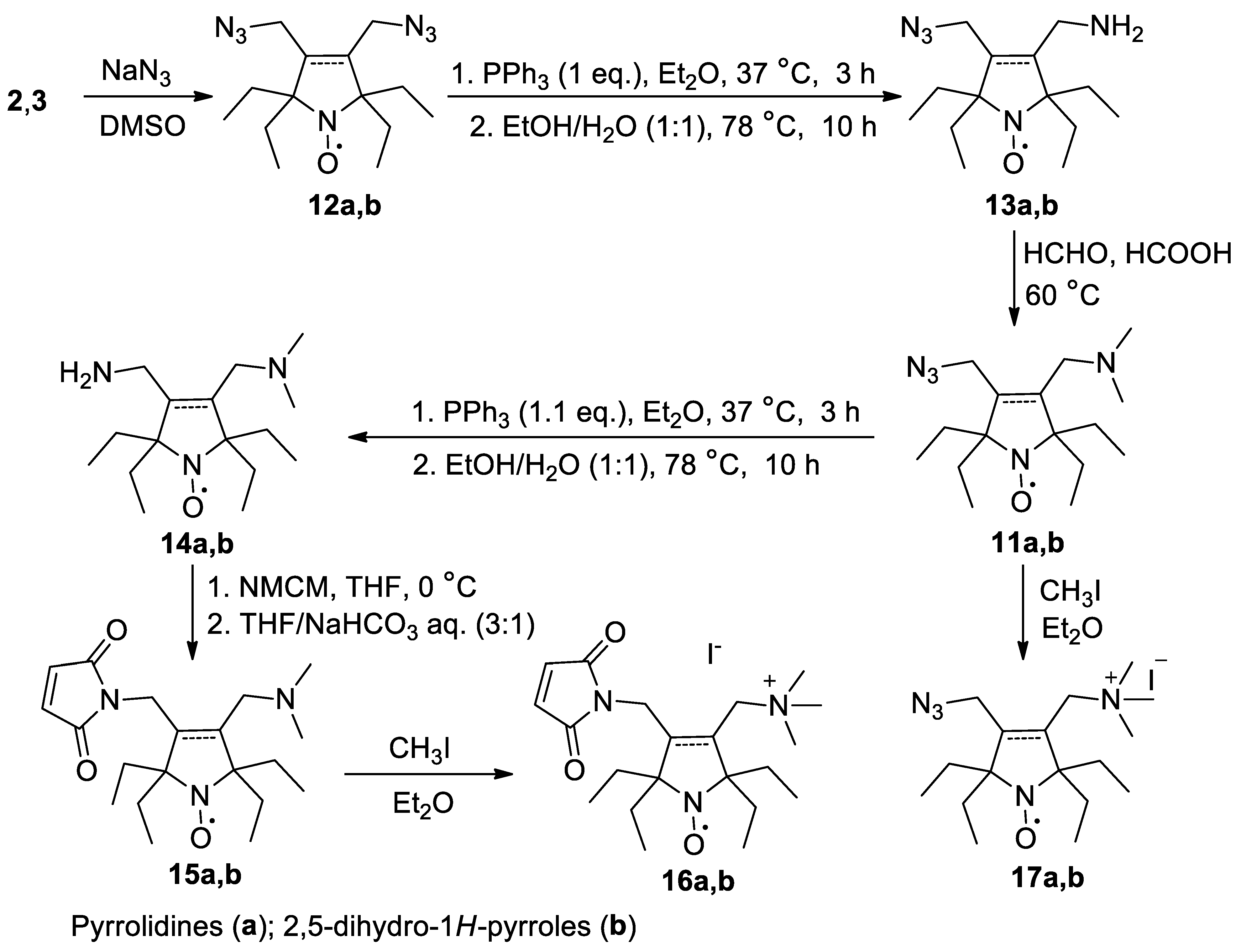
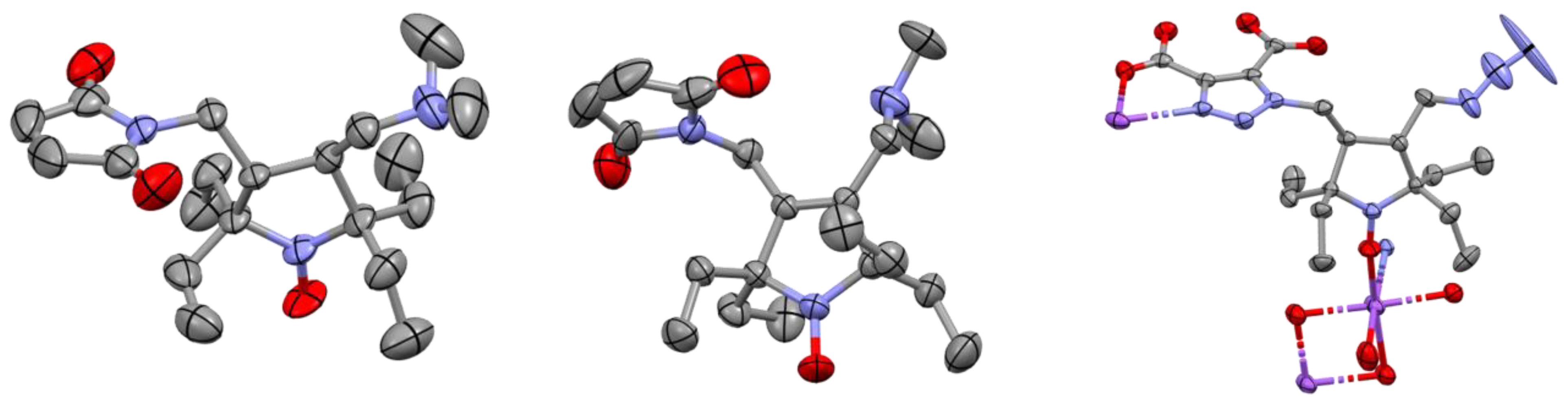
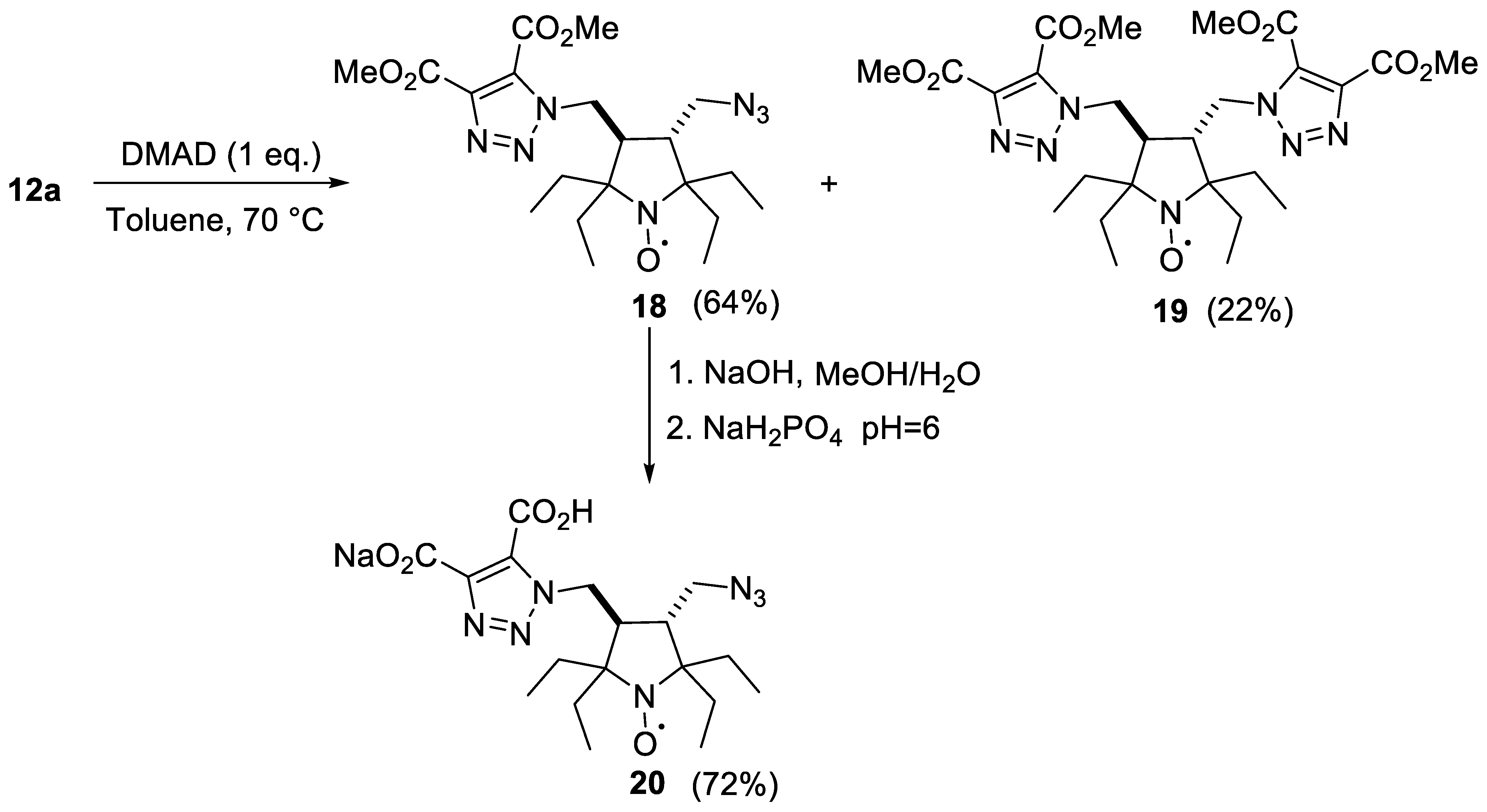
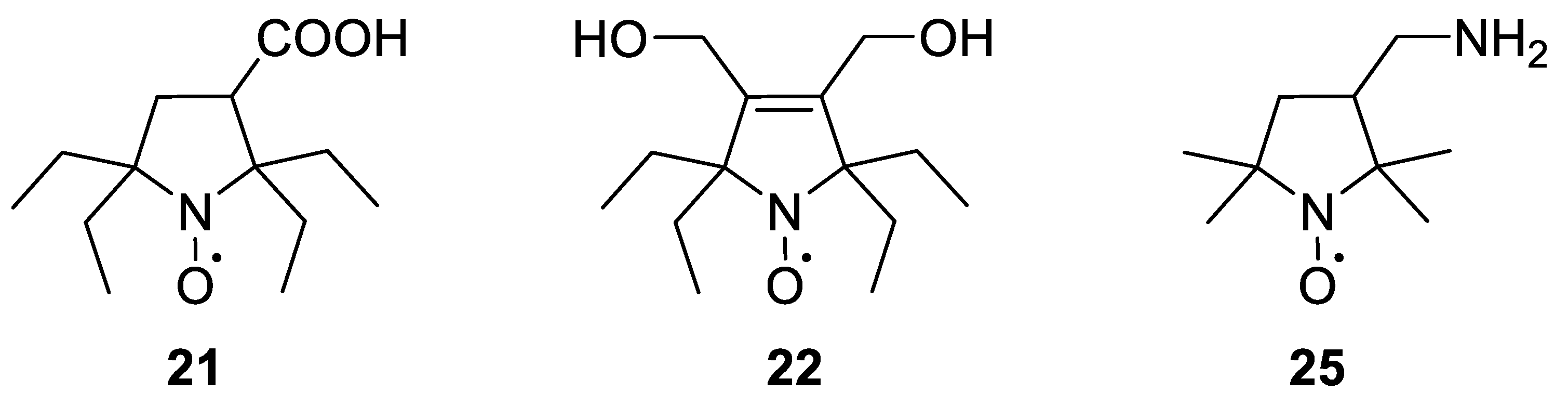
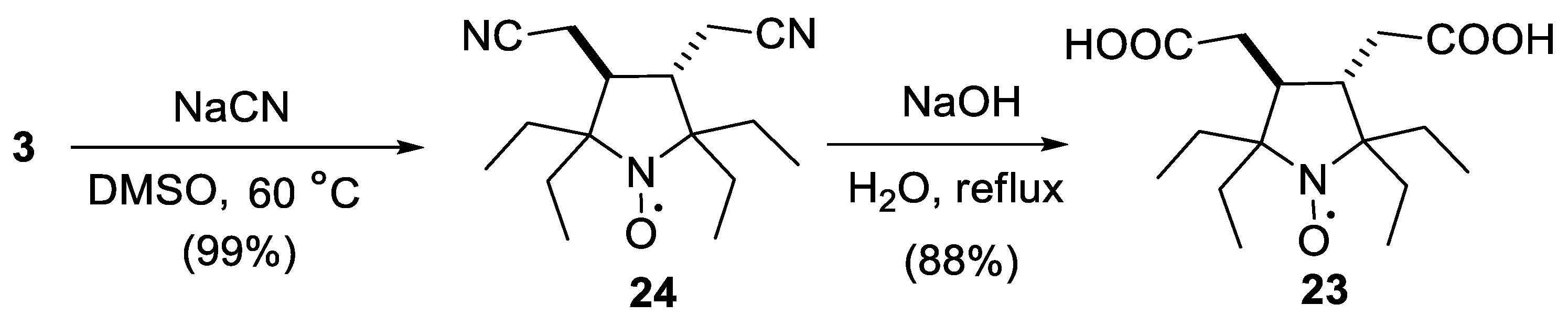
2.1. Synthesis of the Hydrophilic Nitroxides
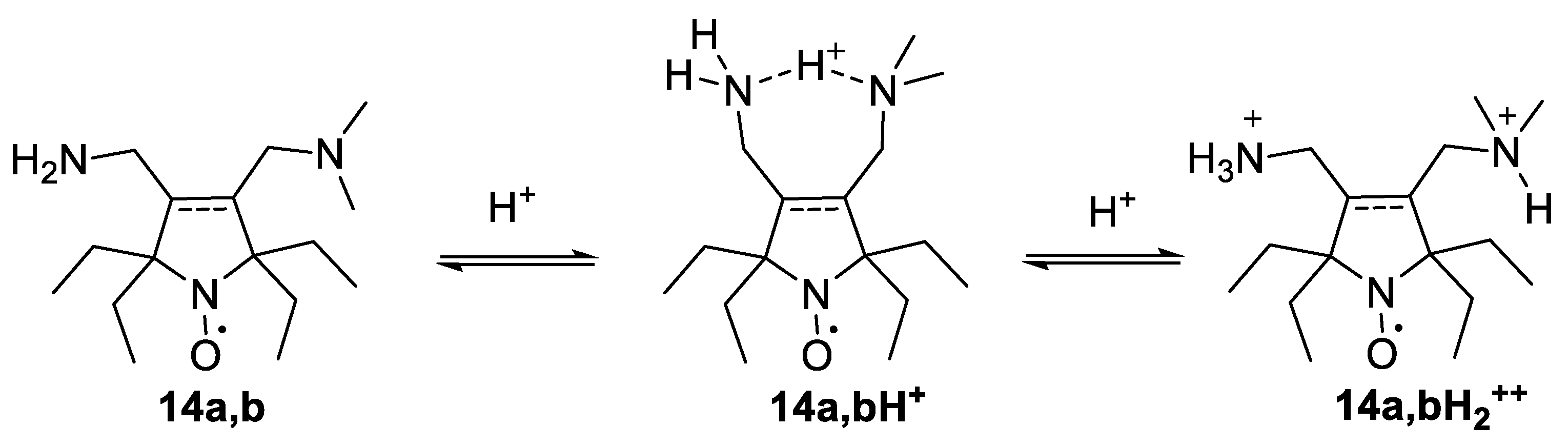
2.2. EPR Studies of the New Nitroxides
2.3. Lipophilicity
2.4. Reduction Rates in Ascorbate-Containing Systems
2.5. Decay in the Rat Liver Homogenates
3. Materials and Methods
3.1. General Information
3.2. Synthesis
3.2.1. A Reaction of Hydroxymethyl Nitroxides 1 and 8 with Methanesulfonyl Chloride
3.2.2. Reaction of Mesylates with Sodium Azide (General Method)
3.2.3. The Eschweiler–Clarke Methylation of Nitroxides 10 and 13a,b (General Method)
3.2.4. General Procedure for Quaternization of Dimethylamino Group
3.2.5. Reaction of Nitroxide 12a with Dimethyl Acetylenedicarboxylate (DMAD)
3.2.6. General Procedure of the Synthesis of Amines 13a,b and 14a,b with PPh3
3.2.7. General Procedure of the Synthesis of Maleimides 7 and 15a,b
3.3. EPR Measurements and Kinetics
3.4. Animals
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Galazzo, L.; Bordignon, E. Electron paramagnetic resonance spectroscopy in structural-dynamic studies of large protein complexes. In Progress in Nuclear Magnetic Resonance Spectroscopy; Neuhaus, D., Bodenhausen, G., Eds.; Elsevier BV: Amsterdam, The Netherlands, 2023; Volume 134–135, pp. 1–19. [Google Scholar] [CrossRef]
- Bonucci, A.; Ouari, O.; Guigliarelli, B.; Belle, V.; Mileo, E. In-Cell EPR: Progress towards Structural Studies Inside Cells. ChemBioChem 2020, 21, 451–460. [Google Scholar] [CrossRef]
- Klug, C.S.; Lerch, M.T.; Feix, J.B. Applications of Nitroxide Spin Labels to Structural Biology. In Nitroxides: Synthesis, Properties and Applications; Ouari, O., Gigmes, D., Eds.; The Royal Society of Chemistry: London, UK, 2021; Chapter 10; pp. 392–419. [Google Scholar] [CrossRef]
- Khramtsov, V.V. Spin probes and imaging using nitroxides. In Nitroxides: Synthesis, Properties and Applications; Ouari, O., Gigmes, D., Eds.; The Royal Society of Chemistry: London, UK, 2021; Chapter 4; pp. 147–186. [Google Scholar] [CrossRef]
