
Can Some Anticancer Drugs Be Repurposed to Treat Amyotrophic Lateral Sclerosis? A Brief Narrative Review

National Centre for Drug Research and Evaluation, Istituto Superiore di Sanità, 00161 Rome, Italy
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2024, 25(3), 1751; https://doi.org/10.3390/ijms25031751
Submission received: 14 December 2023
/
Revised: 25 January 2024
/
Accepted: 30 January 2024
/
Published: 1 February 2024
(This article belongs to the Special Issue Amyotrophic Lateral Sclerosis as a Systemic Disease 2.0)
Abstract
:Amyotrophic lateral sclerosis (ALS) is a rare progressive motor neuron disease that, due to its high complexity, still lacks effective treatments. Development of a new drug is a highly costly and time-consuming process, and the repositioning of approved drugs can represent an efficient strategy to provide therapeutic opportunities. This is particularly true for rare diseases, which are characterised by small patient populations and therefore attract little commercial interest. Based on the overlap between the biological background of cancer and neurodegeneration, the repurposing of antineoplastic drugs for ALS has been suggested. The objective of this narrative review was to summarise the current experimental evidence on the use of approved anticancer drugs in ALS. Specifically, anticancer drugs belonging to different classes were found to act on mechanisms involved in the ALS pathogenesis, and some of them proved to exert beneficial effects in ALS models. However, additional studies are necessary to confirm the real therapeutic potential of anticancer drugs for repositioning in ALS treatment.
1. Introduction
Amyotrophic lateral sclerosis (ALS) is a rare neurodegenerative disease characterised by selective damage to upper and lower motor neurons, leading to death, usually as a consequence of respiratory failure, approximately 3–5 years after symptom onset [1,2]. The prevalence of ALS has been reported as between 4.1 and 8.4 per 100,000 and it is expected to grow, mainly due to the ageing population [3].
The pathophysiological mechanisms underlying the disease are still poorly understood [4]. As is the case with other neurodegenerative diseases, ALS genesis appears to be regulated from a complex interaction between individual genetic risks, aging and environmental factors [5]. More than 90% of ALS cases are sporadic, whereas about 5–10% are familial [1]. About 60% of familiar and 10% of sporadic ALS cases are due to pathogenic mutation in superoxide dismutase 1 (SOD1), TAR DNA-binding protein (TARDBP), fused in sarcoma (FUS) and chromosome 9 open reading frame 72 (C9orf72), the four most common ALS-associated genes [6].
Many ALS patients also show cognitive disturbances, extrapyramidal deficits and neuropathological findings, which reveal the multisystem nature of the disease [7]. Such a multifactorial nature partially explains why, in spite of intense basic research efforts, effective treatments remain elusive and ALS still represents an unmet medical need. Indeed, currently only two drugs, riluzole and edaravone, have currently received marketing authorisation for ALS treatment; moreover, their efficacy is rather limited [8].
The discovery of a new drug is a highly costly and time-consuming process, and the propensity of pharmaceutical companies to allocate their resources depends on the commercial potential of the future drug. Thus, in the case of rare diseases, the interest of private industry is often limited. The repositioning of drugs already approved for alternative indications could represent an efficient strategy to provide therapeutic opportunities for orphan diseases [9]. The advantages of this approach vs. de novo drug development are obvious. Indeed, for approved drugs, pharmacodynamics, pharmacokinetics and safety in the clinical setting are already established, making the research process quicker and less costly, subsequently allowing their rapid implementation into new medical applications. Repositioning may typically be considered when a drug acts on a pathogenetic mechanism that is shared by different diseases. From a more complex point of view, repositioning may rely on the “promiscuous” nature of the drugs, as they often interact with multiple targets, each of which may have a role in the pathogenesis of different diseases [10].
Some evidence has highlighted the existence of intriguing relationships between neurodegeneration and cancer [11,12]. In particular, despite the opposite hallmarks of the two conditions (excessive cell proliferation vs. cell loss), it has been suggested that some anticancer drugs might be repurposed for the treatment of neurodegenerative diseases, including ALS [13,14]. In agreement, we proposed in a previous article that fenretinide, an analogue of retinol endowed with antineoplastic activity, could additionally be considered as well for the treatment of ALS and other neurological disorders [15].
As far as ALS is specifically concerned, its possible interconnections with cancer have been explored by several studies supporting mutual links between these two age-related diseases [16]. Indeed, microarray analysis of ALS patient samples showed that candidate genes for ALS biomarkers are related to cancer development [17,18,19]. Other studies revealed common signalling pathways between ALS and cancer, such as Scr/c–abl, which was found to be overactivated during both cancer and ALS progression [20], and the P38 mitogen-activated protein kinase (p38MAPK) pathway [21], whose inhibition rescued the axonal transport defects in ALS mice [22].
Epidemiological studies on the possible association between cancer and ALS have reported discordant results. Fang et al. [23] reported no overall association of cancer and risk of ALS, while more cases of some specific tumours (specifically, prostate and brain) were diagnosed in ALS patients. Freedman et al. [24] reported that ALS mortality was not associated with total cancers, while the risk of ALS death was found to be increased or decreased when specific tumours were considered. Moreover, a longitudinal study showed a reduced overall risk of cancer, but an increased risk for salivary and testicular cancer, in ALS patients [25].
Such discrepancies may well be explained by the fact that speaking in terms of “cancer” as though it were a single disease can be misleading, since even cancers with the same histological origin can dramatically differ from one another in terms of clinical, prognostic and therapeutic issues according to their specific molecular profiles. Similarly, “anticancer drugs” is an umbrella term including very different molecules (and related mechanisms of action) that should not be regarded as a whole.
In the present review, we examine anticancer drugs belonging to different classes and discuss the mechanisms underlying their possible therapeutic roles on ALS. A detailed literature search of PubMed was performed to identify publications (including clinical studies, animal studies and in vitro studies) reporting use of already approved anticancer drugs in ALS. Clinical.trial.gov and DrugBank databases were also interrogated.
2. Miscellaneous
Fenretinide (DrugBank Accession Number DB05076, N-(4-hydroxyphenyl)retinamide) is a semisynthetic derivative of all-trans retinoic acid produced in the USA in the 1960s and was first proposed as an anticancer treatment due to its significant antitumour activity and favourable toxicological profile [26]. The antitumour effect of fenretinide is very complex and includes different mechanisms of action [15].
Even if it has not yet been approved by the EMA, fenretinide obtained orphan designation by the European Commission for the treatment of primary malignant bone tumours [27] and cutaneous T-cell lymphoma [28].
Very recently, we demonstrated that low doses (10 mg/kg) of a new fenretinide formulation significantly attenuates the neurological phenotype and extends the survival of mice expressing the mutated form of human SOD1 protein (mSOD1G93A ALS mice), even when administered after the onset of motor symptoms [29]. We also demonstrated that in cultured motoneurons the expression of ALS-linked SOD1 mutation resulted in mitochondrial dysfunction, which can be reversed by treatment with fenretinide. The ability of FEN to protect myotubes from “in vitro” mSOD1 toxicity could partially explain the attenuation of the progression of neurological symptoms observed in mSOD1G93A mice chronically treated with the drug [29].
Our results extended the neuroprotective potential of this anticancer drug to ALS treatment already reported for other neurological diseases like multiple sclerosis and Alzheimer’s disease [15]. The neuroprotective effects of fenretinide occurred at much lower doses than those required for its antitumour activity. Indeed, high doses of fenretinide can activate acute responses leading to ROS increase and cell death [30], while, at subtoxic concentrations, the drug can stimulate an adaptive stress response [31].
3. Alkylating Agents
The platinum complexes have revolutionised cancer therapy and the majority of chemotherapic regimens routinely applied in the clinical setting are still platinum-based [32]. These inorganic compounds are classified as alkylating agents and are used in the treatment of different forms of cancer, including sarcomas, carcinomas, lymphomas and germ cell tumours [33].
Cisplatin (DrugBank Accession Number DB00515) was the first member of its class, which now also includes carboplatin and oxaliplatin. As with all the other alkylating agents, cisplatin prevents the cell from dividing by adding an alkyl group to the DNA. However, only a small fraction of the administered dose reacts with the DNA to induce cytotoxicity, whereas the largest amount binds cellular proteins, thereby influencing other potential targets [34]. Calderone et al. showed that cisplatin selectively binds to His-19 residue located on the surface of the bovine SOD protein Cu/Zn superoxide dismutase (SOD1) [35]. SOD1 is an antioxidant enzyme that catalyses the dismutation of superoxide radicals; approximately 20% of familial ALS (FALS) cases are due to mutations in the SOD1 gene and, albeit at a very low frequency, SOD1 mutations are also observed in the sporadic form of the disease (SALS) [36]. Furthermore, not only the mutation but also the aggregation of the wild type SOD1 protein may play a role in modulating disease initiation [37]. In 2012, Banci and colleagues showed that cisplatin also interacts with the human form of SOD1, binding two cysteines (Cys6 and Cys111) onto the protein surface [38]. Cys6 and Cys111 residues were implicated in the aberrant aggregation of the mutated form of SOD1 [39], which are deemed to be essential in inducing endoplasmic reticulum (ER) stress related to SOD1 protein misfolding in ALS [40]. The potential use of cisplatin in the treatment of ALS was thus proposed [41].
Carboplatin (DrugBank Accession Number DB00958) is another platinum-based drug already approved to treat different forms of cancer. Due to its hydrophilic nature, carboplatin is longer retained longer within brain tissue; interestingly, it was found to be highly effective against glioblastoma while being nontoxic to normal brain tissue [42]. In breast cancer cells, carboplatin induced the expression of the omega class of cytosolic glutathione S-transferase (GSTO1) [43], an enzyme that is significantly reduced in peripheral blood mononuclear cells and in the spinal cord from ALS patients [44]. The glutathione S-transferase omega 1 (GSTO1) and 2 (GstO2, the Drosophila homolog of human GSTO1) were found to be involved in the oxidative damage underlying the pathogenesis of neurodegenerative diseases [45]. The overexpression of GSTO was shown to reduce the citoplasmatic accumulation of two proteins whose abnormal aggregations are characteristics of ALS and frontotemporal dementia [46], namely, the fused in sarcoma (FUS) DNA/RNA-binding protein and the Transactive response DNA-binding protein-43 (TDP-43) [47]. Specifically, Cha et al. showed that FUS neurotoxicity is sustained by impaired protein solubility induced by glutathionylation and that the overexpression of glutathione transferase omega 2 (GstO2) reduces abnormal protein aggregates in both TDP43 and FUS transgenic Drosophila, thus highlighting the therapeutic potential of carboplatin in ALS. Indeed, the drug rescued the mitochondrial disfunction and dose-dependently reduced locomotor and eye deficits in the FUS-ALS fly model [48].
4. Antimetabolites
Antimetabolites are nucleoside analogues interfering or competing with nucleoside triphosphates in the synthesis of DNA (antimitotic) or RNA or both. The fluoropyrimidine 5-fluorouracil (5-FU) (DrugBank Accession Number DB00544) is a pyrimidine analogue used as a palliative cancer treatment or to treat basal cell carcinomas. Besides its antimitotic effect, 5-FU can also induce striking alterations in RNA metabolism, splicing and post-transcriptional modification [49], suggesting the possible occurrence of several off-target effects. In a preclinical study designed to evaluate stem cell mobilisation in the murine ALS model, Rando et al. administered 5-FU as a negative control. As expected for an anticancer drug, the 5-FU administration induced a reduction of cellular component with a rapid turnover, as blood cells, an effect fully recovered after two weeks of repeated treatment. Surprisingly, however, when chronically administered in the pre-symptomatic phase, 5-FU also delayed the disease onset, improved the motor performance and increased the lifespan of ALS-treated animals, while it did not exert major effects on the myogenic, apoptotic or autophagic markers commonly elevated in mSOD1G93A muscles [50]. Although no mechanistic data were provided by the above study, 5-FU has been reported to reduce tryptophan-induced SOD1 aggregation in cells [51]. This can be very relevant to the possible therapeutic effects of 5FU on human ALS, since tryptophan residue at position 32 has a critical influence on human SOD1 toxicity to motor neurons [52].
5. Hormone Antagonists
Tamoxifen (DrugBank Accession NumberDB00675) is a selective estrogen receptor modulator with both estrogenic and anti-estrogenic effects. In breast tissue, tamoxifen exerts anti-estrogenic and antitumour effects by blocking estrogens from entering cancer cells and thus reducing or eliminating the cells’ ability to grow and spread [53]. It is generally used to treat breast cancer in men and women and as a prophylactic treatment against breast cancer in women. Besides its antitumour activity, tamoxifen also showed neuroprotective effects in some preclinical models of neurological disease [54]. In experimental brain injury, the drug reduced neuroinflammation through TLR4/NF-kappaB pathways [55], while in a murine model of spinal cord injury it reduced microglia activation and the apoptotic death of neural cells [54,56]. Interestingly, in mice overexpressing TDP-43 DNA/RNA-binding protein (identified as the major component of the cytoplasmic inclusions in frontotemporal dementia and ALS), tamoxifen treatment was associated with an improvements in motor functions. The behavioural effect was accompanied by a reduction in the neuronal loss and TDP-43 inclusion in the forebrain of mice. Furthermore, in this murine model, tamoxifen also increased MTOR-dependent autophagy through AKT/PKB inhibition [56]. On the basis of the above results, a placebo-controlled randomised clinical trial was conducted in ALS patients without mutations in superoxide dismutase-1 (SOD1) or fused in sarcoma (FUS) genes [57]. Tamoxifen only modestly attenuated disease progression without exerting any significant effect on the primary clinical endpoint (time to death or dependence on mechanical ventilation, and tracheostomy with continuous mechanical ventilation and noninvasive ventilation for more than 12 h per day). This study must, however, be considered inconclusive; due to the extremely limited sample size (10 patients on tamoxifen and 8 on placebo), it was dramatically underpowered to detect a statistical significance in the clinical endpoint. Considering both the preclinical evidence and the inverse correlation between tamoxifen treatments and ALS risk reported in a population-based case–control study of >10,000 US cases [58], the potential therapeutic role of tamoxifen on human ALS seems worthy of further investigation in larger clinical studies.
6. Protein Kinase (PK) Inhibitors
Protein kinase (PK) inhibitors are a large group of antineoplastic agents that exert antiproliferative and cytotoxic effects through the inhibition of different pro-survival protein kinase activity. These drugs are generally categorised according to the amino acid that they phosphorylate and, among these, the tyrosine kinase receptor inhibitors remain the best characterised.
The c-Src and c-Abl tyrosine kinases are normally activated only in response to external signals such as molecules released following brain injury (adenosine, cytokines and ROS), and their activation was found abnormally elevated in neurodegenerative diseases [59]. Specifically, Src overactivation can trigger neuronal entry into aberrant cell cycles and induces post-mitotic death [13], whereas c-Abl mainly acts through mechanisms such as neuroinflammation and oxidative stress [60].
A significant increase in c-Abl mRNA was detected in the motor neurons of patients with a sporadic form of ALS [61] and in the spinal cord of transgenic ALS mice mSOD1G93A as compared with WT littermates [62]. In mSOD1G93A mice, the increased expression of c-Abl protein was accompanied by an increase in its active phosphorylated form, thus suggesting that c-Abl could represent a potential therapeutic target for ALS.
Imatinib (DrugBank Accession Number DB00619, Gleevec—STI571), an inhibitor of the oncogenic Bcr-Abelson (Bcr–Abl) which arises from a chromosomal rearrangement (Philadelpha chromosome) in acute lymphoblastic leukaemia, was the first-in-class compound to receive regulatory approval [63] and was then followed by many others within the next 10 years. The effectiveness of imatinib in preventing neuronal death was subsequently explored in several diseases affecting the central nervous system, including Alzheimer’s disease, multiple sclerosis, Parkinson’s disease and spinal cord injury [64]. As for ALS, imatinib was the only kinase inhibitor in the group that decreased mutant SOD1 protein levels. In 2015, Rojas et al. showed that c-Abl activation induced by oxidative stress can be counteracted by imatinib, and that the drug protected motoneurons from mSOD1 astrocyte-mediated toxicity [65]. Interestingly, one ALS patient treated for 7 years with imatinib for chronic myeloid leukaemia manifested a clear worsening of their ALS symptoms shortly after TKIs were withdrawn [20]. Furthermore, imatinib also inhibits the PDGFA receptor tyrosine protein kinases, thus potentially affecting multiple ALS targets [66].
The potential role of c-Abl inhibition in ALS is strengthened by the finding that another c-Abl inhibitor, dasatinib (DrugBank Accession Number DB01254), was able to attenuate motoneuron loss, delay disease progression and extend the survival of mSOD1G93A ALS mice. Administration of dasatinib also induced a dose-dependent reduction in both c-Abl phosphorylation and caspase-3 activation, thus suggesting that a suppression of apoptotic cell death of motor neurons played a role in the neuroprotective effects of the drug [62].