- Kovaleva, E.; Molochnikov, L. pH-Sensitive Nitroxide Radicals for Studying Inorganic and Organo-Inorganic Materials and Systems. In Nitroxides: Applications in Chemistry, Biomedicine and Material Science; Likhtenshtein, G.I., Yamauchi, J., Nakatsuji, S., Smirnov, A., Tamura, R., Eds.; Wiley—VCH Verlag, GmbH & Co. KGaA: Weinheim, Germany, 2008; Chapter 7; pp. 211–246. [Google Scholar] [CrossRef]
- Voinov, M.A.; Smirnov, A.I. Spin labels and spin probes for measurements of local pH and electrostatics by EPR. In Electron Paramagnetic Resonance; Gilbert, B.C., Murphy, D.M., Chechik, V., Eds.; RSC Publishing: Cambridge, UK, 2010; Volume 22, pp. 71–106. [Google Scholar]
- Menzildjian, G.; Schlagnitweit, J.; Casano, G.; Ouari, O.; Gajan, D.; Lesage, A. Polarizing agents for efficient high field DNP solid-state NMR spectroscopy under magic-angle spinning: From design principles to formulation strategies. Chem. Sci. 2023, 14, 6120–6148. [Google Scholar] [CrossRef]
- Jaudzems, K.; Polenova, T.; Pintacuda, G.; Oschkinat, H.; Lesage, A. DNP NMR of biomolecular assemblies. J. Struct. Biol. 2019, 206, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, K.; Yasukawa, K. Imaging in vivo redox status in high spatial resolution with OMRI. Free Radic. Res. 2012, 46, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- Boudries, D.; Massot, P.; Parzy, E.; Seren, S.; Mellet, P.; Franconi, J.-M.; Miraux, S.; Bezançon, E.; Marque, S.R.A.; Audran, G.; et al. A system for in vivo on-demand ultra-low field Overhauser-enhanced 3D-Magnetic resonance imaging. J. Magn. Reson. 2023, 348, 107383. [Google Scholar] [CrossRef] [PubMed]
- Pierro, A.; Drescher, M. Dance with Spins: Site-Directed Spin Labeling Coupled to Electron Paramagnetic Resonance Spectroscopy Directly inside Cells. Chem. Commun. 2023, 59, 1274–1284. [Google Scholar] [CrossRef] [PubMed]