A phenotypic-based drug screening carried on induced pluripotent stem cells (iPSCs) from sporadic ALS patients demonstrated that more than half of the screened hits targeted the Src/c-Abl signalling pathway. The authors provide evidence that the selected Src/c-Abl kinases inhibitor bosutinib (DrugBank Accession Number DB06616, which is approved for chronic myelogenous leukaemia) promoted autophagy and rescued ALS patient motor neurons from degeneration [67], whereas in an innovative 3D model of human ALS motor unit its co-application significantly increased the beneficial effects of rapamycin towards muscle contraction deficit [68]. The neuroprotective effects of bosutinib were confirmed in vivo in the mSOD1 mice model. When administered intraperitoneally (5 mg/kg per day) to ALS mice, the drug slightly delayed disease onset and extended survival. Such effects were accompanied by a decrease in misfolded SOD1 protein and motor neuron death in the spinal cords of treated mice [67]). The addition of a good brain penetration after systemic administration [69] further enhances the interest of the potential role of bosutinib in the treatment of neurological diseases. On these bases, a phase I clinical trial was approved by the Japanese Pharmaceuticals and Medical Devices Agency (Trial registration number UMIN000036295) to evaluate bosutinib for ALS patients (iDReAM) [70]. Although the study was mainly focused on the safety and tolerability of the drug and its efficacy only represented an exploratory endpoint, >50% of bosutinib-treated patients appeared to maintain clinical stability [71]. Considering the very limited number of patients enrolled and the short period of treatment, however, further clinical trials will be required to support the possible use of bosutinib in ALS patients.
Masitinib (DrugBank Accession Number DB11526) is the first anticancer therapy approved in veterinary medicine for the treatment of unresectable canine mast cell tumours. Masitinib selectively inhibits the c-KIT receptor, reducing the adverse effects of an overall TK inhibition [72]. The c-KIT receptor in humans is mainly involved in cancer and inflammation and masitinib was found to inhibit neuroinflammation-related symptoms by acting on mast cells and microglia [73]. In agreement, masitinib treatment was found beneficial in multiple sclerosis [73] and Alzheimer’s disease [74], two neurodegenerative diseases in which, as is the case with ALS, neuroinflammation plays a major role. Trias et al. firstly reported that oral masitinib decreased microgliosis and increased survival time of transgenic mSOD1-rats even when delivered after the onset of paralysis [75]. Subsequently, the same group demonstrated that the effects of masitinib in ALS also involved mast cells accumulated around degenerating motor axons and contributing to distal axonopathy and paralysis progression [76,77,78], thereby indicating that, besides microglia, mast cells represent a target of masatinib in ALS pathology. Accordingly, the drug was very recently reported to prevent mast cell infiltration and accumulation around spinal motor neurons of symptomatic mSOD1G93A ALS mice [79]. As for ALS patients, a phase 2/3 randomised, double-blind placebo-controlled clinical trial showed that masitinib plus riluzole significantly reduced decline in the ALS Functional Rating Scale–Revised (ALSFRS-R) as compared to riluzole alone [80,81]. The European Medicines Agency (EMA) is now expected to issue a decision on the conditional approval of Alsitek (masitinib) as an add-on oral therapy for ALS.
7. Monoclonal Antibodies
Monoclonal antibody (mAb)-based immunotherapy is currently regarded as a crucial option in cancer treatment.
After its identification, the CD20 protein was found to be highly expressed on cancerous B cells in non-Hodgkin’s lymphoma, whereas it was not present on healthy immature B cells. Consequently, in the 90s, CD20 became the first target for mAb therapy and the anti-CD20 mAb rituximab (DrugBank Accession Number DB00073) was originally approved by the US FDA as a single agent to treat B-cell non-Hodgkin’s Lymphoma [82].
Rituximab and second-generation anti-CD20 are now used in the treatment of different malignant and non-malignant diseases, including neuroinflammatory conditions such as multiple sclerosis [83,84].
Unpublished data from Lichtenstein’s Lab suggested that reducing B cell counts by treating pre-symptomatic mSOD1G93A mice with rituximab extended ALS mice survival. However, preclinical or clinical trials examining rituximab’s efficacy in ALS are still lacking [85].
8. Vinca Alkaloids
Vincristine (DrugBank Accession Number DB00541) is a vinca alkaloid isolated from Vinca Rosea and it is approved and marketed under several brand names for the treatment of acute leukaemia, malignant lymphoma, Hodgkin’s disease, acute erythraemia and acute panmyelosis.
The antitumour activity of vincristine is due primarily to the inhibition of mitosis by binding the β-subunit of tubulin heterodimers, leading to the arrest of division at metaphases and subsequently to cell death. Binding to the β-tubulin of axon microtubules leads to peripheral neuropathy, representing the major side effect of the long-term administration of vincristine [86]. However, at lower doses or in the presence of a higher clearance rate, vincristine binds tubulin in a reversible manner; its antitumour efficacy as well as its neurotoxicity are thus strictly dependent on the dose and the length of exposure [87].
Vincristine doses lower than those typically used in cancer chemotherapy have been proven to delay the onset of motor deficit and improve the lifespan of mSOD1G93A ALS mice; these beneficial effects were associated with a reduction in microglial cell proliferation, which in ALS sustains neuroinflammation by the release of pro-inflammatory cytokines [88].
Due to its ability to increase platelet counts at low doses, vincristine is also used to treat patients with immune thrombocytopenia [89,90]. As ALS patients’ platelets were found to be more aggregated or grouped than controls [91,92], the effects of vincristine on platelet activation should also be taken into account when considering its possible use in ALS treatment.
9. Immunomodulating Agents
Thalidomide (Thalomid® DrugBank Accession Number DB01041), a glutamic acid derivative, was first proposed as a sedative in the late 1950s and subsequently withdrawn in 1961 when it was shown to be teratogenic, causing severe birth defects (phocomelia) when given to pregnant women.
Several decades later, thalidomide was additionally found to exert immunosuppressive, anti-inflammatory and anti-angiogenic effects, suggesting a therapeutic use in inflammatory and malignant diseases. Indeed, in 1998, the Food and Drug Administration approved thalidomide as a therapy for erythema nodosum leprosum, and subsequently the drug was also approved for the treatment of multiple myeloma [93].
Mechanistically, one of the primary effects of thalidomide is the selective inhibition of TNF-α synthesis by activated monocytes and microglia [94]. Such a mechanism makes thalidomide potentially useful to mitigate neuroinflammatory-associated diseases [95].
In 2006, Kiaei et al. showed that the oral administration of thalidomide or its immunomodulatory imide drug analogue lenalidomide (Revlimid®, DrugBank Accession Number DB00480), used to treat multiple myeloma and anaemia in low- to intermediate-risk myelodysplastic syndrome, was able to modify the disease course and to extend the lifespan of mSOD1G93A ALS mice. Such effects were associated with a reduction in TNF-α levels and motor neuron death [96]. A subsequent study showed clear protective effects of lenalidomide even if administered (orally) in already symptomatic mice [97].
Unfortunately, however, the available clinical evidence does not support the use of these compounds to treat ALS, as in a phase II clinical trial thalidomide not only failed to improve the ALS Functional Rating Scale (ALSFRS) and/or the pulmonary function of patients, but also induced several side effects [98].
10. Concluding Remarks
Building upon the consideration that neurodegeneration and cancer share some common mechanisms, it has been suggested that anticancer drugs might be repurposed for the treatment of neurodegenerative diseases [13,14]. Drug repurposing is particularly interesting for diseases that are at the same time rare and very serious, and for which no effective treatments are available. ALS represents a paradigmatic example of the above class.