- Nakaoka, R.; Kato, K.; Yamamoto, K.; Yasui, H.; Matsumoto, S.; Kirilyuk, I.A.; Khramtsov, V.V.; Inanami, O.; Hirata, H. Electron Paramagnetic Resonance Implemented with Multiple Harmonic Detections Successfully Maps Extracellular pH In Vivo. Anal. Chem. 2023, 95, 3940–3950. [Google Scholar] [CrossRef] [PubMed]
- Audran, G.; Bosco, L.; Brémond, P.; Franconi, J.-M.; Koonjoo, N.; Marque, S.R.A.; Massot, P.; Mellet, P.; Parzy, E.; Thiaudiere, E. Enzymatically Shifting Nitroxides for EPR Spectroscopy and Overhauser-Enhanced Magnetic Resonance Imaging. Angew. Chem. Int. Ed. 2015, 54, 13379–13384. [Google Scholar] [CrossRef]
- Kosem, N.; Naganuma, T.; Ichikawa, K.; Morales, N.P.; Yasukawa, K.; Hyodo, F.; Yamada, R.; Utsumi, H. Whole-body kinetic image of a redox probe in mice using Overhauser-enhanced MRI. Free Rad. Biol. Med. 2012, 53, 328–336. [Google Scholar] [CrossRef]
- Kocherginsky, N.; Swartz, H. Nitroxide Spin Labels—Reactions in Biology and Chemistry; CRC Press, Inc.: Boca Raton, FL, USA, 1995. [Google Scholar]
- Paletta, J.T.; Pink, M.; Foley, B.; Rajca, S.; Rajca, A. Synthesis and Reduction Kinetics of Sterically Shielded Pyrrolidine Nitroxides. Org. Lett. 2012, 14, 5322–5325. [Google Scholar] [CrossRef]
- Wang, Y.; Paletta, J.T.; Berg, K.; Reinhart, E.; Rajca, S.; Rajca, A. Synthesis of unnatural amino acids functionalized with sterically shielded pyrroline nitroxides. Org. Lett. 2014, 16, 5298–5300. [Google Scholar] [CrossRef] [PubMed]
- Jagtap, A.P.; Krstic, I.; Kunjir, N.C.; Hänsel, R.; Prisner, T.F.; Sigurdsson, S.T. Sterically Shielded Spin Labels for In-Cell EPR Spectroscopy: Analysis of Stability in Reducing Environment. Free Radic. Res. 2015, 49, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Ketter, S.; Dajka, M.; Rogozhnikova, O.; Dobrynin, S.A.; Tormyshev, V.M.; Bagryanskaya, E.G.; Joseph, B. In situ distance measurements in a membrane transporter using maleimide functionalized orthogonal spin labels and 5-pulse electron-electron double resonance spectroscopy. J. Magn. Reson. Open 2022, 10, 100041. [Google Scholar] [CrossRef]
- Bleicken, S.; Assafa, T.E.; Zhang, H.; Elsner, C.; Ritsch, I.; Pink, M.; Rajca, S.; Jeschke, G.; Rajca, A.; Bordignon, E. gem-Diethyl Pyrroline Nitroxide Spin Labels: Synthesis, EPR Characterization, Rotamer Libraries and Biocompatibility. ChemistryOpen 2019, 8, 1035. [Google Scholar] [CrossRef] [PubMed]
- Karthikeyan, G.; Bonucci, A.; Casano, G.; Gerbaud, G.; Abel, S.; Thomé, V.; Kodjabachian, L.; Magalon, A.; Guigliarelli, B.; Belle, V.; et al. Bioresistant Nitroxide Spin Label for In-Cell EPR Spectroscopy: In Vitro and In Oocytes Protein Structural Dynamics Studies. Angew. Chem. Int. Ed. Engl. 2018, 57, 1366–1370. [Google Scholar] [CrossRef] [PubMed]
- Dobrynin, S.A.; Usatov, M.S.; Zhurko, I.F.; Morozov, D.A.; Polienko, Y.F.; Glazachev, Y.I.; Parkhomenko, D.A.; Tyumentsev, M.A.; Gatilov, Y.V.; Chernyak, E.I.; et al. A simple method of synthesis of 3-carboxy-2,2,5,5-tetraethylpyrrolidine-1-oxyl and preparation of reduction-resistant spin labels and probes of pyrrolidine series. Molecules 2021, 26, 5761. [Google Scholar] [CrossRef] [PubMed]
- Ovcherenko, S.S.; Chinak, O.A.; Chechushkov, A.V.; Dobrynin, S.A.; Kirilyuk, I.A.; Krumkacheva, O.A.; Richter, V.A.; Bagryanskaya, E.G. Uptake of Cell-Penetrating Peptide RL2 by Human Lung Cancer Cells: Monitoring by Electron Paramagnetic Resonance and Confocal Laser Scanning Microscopy. Molecules 2021, 26, 5442. [Google Scholar] [CrossRef] [PubMed]
- Rančić, A.; Babić, N.; Orio, M.; Peyrot, F. Structural features governing the metabolic stability of tetraethyl-substituted nitroxides in rat liver microsomes. Antioxidants 2023, 12, 402. [Google Scholar] [CrossRef]
- Babić, N.; Orio, M.; Peyrot, F. Unexpected rapid aerobic transformation of 2,2,6,6-tetraethyl-4-oxo(piperidin-1-yloxyl) radical by cytochrome P450 in the presence of NADPH: Evidence against a simple reduction of the nitroxide moiety to the hydroxylamine. Free Radic. Biol. Med. 2020, 156, 144–156. [Google Scholar] [CrossRef]
- Dobrynin, S.A.; Glazachev, Y.I.; Gatilov, Y.V.; Chernyak, E.I.; Salnikov, G.E.; Kirilyuk, I.A. Synthesis of 3,4-bis(hydroxymethyl)-2,2,5,5-tetraethylpyrrolidin-1-oxyl via 1,3-dipolar cycloaddition of azomethine ylide to activated alkene. J. Org. Chem. 2018, 83, 5392–5397. [Google Scholar] [CrossRef]