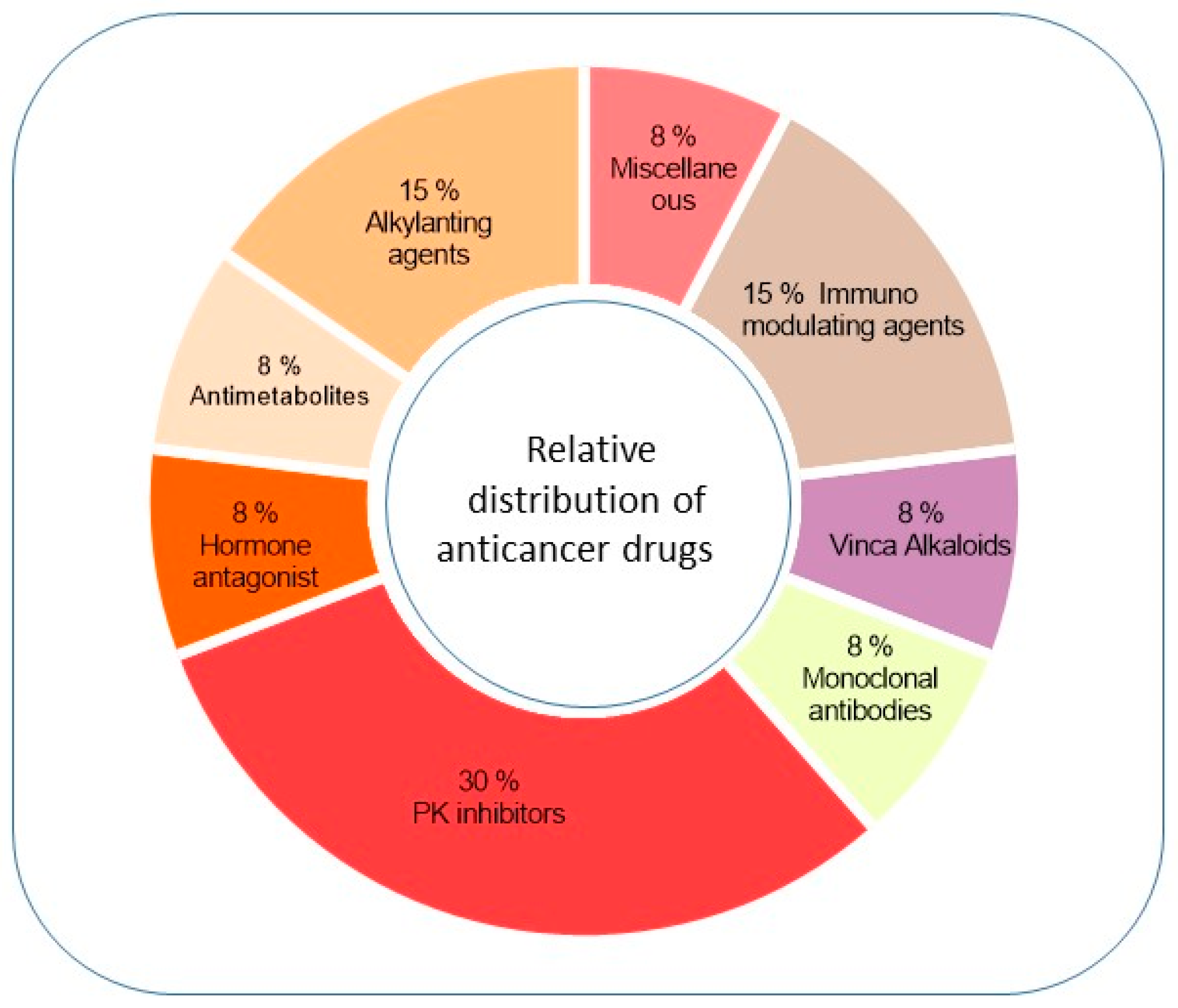
In the present review, we examined anticancer drugs belonging to different classes (Figure 1) and discussed the mechanisms underlying their possible therapeutic role in ALS (Table 1).
This seems at odds with the obvious observation that—since anticancer and anti-ALS treatments should operate in opposite directions (i.e., counteracting vs. promoting cell survival), thus having opposite effects—there is no way the same drugs could be used to treat both conditions.
An effective example arises from autophagy and apoptosis, which play a central role in both conditions. Anticancer therapies are designed to decrease uncontrolled cell proliferation by boosting apoptosis, whereas the inhibition of apoptosis and promotion of autophagy, whose failure leads to neurodegeneration and cell death, are attempted to treat neurodegenerative diseases [99]. However, depending on the administered dosage, the same drug can have opposite effects; a behaviour known as hormesis (namely, the phenomenon in which small doses of toxins and other stressors show stimulative effects [100]). Hormetic behaviour could explain the effectiveness of some anticancer drugs in ALS disease. For example, the anticancer drug fenretinide can act as a pro-apoptotic/pro-autophagic drug in a concentration-dependent manner [15]. Used at a sub-toxic dosage, chronic fenretinide administration was able to delay locomotor deficit progression in ALS mice, preserving muscle cells and motor neurons from mutant SOD1 toxicity [29]. Hormesis could also explain the effects elicited by the anticancer drug 5-FU in ALS mice [50], since it was reported to show hormetic reactions on cultured cancer cells [101]. Again, vincristine, used at doses lower than those normally employed as anticancer treatments, was found to delay disease progression in ALS mice [88].
Besides hormesis, another possible explanation for the apparent effectiveness of anticancer drugs in neurodegenerative conditions lies in their multitarget activities. This may be particularly relevant for a multifactorial disease like ALS, in which a traditional drug discovery approach (single target—single drug) may not be enough to fill the unmet need. Indeed, a multitarget pharmacology has been described for the majority of small molecule anticancer drugs [102]. Although such mechanisms are still poorly studied, their possible role in drug repositioning deserves further investigation.
Of interest within this review appears to be the polypharmacology of immunomodulatory imide drugs (thalidomide and its derivate lenalidomide), which exert their theratogenic and anticancer activity primarily by targeting the human cerebral protein with the Ion protease, Cereblon (CRBN) [103]. However, both thalidomide and its analogue lenalidomide were proven to down-regulate tumour necrosis factor-α (TNF-α) using a Cereblon-independent mechanism, suggesting a new therapeutic use for these drugs in disorders in which neuroinflammation exerts a pivotal role [95].
However interesting it may be, drug repositioning based on multitarget pharmacology is a very complex—and often unpredictable—approach. Indeed, to reduce the risk of unwanted effects in humans, almost every target should be identified and characterised. When repositioning is based on a pharmacological target that is never exploited within the clinical setting, further dose-response data may be required, and this might sometimes result in the need to return to the preclinical stages of regulatory approval.
Furthermore, proposing the use of cancer drugs as a treatment for neurological diseases requires a precise evaluation of cognitive function, as one of their major side effects affecting patients’ quality of life is a decline in learning, attention, executive functions, memory, multitasking and processing speed [104]. This condition, known as “chemobrain” or “chemofog”, on one hand confirms that anticancer drugs may influence central nervous system processes while also cautioning on the other hand about the possible neurotoxic effects of these drugs.
As a whole, the interesting evidence collected and discussed in this review seems to support the fascinating idea of repurposing anticancer drugs for ALS treatment. Hormesis and multitarget pharmacology could partially explain why drugs designed to block cell growth and survival should be used in neurodegenerative diseases.
The application of a network-based approach, which has already been successfully used for the repositioning of other pharmacological classes in ALS, has the potential to yield new knowledge [105]. Indeed, the evidence reported in this narrative review is mainly based on clinical and epidemiological evaluation in human studies and phenotypic change exerted by anticancer drugs in animal models. Combining a systematic computational approach based on the network medicine construct could lead to additional information on the potential of proposed repositioning [106].
However, the complexity of these phenomena, together with the poor knowledge of the CNS effects of anticancer drugs, dictates the need for a deeper mechanistic characterisation before considering this potential therapeutic option.
Author Contributions
Conceptualisation and original draft preparation, R.L.P.; review and editing, R.L.P., M.A. and P.P. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Istituto Superiore di Sanità, intramural funding “Ricerca Corrente”.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef]
- Bianchi, E.; Pupillo, E.; De Feudis, A.; Enia, G.; Vitelli, E.; Beghi, E. Trends in survival of ALS from a population-based registry. Amyotroph. Lateral Scler. Front. Degener. 2022, 23, 344–352. [Google Scholar] [CrossRef]
- Xu, L.; Liu, T.; Liu, L.; Yao, X.; Chen, L.; Fan, D.; Zhan, S.; Wang, S. Global variation in prevalence and incidence of amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. 2020, 267, 944–953. [Google Scholar] [CrossRef]
- Keon, M.; Musrie, B.; Dinger, M.; Brennan, S.E.; Santos, J.; Saksena, N.K. Destination Amyotrophic Lateral Sclerosis. Front. Neurol. 2021, 12, 596006. [Google Scholar] [CrossRef] [PubMed]
- Riancho, J.; Gil-Bea, F.J.; Santurtun, A.; López de Munaín, A. Amyotrophic lateral sclerosis: A complex syndrome that needs an integrated research approach. Neural Regen. Res. 2019, 14, 193–196. [Google Scholar] [CrossRef]
- Akçimen, F.; Lopez, E.R.; Landers, J.E.; Nath, A.; Chiò, A.; Chia, R.; Traynor, B.J. Amyotrophic lateral sclerosis: Translating genetic discoveries into therapies. Nat. Rev. Genet. 2023, 24, 642–658. [Google Scholar] [CrossRef] [PubMed]
- Silani, V.; Ludolph, A.; Fornai, F. The emerging picture of ALS: A multisystem, not only a “motor neuron disease”. Arch. Ital. Biol. 2017, 155, 99–109. [Google Scholar] [CrossRef]
- Jaiswal, M.K. Riluzole and edaravone: A tale of two amyotrophic lateral sclerosis drugs. Med. Res. Rev. 2019, 39, 733–748. [Google Scholar] [CrossRef]