- Dobrynin, S.A.; Gulman, M.M.; Morozov, D.A.; Zhurko, I.F.; Taratayko, A.I.; Sotnikova, Y.S.; Glazachev, Y.I.; Gatilov, Y.V.; Kirilyuk, I.A. Synthesis of Sterically Shielded Nitroxides Using the Reaction of Nitrones with Alkynylmagnesium Bromides. Molecules 2022, 27, 7626. [Google Scholar] [CrossRef] [PubMed]
- Dobrynin, S.; Kutseikin, S.; Morozov, D.; Krumkacheva, O.; Spitsyna, A.; Gatilov, Y.; Silnikov, V.; Angelovski, G.; Bowman, M.K.; Kirilyuk, I. Human serum albumin labelled with sterically-hindered nitroxides as potential MRI contrast agents. Molecules 2020, 25, 1709. [Google Scholar] [CrossRef] [PubMed]
- Tsai, P.; Cao, G.L.; Merkel, T.J.; Rosen, G.M. Spin labelling of Bacillus anthracis endospores: A model for in vivo tracking by EPR imaging. Free Radic. Res. 2008, 42, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Hideg, K.; Sár, C.P.; Hankovszky, O.H.; Tamás, T.; Jerkovich, G. Synthesis of new 3,4-disubstituted 2,5-dihydro-1H-pyrrol-1-yloxyl spin label reagents via allylic rearrangements. Synthesis 1993, 1993, 390–394. [Google Scholar] [CrossRef]
- Dikalov, S.I.; Dikalova, A.E.; Morozov, D.A.; Kirilyuk, I.A. Cellular accumulation and antioxidant activity of acetoxymethoxycarbonyl pyrrolidine nitroxides. Free Radic. Res. 2018, 52, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Kálai, T.; Bognár, B.; Zsolnai, D.; Berente, Z.; Hideg, K. Synthesis of nitroxide-annulated carbocycles and heteroicycles. Synthesis 2012, 44, 3655–3660. [Google Scholar] [CrossRef]
- Fawzi, N.L.; Fleissner, M.R.; Anthis, N.J.; Kálai, T.; Hideg, K.; Hubbell, W.L.; Clore, G.M. A rigid disulfide-linked nitroxide side chain simplifies the quantitative analysis of PRE data. J. Biomol. NMR. 2011, 51, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Columbus, L.; Kálai, T.; Jekö, J.; Hideg, K.; Hubbell, W.L. Molecular motion of spin labeled side chains in alpha-helices: Analysis by variation of side chain structure. Biochemistry 2001, 40, 3828–3846. [Google Scholar] [CrossRef]
- Lösel, R.M.; Philipp, R.; Kálai, T.; Hideg, K.; Trommer, W.E. Synthesis and application of novel bifunctional spin labels. Bioconjug. Chem. 1999, 10, 578–582. [Google Scholar] [CrossRef]
- Widder, P.; Schuck, J.; Summerer, D.; Drescher, M. Combining site-directed spin labeling in vivo and in-cell EPR distance determination. Phys. Chem. Chem. Phys. 2020, 22, 4875–4879. [Google Scholar] [CrossRef]
- Chang, P.V.; Prescher, J.A.; Sletten, E.M.; Baskin, J.M.; Miller, I.A.; Agard, N.J.; Lo, A.; Bertozzi, C.R. Copper-free click chemistry in living animals. Proc. Natl. Acad. Sci. USA 2010, 107, 1821–1826. [Google Scholar] [CrossRef] [PubMed]
- De Bruin, B.; Kuhnast, B.; Hinnen, F.; Yaouancq, L.; Amessou, M.; Johannes, L.; Samson, A.; Boisgard, R.; Tavitian, B.; Dollé, F. 1-[3-(2-[18F]fluoropyridin-3-yloxy)propyl]pyrrole-2,5-dione: Design, synthesis, and radiosynthesis of a new [18F]fluoropyridine-based maleimide reagent for the labeling of peptides and proteins. Bioconjug. Chem. 2005, 16, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Keller, O.; Rudinger, J. Preparation and some properties of maleimido acids and maleoyl derivatives of peptides. Helv. Chim. Acta 1975, 58, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Kálai, T.; Hubbell, W.L.; Hideg, K. Click Reactions with Nitroxides. Synthesis 2009, 2009, 1336–1340. [Google Scholar] [CrossRef]
- Polienko, Y.F.; Dobrynin, S.A.; Lomanovich, K.A.; Brovko, A.O.; Bagryanskaya, E.G.; Kirilyuk, I.A. Origin of Long-Range Hyperfine Couplings in the EPR Spectra of 2,2,5,5-Tetraethylpyrrolidine-1-oxyls. ACS Omega 2023, 8, 38723–38732. [Google Scholar] [CrossRef] [PubMed]
- Kirilyuk, I.; Polovyanenko, D.; Semenov, S.; Grigor’ev, I.; Gerasko, O.; Fedin, V.; Bagryanskaya, E. Inclusion Complexes of Nitroxides of Pyrrolidine and Imidazoline Series with Cucurbit[7]uril. J. Phys. Chem. B 2010, 114, 1719–1728. [Google Scholar] [CrossRef] [PubMed]