- Sardana, D.; Zhu, C.; Zhang, M.; Gudivada, R.C.; Yang, L.; Jegga, A.G. Drug repositioning for orphan diseases. Brief. Bioinform. 2011, 12, 346–356. [Google Scholar] [CrossRef] [PubMed]
- von Eichborn, J.; Murgueitio, M.S.; Dunkel, M.; Koerner, S.; Bourne, P.E.; Preissner, R. PROMISCUOUS: A database for network-based drug-repositioning. Nucleic Acids Res. 2011, 39, D1060–D1066. [Google Scholar] [CrossRef]
- Mogavero, M.P.; Silvani, A.; DelRosso, L.M.; Salemi, M.; Ferri, R. Focus on the Complex Interconnection between Cancer, Narcolepsy and Other Neurodegenerative Diseases: A Possible Case of Orexin-Dependent Inverse Comorbidity. Cancers 2021, 13, 2612. [Google Scholar] [CrossRef]
- Seo, J.; Park, M. Molecular crosstalk between cancer and neurodegenerative diseases. Cell. Mol. Life Sci. 2020, 77, 2659–2680. [Google Scholar] [CrossRef]
- Liu, D.Z. Repurposing cancer drugs to treat neurological diseases—Src inhibitors as examples. Neural Regen. Res. 2017, 12, 910–911. [Google Scholar] [CrossRef]
- Advani, D.; Gupta, R.; Tripathi, R.; Sharma, S.; Ambasta, R.K.; Kumar, P. Protective role of anticancer drugs in neurodegenerative disorders: A drug repurposing approach. Neurochem. Int. 2020, 140, 104841. [Google Scholar] [CrossRef]
- Potenza, R.L.; Lodeserto, P.; Orienti, I. Fenretinide in Cancer and Neurological Disease: A Two-Face Janus Molecule. Int. J. Mol. Sci. 2022, 23, 7426. [Google Scholar] [CrossRef]
- Riancho, J.; Delgado-Alvarado, M.; Andreu, M.D.; Paz-Fajardo, L.; Arozamena, S.; Gil-Bea, F.J.; López de Munaín, A. Amyotrophic lateral sclerosis (ALS), cancer, autoimmunity and metabolic disorders: An unsolved tantalizing challenge. Br. J. Pharmacol. 2021, 178, 1269–1278. [Google Scholar] [CrossRef]
- Taguchi, Y.H.; Wang, H. Genetic Association between Amyotrophic Lateral Sclerosis and Cancer. Genes 2017, 8, 243. [Google Scholar] [CrossRef]
- Papa, L.; Hahn, M.; Marsh, E.L.; Evans, B.S.; Germain, D. SOD2 to SOD1 switch in breast cancer. J. Biol. Chem. 2014, 289, 5412–5416. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, I.; Azuma, Y.; Yamaguchi, M. Cancer-related genes and ALS. Front. Biosci. (Landmark Ed.) 2019, 24, 1241–1258. [Google Scholar] [CrossRef] [PubMed]
- Riancho, J.; Gil-Bea, F.J.; Castanedo-Vazquez, D.; Sedano, M.J.; Zufiría, M.; de Eulate, G.F.G.; Poza, J.J.; Lopez de Munain, A. Clinical evidences supporting the Src/c-Abl pathway as potential therapeutic target in amyotrophic lateral sclerosis. J. Neurol. Sci. 2018, 393, 80–82. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Choi, E.J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 2010, 1802, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, K.L.; Kalmar, B.; Rhymes, E.R.; Fellows, A.D.; Ahmed, M.; Whiting, P.; Davies, C.H.; Greensmith, L.; Schiavo, G. Inhibiting p38 MAPK alpha rescues axonal retrograde transport defects in a mouse model of ALS. Cell Death Dis. 2018, 9, 596. [Google Scholar] [CrossRef]
- Fang, F.; Al-Chalabi, A.; Ronnevi, L.O.; Turner, M.R.; Wirdefeldt, K.; Kamel, F.; Ye, W. Amyotrophic lateral sclerosis and cancer: A register-based study in Sweden. Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Freedman, D.M.; Curtis, R.E.; Daugherty, S.E.; Goedert, J.J.; Kuncl, R.W.; Tucker, M.A. The association between cancer and amyotrophic lateral sclerosis. Cancer Causes Control 2013, 24, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Gibson, S.B.; Abbott, D.; Farnham, J.M.; Thai, K.K.; McLean, H.; Figueroa, K.P.; Bromberg, M.B.; Pulst, S.M.; Cannon-Albright, L. Population-based risks for cancer in patients with ALS. Neurology 2016, 87, 289–294. [Google Scholar] [CrossRef]
- Rotmensz, N.; De Palo, G.; Formelli, F.; Costa, A.; Marubini, E.; Campa, T.; Crippa, A.; Danesini, G.; Grottaglie, M.D.; Di Mauro, M.; et al. Long-term tolerability of fenretinide (4-HPR) in breast cancer patients. Eur. J. Cancer Clin. Oncol. 1991, 27, 1127–1131. [Google Scholar] [CrossRef]
- European Medicines Agency. EU/3/06/426: Public Summary of Positive Opinion for Orphan Designation of Fenretinide for the Treatment of Primary Malignant Bone Tumours. Available online: https://www.ema.europa.eu/en/documents/orphan-designation/eu306426-public-summary-positive-opinion-orphan-designation-fenretinide-treatment-primary-malignant-bone-tumours_en.pdf (accessed on 15 January 2024).
- European Medicines Agency. EU/3/16/1751: Public Summary of Positive Opinion for Orphan Designation of Fenretinide for the Treatment of Peripheral T-Cell Lymphoma. Available online: https://ec.europa.eu/health/documents/community-register/2016/20161014136138/dec_136138_en.pdf (accessed on 15 January 2024).
- Orienti, I.; Armida, M.; Dobrowolny, G.; Pepponi, R.; Sollazzini, G.; Pezzola, A.; Casola, I.; Musarò, A.; Popoli, P.; Potenza, R.L. Fenretinide Beneficial Effects on Amyotrophic Lateral Sclerosis-associated SOD1G93A Mutant Protein Toxicity: In Vitro and In Vivo Evidences. Neuroscience 2021, 473, 1–12. [Google Scholar] [CrossRef]
- Cao, J.; Ying, M.; Xie, N.; Lin, G.; Dong, R.; Zhang, J.; Yan, H.; Yang, X.; He, Q.; Yang, B. The Oxidation States of DJ-1 Dictate the Cell Fate in Response to Oxidative Stress Triggered by 4-HPR: Autophagy or Apoptosis? Antioxid. Redox Signal. 2014, 21, 1443–1459. [Google Scholar] [CrossRef]
- Kim, Y.-K.; Hammerling, U. The mitochondrial PKCδ/retinol signal complex exerts real-time control on energy homeostasis. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158614. [Google Scholar] [CrossRef]
- Zhang, C.; Xu, C.; Gao, X.; Yao, Q. Platinum-based drugs for cancer therapy and anti-tumor strategies. Theranostics 2022, 12, 2115–2132. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Jamieson, E.R.; Lippard, S.J. Structure, Recognition, and Processing of Cisplatin-DNA Adducts. Chem. Rev. 1999, 99, 2467–2498. [Google Scholar] [CrossRef] [PubMed]
- Calderone, V.; Casini, A.; Mangani, S.; Messori, L.; Orioli, P.L. Structural investigation of cisplatin-protein interactions: Selective platination of His19 in a cuprozinc superoxide dismutase. Angew. Chem. Int. Ed. Engl. 2006, 45, 1267–1269. [Google Scholar] [CrossRef]
- Gruzman, A.; Wood, W.L.; Alpert, E.; Prasad, M.D.; Miller, R.G.; Rothstein, J.D.; Bowser, R.; Hamilton, R.; Wood, T.D.; Cleveland, D.W.; et al. Common molecular signature in SOD1 for both sporadic and familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2007, 104, 12524–12529. [Google Scholar] [CrossRef] [PubMed]
- Prudencio, M.; Durazo, A.; Whitelegge, J.P.; Borchelt, D.R. An examination of wild-type SOD1 in modulating the toxicity and aggregation of ALS-associated mutant SOD1. Hum. Mol. Genet. 2010, 19, 4774–4789. [Google Scholar] [CrossRef]
- Banci, L.; Bertini, I.; Cantini, F.; Kozyreva, T.; Massagni, C.; Palumaa, P.; Rubino, J.T.; Zovo, K. Human superoxide dismutase 1 (hSOD1) maturation through interaction with human copper chaperone for SOD1 (hCCS). Proc. Natl. Acad. Sci. USA 2012, 109, 13555–13560. [Google Scholar] [CrossRef]
- Cozzolino, M.; Amori, I.; Pesaresi, M.G.; Ferri, A.; Nencini, M.; Carrì, M.T. Cysteine 111 affects aggregation and cytotoxicity of mutant Cu,Zn-superoxide dismutase associated with familial amyotrophic lateral sclerosis. J. Biol. Chem. 2008, 283, 866–874. [Google Scholar] [CrossRef]
- Perri, E.R.; Parakh, S.; Vidal, M.; Mehta, P.; Ma, Y.; Walker, A.K.; Atkin, J.D. The Cysteine (Cys) Residues Cys-6 and Cys-111 in Mutant Superoxide Dismutase 1 (SOD1) A4V Are Required for Induction of Endoplasmic Reticulum Stress in Amyotrophic Lateral Sclerosis. J. Mol. Neurosci. 2020, 70, 1357–1368, Erratum in J. Mol. Neurosci. 2020, 70, 1369. [Google Scholar] [CrossRef] [PubMed]
- Banci, L.; Bertini, I.; Blaževitš, O.; Calderone, V.; Cantini, F.; Mao, J.; Trapananti, A.; Vieru, M.; Amori, I.; Cozzolino, M.; et al. Interaction of cisplatin with human superoxide dismutase. J. Am. Chem. Soc. 2012, 134, 7009–7014. [Google Scholar] [CrossRef]
- Arbab, A.S. New Targeting in the Reversal of Resistant Glioblastomas. In Cancer Sensitizing Agents for Chemotherapy, 1st ed.; Elsevier Science: Amsterdam, The Netherlands, 2021; Volume 14, pp. 145–160. [Google Scholar]
- Lu, H.; Chen, I.; Shimoda, L.A.; Park, Y.; Zhang, C.; Tran, L.; Zhang, H.; Semenza, G.L. Chemotherapy-Induced Ca2+ Release Stimulates Breast Cancer Stem Cell Enrichment. Cell Rep. 2017, 18, 1946–1957, Erratum in Cell Rep. 2021, 34, 108605. [Google Scholar] [CrossRef]
- Nardo, G.; Pozzi, S.; Pignataro, M.; Lauranzano, E.; Spano, G.; Garbelli, S.; Mantovani, S.; Marinou, K.; Papetti, L.; Monteforte, M.; et al. Amyotrophic lateral sclerosis multiprotein biomarkers in peripheral blood mononuclear cells. PLoS ONE 2011, 6, e25545. [Google Scholar] [CrossRef]
- van de Giessen, E.; Fogh, I.; Gopinath, S.; Smith, B.; Hu, X.; Powell, J.; Andersen, P.; Nicholson, G.; Al Chalabi, A.; Shaw, C.E. Association study on glutathione S-transferase omega 1 and 2 and familial ALS. Amyotroph. Lateral Scler. 2008, 9, 81–84. [Google Scholar] [CrossRef]
- Mackenzie, I.R.; Rademakers, R.; Neumann, M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010, 9, 995–1007. [Google Scholar] [CrossRef]
- Cha, S.J.; Han, Y.J.; Choi, H.J.; Kim, H.J.; Kim, K. Glutathione S-Transferase Rescues Motor Neuronal Toxicity in Fly Model of Amyotrophic Lateral Sclerosis. Antioxidants 2020, 9, 615. [Google Scholar] [CrossRef]
- Cha, S.J.; Lee, S.; Choi, H.J.; Han, Y.J.; Jeon, Y.M.; Jo, M.; Lee, S.; Nahm, M.; Lim, S.M.; Kim, S.H.; et al. Therapeutic modulation of GSTO activity rescues FUS-associated neurotoxicity via deglutathionylation in ALS disease models. Dev. Cell 2022, 57, 783–798.e8. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, K.; Jacob, S.T. An alternative molecular mechanism of action of 5-fluorouracil, a potent anticancer drug. Biochem. Pharmacol. 1997, 53, 1569–1575. [Google Scholar] [CrossRef] [PubMed]
- Rando, A.; de la Torre, M.; Martinez-Muriana, A.; Zaragoza, P.; Musaro, A.; Hernández, S.; Navarro, X.; Toivonen, J.M.; Osta, R. Chemotherapeutic agent 5-fluorouracil increases survival of SOD1 mouse model of ALS. PLoS ONE 2019, 14, e0210752. [Google Scholar] [CrossRef] [PubMed]
- Pokrishevsky, E.; Hong, R.H.; Mackenzie, I.R.; Cashman, N.R. Spinal cord homogenates from SOD1 familial amyotrophic lateral sclerosis induce SOD1 aggregation in living cells. PLoS ONE 2017, 12, e0184384. [Google Scholar] [CrossRef] [PubMed]
- DuVal, M.G.; Hinge, V.K.; Snyder, N.; Kanyo, R.; Bratvold, J.; Pokrishevsky, E.; Cashman, N.R.; Blinov, N.; Kovalenko, A.; Allison, W.T. Tryptophan 32 mediates SOD1 toxicity in a in vivo motor neuron model of ALS and is a promising target for small molecule therapeutics. Neurobiol. Dis. 2019, 124, 297–310. [Google Scholar] [CrossRef]
- Lee, W.L.; Cheng, M.H.; Chao, H.T.; Wang, P.H. The role of selective estrogen receptor modulators on breast cancer: From tamoxifen to raloxifene. Taiwan J. Obstet. Gynecol. 2008, 47, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Colón, J.M.; Miranda, J.D. Tamoxifen: An FDA approved drug with neuroprotective effects for spinal cord injury recovery. Neural Regen. Res. 2016, 11, 1208–1211. [Google Scholar] [CrossRef]
- Sun, X.; Ji, C.; Hu, T.; Wang, Z.; Chen, G. Tamoxifen as an effective neuroprotectant against early brain injury and learning deficits induced by subarachnoid hemorrhage: Possible involvement of inflammatory signaling. J. Neuroinflamm. 2013, 10, 157. [Google Scholar] [CrossRef]
- Wang, I.F.; Guo, B.S.; Liu, Y.C.; Wu, C.C.; Yang, C.H.; Tsai, K.J.; Shen, C.K. Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA-binding protein 43. Proc. Natl. Acad. Sci. USA 2012, 109, 15024–15029. [Google Scholar] [CrossRef]
- Chen, P.C.; Hsieh, Y.C.; Huang, C.C.; Hu, C.J. Tamoxifen for amyotrophic lateral sclerosis: A randomized double-blind clinical trial. Medicine 2020, 99, e20423. [Google Scholar] [CrossRef]
- Pfeiffer, R.M.; Mayer, B.; Kuncl, R.W.; Check, D.P.; Cahoon, E.K.; Rivera, D.R.; Freedman, D.M. Identifying potential targets for prevention and treatment of amyotrophic lateral sclerosis based on a screen of medicare prescription drugs. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 235–245. [Google Scholar] [CrossRef]
- Schlatterer, S.D.; Acker, C.M.; Davies, P. c-Abl in neurodegenerative disease. J. Mol. Neurosci. 2011, 45, 445–452. [Google Scholar] [CrossRef]
- Feng, L.; Fu, S.; Yao, Y.; Li, Y.; Xu, L.; Zhao, Y.; Luo, L. Roles for c-Abl in postoperative neurodegeneration. Int. J. Med. Sci. 2022, 19, 1753–1761. [Google Scholar] [CrossRef]
- Jiang, Y.M.; Yamamoto, M.; Kobayashi, Y.; Yoshihara, T.; Liang, Y.; Terao, S.; Takeuchi, H.; Ishigaki, S.; Katsuno, M.; Adachi, H.; et al. Gene expression profile of spinal motor neurons in sporadic amyotrophic lateral sclerosis. Ann. Neurol. 2005, 57, 236–251. [Google Scholar] [CrossRef] [PubMed]
- Katsumata, R.; Ishigaki, S.; Katsuno, M.; Kawai, K.; Sone, J.; Huang, Z.; Adachi, H.; Tanaka, F.; Urano, F.; Sobue, G. c-Abl inhibition delays motor neuron degeneration in the G93A mouse, an animal model of amyotrophic lateral sclerosis. PLoS ONE 2012, 7, e46185. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Cross, D.; Jänne, P.A. Kinase drug discovery 20 years after imatinib: Progress and future directions. Nat. Rev. Drug Discov. 2021, 20, 551–569. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Kulshrestha, R.; Singh, N.; Jaggi, A.S. Expanding spectrum of anticancer drug, imatinib, in the disorders affecting brain and spinal cord. Pharmacol. Res. 2019, 143, 86–96. [Google Scholar] [CrossRef]
- Rojas, F.; Gonzalez, D.; Cortes, N.; Ampuero, E.; Hernández, D.E.; Fritz, E.; Abarzua, S.; Martinez, A.; Elorza, A.A.; Alvarez, A.; et al. Reactive oxygen species trigger motoneuron death in non-cell-autonomous models of ALS through activation of c-Abl signaling. Front. Cell. Neurosci. 2015, 9, 203. [Google Scholar] [CrossRef]
- McGary, E.C.; Onn, A.; Mills, L.; Heimberger, A.; Eton, O.; Thomas, G.W.; Shtivelband, M.; Bar-Eli, M. Imatinib mesylate inhibits platelet-derived growth factor receptor phosphorylation of melanoma cells but does not affect tumorigenicity in vivo. J. Investig. Dermatol. 2004, 122, 400–405. [Google Scholar] [CrossRef]