- Kirilyuk, I.A.; Bobko, A.A.; Khramtsov, V.V.; Grigor’ev, I.A. Nitroxides with two pK values–Useful spin probes for pH monitoring within a broad range. Org. Biomol. Chem. 2005, 3, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Goldman, S.D.B.; Funk, R.S.; Rajewski, R.A.; Krise, J.P. Mechanisms of amine accumulation in, and egress from, lysosomes. Bioanalysis 2009, 1, 1445–1459. [Google Scholar] [CrossRef]
- Davis, R.M.; Sowers, A.L.; DeGraff, W.; Bernardo, M.; Thetford, A.; Krishna, M.C.; Mitchell, J.B. A novel nitroxide is an effective brain redox imaging contrast agent and in vivo radioprotector. Free Radic. Biol. Med. 2011, 51, 780–790. [Google Scholar] [CrossRef]
- Kálai, T.; Balog, M.; Jekö, J.; Hideg, K. Synthesis and Reactions of a Symmetric Paramagnetic Pyrrolidine Diene. Synthesis 1999, 1999, 973–980. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nitroxide | aN, mT ±0.005 | aH, mT ±0.005 | Hp-p, mT ±0.005 | g-Factor ±0.00002 | Kp | kred × 104, M−1 s−1 |
|---|---|---|---|---|---|---|
| 1 | 1.57 | 0.22; 0.22 | 0.125 | 2.00563 [27] | 41 ± 2 | 3.3 ± 0.8 [26] |
| 5 | 1.52 | 0.22; 0.24 | 0.27 | 2.00562 | ||
| 6 | 1.51 | 0.22; 0.23 | 0.26 | 2.00562 | ||
| 7 | 1.52 | 0.24; 0.26 | 0.28 | 2.00562 | 84 ± 2 | |
| 8 | 1.52 | 0.24; 0.25 | 0.27 | 2.00562 | 6.0 ± 2.0 | |
| 9 | 1.51 | 0.23; 0.27 | 0.28 | 2.00562 | 3.0 ± 1.0 | |
| 10 | 1.50 | 0.23; 0.25 | 0.27 | 2.00562 | 3.0 ± 1.0 | |
| 11a | 1.49 | 0.23; 0.27 | 0.26 | 2.00563 | 52 ± 5 | 4.0 ± 1.0 |
| 11b | 1.51 | 0.21 | 2.00562 | 15 ± 1 | 15 ± 4.0 | |
| 13a | 1.49 | 0.22; 0.28 | 0.26 | 2.00563 | 350 ± 35 | 6.3 ± 0.9 |
| pH = 1.5 * | 1.51 | |||||
| pH = 13 * | 1.53 | |||||
| 13b | 1.51 | 0.18 | 2.00562 | 45 ± 2 | 7.6 ± 0.3 | |
| pH = 1.5 * | 1.53 | |||||
| pH = 13 * | 1.55 | |||||
| 14a | 1.51 | 0.20; 0.28 | 0.26 | 2.00562 | 6.4 ± 1.0 | |
| pH = 7.4 * | 1.52 | 0.23; 0.23 | ||||
| pH = 1.5 * | 1.46 | 0.31; 0.31 | ||||
| pH = 13 * | 1.54 | 0.25; 0.25 | ||||
| 14b | 1.52 | 0.19 | 2.00563 | 7 ± 0.3 | 8 ± 1.0 | |
| pH = 7.4 * | 1.53 | |||||
| pH = 1.5 * | 1.49 | |||||
| pH = 13 * | 1.55 | |||||
| 15a | 1.48 | 0.26; 0.31 | 2.00562 | |||