- Imamura, K.; Izumi, Y.; Watanabe, A.; Tsukita, K.; Woltjen, K.; Yamamoto, T.; Hotta, A.; Kondo, T.; Kitaoka, S.; Ohta, A.; et al. The Src/c-Abl pathway is a potential therapeutic target in amyotrophic lateral sclerosis. Sci. Transl. Med. 2017, 9, eaaf3962. [Google Scholar] [CrossRef]
- Osaki, T.; Uzel, S.G.M.; Kamm, R.D. Microphysiological 3D model of amyotrophic lateral sclerosis (ALS) from human iPS-derived muscle cells and optogenetic motor neurons. Sci. Adv. 2018, 4, eaat5847. [Google Scholar] [CrossRef]
- Lonskaya, I.; Hebron, M.L.; Desforges, N.M.; Franjie, A.; Moussa, C.E. Tyrosine kinase inhibition increases functional parkin-Beclin-1 interaction and enhances amyloid clearance and cognitive performance. EMBO Mol. Med. 2013, 5, 1247–1262. [Google Scholar] [CrossRef]
- Imamura, K.; Izumi, Y.; Banno, H.; Uozumi, R.; Morita, S.; Egawa, N.; Ayaki, T.; Nagai, M.; Nishiyama, K.; Watanabe, Y.; et al. Induced pluripotent stem cell-based Drug Repurposing for Amyotrophic lateral sclerosis Medicine (iDReAM) study: Protocol for a phase I dose escalation study of bosutinib for amyotrophic lateral sclerosis patients. BMJ Open 2019, 9, e033131. [Google Scholar] [CrossRef]
- Imamura, K.; Izumi, Y.; Nagai, M.; Nishiyama, K.; Watanabe, Y.; Hanajima, R.; Egawa, N.; Ayaki, T.; Oki, R.; Fujita, K.; et al. Safety and tolerability of bosutinib in patients with amyotrophic lateral sclerosis (iDReAM study): A multicentre, open-label, dose-escalation phase 1 trial. eClinicalMedicine 2022, 53, 101707. [Google Scholar] [CrossRef] [PubMed]
- Dubreuil, P.; Letard, S.; Ciufolini, M.; Gros, L.; Humbert, M.; Castéran, N.; Borge, L.; Hajem, B.; Lermet, A.; Sippl, W.; et al. Masitinib (AB1010), a potent and selective tyrosine kinase inhibitor targeting KIT. PLoS ONE 2009, 4, e7258. [Google Scholar] [CrossRef] [PubMed]
- Vermersch, P.; Benrabah, R.; Schmidt, N.; Zéphir, H.; Clavelou, P.; Vongsouthi, C.; Dubreuil, P.; Moussy, A.; Hermine, O. Masitinib treatment in patients with progressive multiple sclerosis: A randomized pilot study. BMC Neurol. 2012, 12, 36. [Google Scholar] [CrossRef] [PubMed]
- Piette, F.; Belmin, J.; Vincent, H.; Schmidt, N.; Pariel, S.; Verny, M.; Marquis, C.; Mely, J.; Hugonot-Diener, L.; Kinet, J.P.; et al. Masitinib as an adjunct therapy for mild-to-moderate Alzheimer’s disease: A randomised, placebo-controlled phase 2 trial. Alzheimers Res. Ther. 2011, 3, 16. [Google Scholar] [CrossRef] [PubMed]
- Trias, E.; Ibarburu, S.; Barreto-Núñez, R.; Babdor, J.; Maciel, T.T.; Guillo, M.; Gros, L.; Dubreuil, P.; Díaz-Amarilla, P.; Cassina, P.; et al. Post-paralysis tyrosine kinase inhibition with masitinib abrogates neuroinflammation and slows disease progression in inherited amyotrophic lateral sclerosis. J. Neuroinflamm. 2016, 13, 177. [Google Scholar] [CrossRef]
- Trias, E.; Ibarburu, S.; Barreto-Núñez, R.; Varela, V.; Moura, I.C.; Dubreuil, P.; Hermine, O.; Beckman, J.S.; Barbeito, L. Evidence for mast cells contributing to neuromuscular pathology in an inherited model of ALS. JCI Insight 2017, 2, e95934. [Google Scholar] [CrossRef]
- Trias, E.; King, P.H.; Si, Y.; Kwon, Y.; Varela, V.; Ibarburu, S.; Kovacs, M.; Moura, I.C.; Beckman, J.S.; Hermine, O.; et al. Mast cells and neutrophils mediate peripheral motor pathway degeneration in ALS. JCI Insight 2018, 3, e123249. [Google Scholar] [CrossRef] [PubMed]
- Trias, E.; Kovacs, M.; King, P.H.; Si, Y.; Kwon, Y.; Varela, V.; Ibarburu, S.; Moura, I.C.; Hermine, O.; Beckman, J.S.; et al. Schwann cells orchestrate peripheral nerve inflammation through the expression of CSF1, IL-34, and SCF in amyotrophic lateral sclerosis. Glia 2020, 68, 1165–1181. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, M.; Alamón, C.; Maciel, C.; Varela, V.; Ibarburu, S.; Tarragó, L.; King, P.H.; Si, Y.; Kwon, Y.; Hermine, O.; et al. The pathogenic role of c-Kit+ mast cells in the spinal motor neuron-vascular niche in ALS. Acta Neuropathol. Commun. 2021, 9, 136. [Google Scholar] [CrossRef]
- Mora, J.S.; Genge, A.; Chio, A.; Estol, C.J.; Chaverri, D.; Hernández, M.; Marín, S.; Mascias, J.; Rodriguez, G.E.; Povedano, M.; et al. AB10015 STUDY GROUP. Masitinib as an add-on therapy to riluzole in patients with amyotrophic lateral sclerosis: A randomized clinical trial. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 5–14. [Google Scholar] [CrossRef]
- Mora, J.S.; Bradley, W.G.; Chaverri, D.; Hernández-Barral, M.; Mascias, J.; Gamez, J.; Gargiulo-Monachelli, G.M.; Moussy, A.; Mansfield, C.D.; Hermine, O.; et al. Long-term survival analysis of masitinib in amyotrophic lateral sclerosis. Ther. Adv. Neurol. Disord. 2021, 14, 17562864211030365. [Google Scholar] [CrossRef] [PubMed]
- Plosker, G.L.; Figgitt, D.P. Rituximab: A review of its use in non-Hodgkin’s lymphoma and chronic lymphocytic leukaemia. Drugs 2003, 63, 803–843. [Google Scholar] [CrossRef]
- Whittam, D.H.; Tallantyre, E.C.; Jolles, S.; Huda, S.; Moots, R.J.; Kim, H.J.; Robertson, N.P.; Cree, B.A.C.; Jacob, A. Rituximab in neurological disease: Principles, evidence and practice. Pract. Neurol. 2019, 19, 5–20. [Google Scholar] [CrossRef]
- Lin, M.; Zhang, J.; Zhang, Y.; Luo, J.; Shi, S. Ocrelizumab for multiple sclerosis. Cochrane Database Syst. Rev. 2022, 5, CD013247. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Armon, C.; Barkhaus, P.; Barnes, B.; Benatar, M.; Bertorini, T.; Bromberg, M.; Carter, G.T.; Crayle, J.; Cudkowicz, M.; et al. ALSUntangled #67: Rituximab. Amyotroph. Lateral Scler. Front. Degener. 2023, 24, 544–547. [Google Scholar] [CrossRef]
- Mora, E.; Smith, E.M.; Donohoe, C.; Hertz, D.L. Vincristine-induced peripheral neuropathy in pediatric cancer patients. Am. J. Cancer Res. 2016, 6, 2416–2430. [Google Scholar]
- Shukla, R.; Singh, A.; Singh, K.K. Vincristine-based nanoformulations: A preclinical and clinical studies overview. Drug Deliv. Transl. Res. 2024, 14, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Bruce, K.M.; Narayan, K.; Kong, H.C.; Larmour, I.; Lopes, E.C.; Turner, B.J.; Bertram, J.F.; Cheema, S.S. Chemotherapy delays progression of motor neuron disease in the SOD1 G93A transgenic mouse. Chemotherapy 2004, 50, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Lien, L.M.; Lu, W.J.; Lin, K.H.; Kang, L.H.; Chen, T.Y.; Lin, B.J.; Lu, Y.C.; Huang, C.Y.; Shih, C.M.; Chen, H.; et al. Influence of Vincristine, Clinically Used in Cancer Therapy and Immune Thrombocytopenia, on the Function of Human Platelets. Molecules 2021, 26, 5340. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.C.; Meyers, K.M. Canine idiopathic thrombocytopenic purpura. J. Vet. Intern. Med. 1996, 10, 207–218. [Google Scholar] [CrossRef]
- Kiktenko, A.I.; Zlobina, G.P.; Brusov, O.S.; Zakharova, M.N. Structure of peripheral blood platelets surface in patients with amyotrophic lateral sclerosis and multiple sclerosis. Zhurnal Nevrologii i Psikhiatrii Imeni SS Korsakova 2005, 105, 40–42. (In Russian) [Google Scholar]
- Shrivastava, M.; Vivekanandhan, S.; Behari, M. Mitochondrial perturbance and execution of apoptosis in platelet mitochondria of patients with amyotrophic lateral sclerosis. Int. J. Neurosci. 2011, 121, 149–158. [Google Scholar] [CrossRef]
- Melchert, M.; List, A. The thalidomide saga. Int. J. Biochem. Cell Biol. 2007, 39, 1489–1499. [Google Scholar] [CrossRef]
- Lokensgard, J.R.; Hu, S.; van Fenema, E.M.; Sheng, W.S.; Peterson, P.K. Effect of thalidomide on chemokine production by human microglia. J. Infect. Dis. 2000, 182, 983–987. [Google Scholar] [CrossRef]
- Kopp, K.O.; Greer, M.E.; Glotfelty, E.J.; Hsueh, S.C.; Tweedie, D.; Kim, D.S.; Reale, M.; Vargesson, N.; Greig, N.H. A New Generation of IMiDs as Treatments for Neuroinflammatory and Neurodegenerative Disorders. Biomolecules 2023, 13, 747. [Google Scholar] [CrossRef]
- Kiaei, M.; Petri, S.; Kipiani, K.; Gardian, G.; Choi, D.K.; Chen, J.; Calingasan, N.Y.; Schafer, P.; Muller, G.W.; Stewart, C.; et al. Thalidomide and lenalidomide extend survival in a transgenic mouse model of amyotrophic lateral sclerosis. J. Neurosci. 2006, 26, 2467–2473. [Google Scholar] [CrossRef]
- Neymotin, A.; Petri, S.; Calingasan, N.Y.; Wille, E.; Schafer, P.; Stewart, C.; Hensley, K.; Beal, M.F.; Kiaei, M. Lenalidomide (Revlimid) administration at symptom onset is neuroprotective in a mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 2009, 220, 191–197. [Google Scholar] [CrossRef]
- Stommel, E.W.; Cohen, J.A.; Fadul, C.E.; Cogbill, C.H.; Graber, D.J.; Kingman, L.; Mackenzie, T.; Channon Smith, J.Y.; Harris, B.T. Efficacy of thalidomide for the treatment of amyotrophic lateral sclerosis: A phase II open label clinical trial. Amyotroph Lateral Scler. 2009, 10, 393–404. [Google Scholar] [CrossRef]
- Ghavami, S.; Shojaei, S.; Yeganeh, B.; Ande, S.R.; Jangamreddy, J.R.; Mehrpour, M.; Christoffersson, J.; Chaabane, W.; Moghadam, A.R.; Kashani, H.H.; et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog. Neurobiol. 2014, 112, 24–49. [Google Scholar] [CrossRef]
- Stebbing, A.R. Hormesis—The stimulation of growth by low levels of inhibitors. Sci. Total Environ. 1982, 22, 213–234. [Google Scholar] [CrossRef] [PubMed]
- Yoshimasu, T.; Ohashi, T.; Oura, S.; Kokawa, Y.; Kawago, M.; Hirai, Y.; Miyasaka, M.; Nishiguchi, H.; Kawashima, S.; Yata, Y.; et al. A Theoretical Model for the Hormetic Dose-response Curve for Anticancer Agents. Anticancer Res. 2015, 35, 5851–5855. [Google Scholar] [PubMed]
- Shi, D.; Khan, F.; Abagyan, R. Extended Multitarget Pharmacology of Anticancer Drugs. J. Chem. Inf. Model. 2019, 59, 3006–3017. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.X.; Kortuem, K.M.; Stewart, A.K. Molecular mechanism of action of immune-modulatory drugs thalidomide, lenalidomide and pomalidomide in multiple myeloma. Leuk. Lymphoma 2013, 54, 683–687. [Google Scholar] [CrossRef]
- Fleming, B.; Edison, P.; Kenny, L. Cognitive impairment after cancer treatment: Mechanisms, clinical characterization, and management. BMJ 2023, 380, e071726. [Google Scholar] [CrossRef] [PubMed]
- Fiscon, G.; Conte, F.; Amadio, S.; Volonté, C.; Paci, P. Drug Repurposing: A Network-based Approach to Amyotrophic Lateral Sclerosis. Neurotherapeutics 2021, 18, 1678–1691. [Google Scholar] [CrossRef] [PubMed]
- Vincent, F.; Nueda, A.; Lee, J.; Schenone, M.; Prunotto, M.; Mercola, M. Phenotypic drug discovery: Recent successes, lessons learned and new directions. Nat. Rev. Drug Discov. 2022, 21, 899–914, Erratum in Nat. Rev. Drug Discov. 2022, 21, 541. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Relative distribution of considered anticancer drugs in different classes. The pie chart shows the percentage distribution into the different classes of antineoplastic drugs of the molecules considered here for a possible repositioning in ALS.
Figure 1.
Relative distribution of considered anticancer drugs in different classes. The pie chart shows the percentage distribution into the different classes of antineoplastic drugs of the molecules considered here for a possible repositioning in ALS.

{kind=link}
Table 1.
Anticancer drugs effects in ALS.
| Pharmaceutical Family | Role in Cancer | Drug | Effects in ALS | References |
|---|---|---|---|---|
| Miscellaneous | Induction of apoptosis | Fenretinide | ↓ oxidative damage in SOD1 motor neuron ↑ female survival time | [29] |
| Alkylating Agents | Antimitotic: interfere with DNA replication in cancer cells by adding an alkyl group to DNA | Cisplatin | ↓ SOD1 protein misfolding linking SOD1 cysteine residues ↓ FUS aggregation and toxicity | [39,40] |
| Carboplatin | ↓ locomotor deficit FUS-ALS fly model | [48] | ||
| Antimetabolites | Antimitotic: interfere with DNA or RNA synthesis | 5-fluorouracil | ↓ mSOD1 aggregation toxicity delayed disease onset, ↑ motor performance ↑ survival time of ALS SOD1G93A mice | [50,51] |
| Hormone Antagonists | Antiproliferative: inhibit the growth of sensitive cancer cell by antagonising hormone receptors | Tamoxifen | ↓ Apoptosis ↓ microgliosis ↓ neuronal loss ↑ motor function | [56,57,58] |
| Protein Kinase Inhibitors | Antiproliferative and cytotoxic: inhibit pro-survival kinases in cancer cells | Imatinib | ↓ oxidative stress in SOD1 motor neuron | [65] |
| Dasatinib | ↓ apoptotic motor neuron cell death ↓ motor deficit ↑ ALS mice survival time | [62] | ||
| Bosutinib | ↑ autophagy ↓ misfolded SOD1 protein ↓ spinal motor neuron death delayed disease onset ↑ survival of transgenic mice | [67,68,70,71] | ||
| Masitinib | ↓ microgliosis ↓ axonopathy ↓ mast cells infiltration ↑ survival time of ALS rats ↓ ALSFRS-R decline | [75,76,77,78,79,80,81] | ||
| Monoclonal Antibodies | Targeting CD20 protein on cancerous B cells | Rituximab | ↓ B cell counts ↑ survival time of ALS SOD1G93A mice | [85] |
| Vinca Alkaloids | Antimitotic; inhibit cancer cell division by inhibiting tubulin polymerisation | Vincristine | ↓ gliosis in the spinal cord delayed disease onset in ALS mice | [88] |
| Immunomodulating Agents | Inhibition of angiongenesis and TNF-α production synthesis | Thalidomide/Lenalidomide | ↑ body weight loss ↑ motor competence ↑ survival time of ALS SOD1G93A mice | [96,97,98] |
Superoxide dismutase 1: SOD1; mutant superoxide dismutase 1: mSOD1; fused in sarcoma: FUS; Tumour necrosis factor alpha: TNF-α; ALS Functional Rating scale–Revised: ALSFRS-R.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Potenza, R.L.; Armida, M.; Popoli, P. Can Some Anticancer Drugs Be Repurposed to Treat Amyotrophic Lateral Sclerosis? A Brief Narrative Review. Int. J. Mol. Sci. 2024, 25, 1751. https://doi.org/10.3390/ijms25031751
AMA Style
Potenza RL, Armida M, Popoli P. Can Some Anticancer Drugs Be Repurposed to Treat Amyotrophic Lateral Sclerosis? A Brief Narrative Review. International Journal of Molecular Sciences. 2024; 25(3):1751. https://doi.org/10.3390/ijms25031751
Chicago/Turabian StylePotenza, Rosa Luisa, Monica Armida, and Patrizia Popoli. 2024. "Can Some Anticancer Drugs Be Repurposed to Treat Amyotrophic Lateral Sclerosis? A Brief Narrative Review" International Journal of Molecular Sciences 25, no. 3: 1751. https://doi.org/10.3390/ijms25031751
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.