| 15b | 1.51 | 0.23 | 2.00563 | |||
| 16a | 1.47 | 0.23; 0.36 | 0.29 | 2.00562 | 0 | |
| 16b | 1.48 | 0.25 | 2.00562 | |||
| 17a | 1.48 | 0.22; 0.27 | 0.26 | 2.00561 | 0 | 3.0 ± 1.0 |
| 17b | 1.49 | 0.23 | 2.00562 | 0 | 16 ± 3.0 | |
| 20 | 1.50 | 0.24; 0.31 | 0.28 | 2.00561 | 0 | 5.9 ± 1.2 |
| 21 | 1.55 | 0.19 | 0.26 | 2.00563 | 270 | 12.5 ± 0.3 [22] |
| 22 | 1.54 | 0.16 | 2.00563 | 37 ± 2 | 3.4 ± 0.1 | |
| 23 | 1.53 | 0.24; 0.29 | 0.27 | 2.00562 |
| No. | 1, g/mmol | MsCl, g/eq. | DIPEA, g/eq. | CHCl3, mL | Conversion of 1, % | Yield | |
|---|---|---|---|---|---|---|---|
| 3, % | 4, % | ||||||
| 1 | 2/7.75 | 1.4/1.58 | 1.4/1.4 | 50 | 85 | 12 | 68 |
| 2 | 4/15.5 | 3/1.68 | 3/1.5 | 50 | >95 | 35 | 55 |
| 3 | 4/15.5 | 5/2.8 | 4.8/2.2 | 50 | 100 | >95 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Usatov, M.S.; Dobrynin, S.A.; Polienko, Y.F.; Morozov, D.A.; Glazachev, Y.I.; An’kov, S.V.; Tolstikova, T.G.; Gatilov, Y.V.; Bagryanskaya, I.Y.; Raizvikh, A.E.; et al. Hydrophilic Reduction-Resistant Spin Labels of Pyrrolidine and Pyrroline Series from 3,4-Bis-hydroxymethyl-2,2,5,5-tetraethylpyrrolidine-1-oxyl. Int. J. Mol. Sci. 2024, 25, 1550. https://doi.org/10.3390/ijms25031550
Usatov MS, Dobrynin SA, Polienko YF, Morozov DA, Glazachev YI, An’kov SV, Tolstikova TG, Gatilov YV, Bagryanskaya IY, Raizvikh AE, et al. Hydrophilic Reduction-Resistant Spin Labels of Pyrrolidine and Pyrroline Series from 3,4-Bis-hydroxymethyl-2,2,5,5-tetraethylpyrrolidine-1-oxyl. International Journal of Molecular Sciences. 2024; 25(3):1550. https://doi.org/10.3390/ijms25031550
Chicago/Turabian StyleUsatov, Mikhail S., Sergey A. Dobrynin, Yuliya F. Polienko, Denis A. Morozov, Yurii I. Glazachev, Sergey V. An’kov, Tatiana G. Tolstikova, Yuri V. Gatilov, Irina Yu. Bagryanskaya, Arthur E. Raizvikh, and et al. 2024. "Hydrophilic Reduction-Resistant Spin Labels of Pyrrolidine and Pyrroline Series from 3,4-Bis-hydroxymethyl-2,2,5,5-tetraethylpyrrolidine-1-oxyl" International Journal of Molecular Sciences 25, no. 3: 1550. https://doi.org/10.3390/ijms25031550