
The Development and Characterisation of ssDNA Aptamers via a Modified Cell-SELEX Methodology for the Detection of Human Red Blood Cells


Abstract
:1. Introduction
2. Results and Discussion
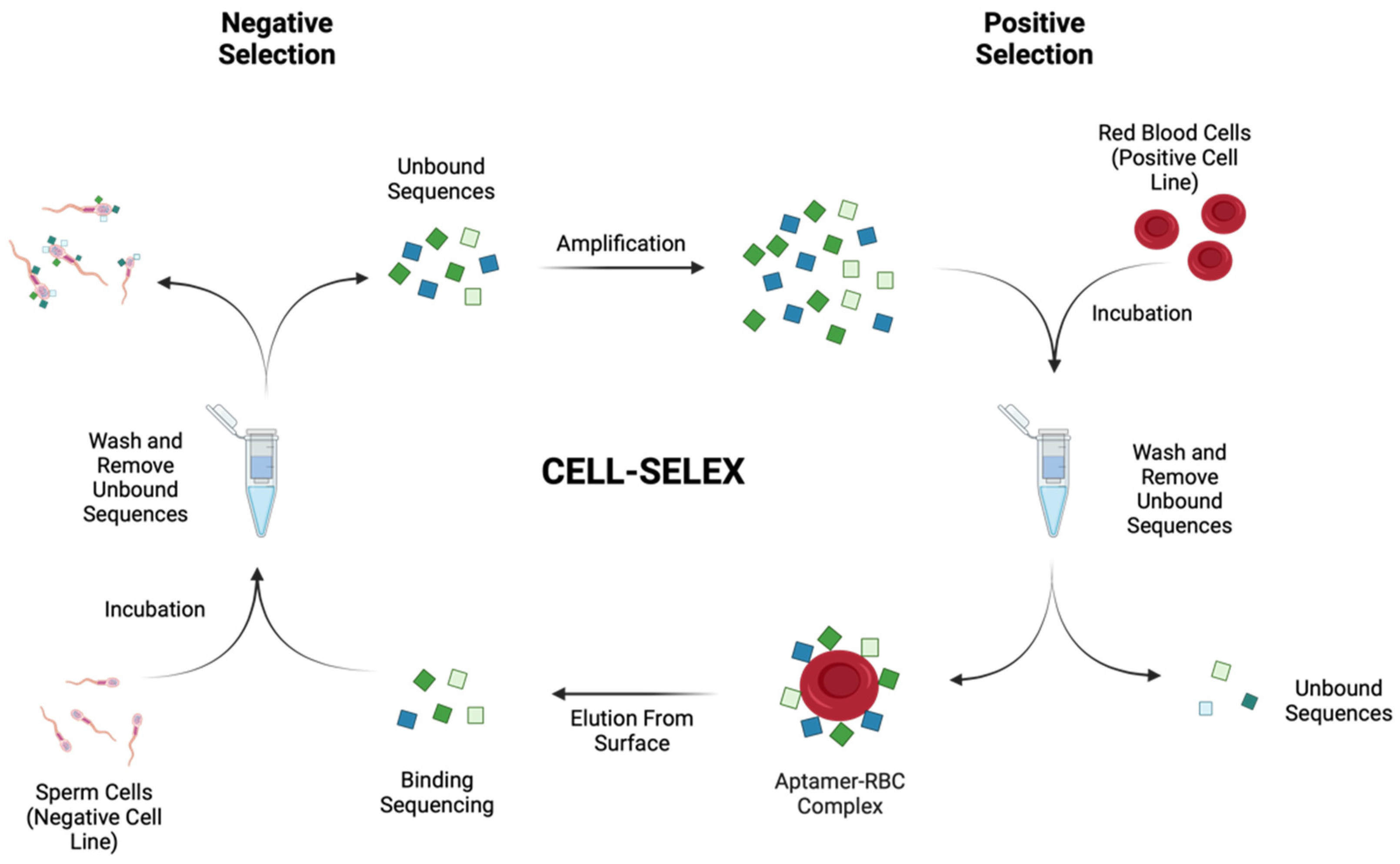
2.1. Cell-SELEX Aptamer Selection
2.2. Massively Parallel Sequencing and Data Processing
2.3. Aptamer Structure Analysis
2.3.1. Secondary Structure Predictions
2.3.2. GQRS Mapper
2.3.3. Circular Dichroism
2.4. Binding Characterisation
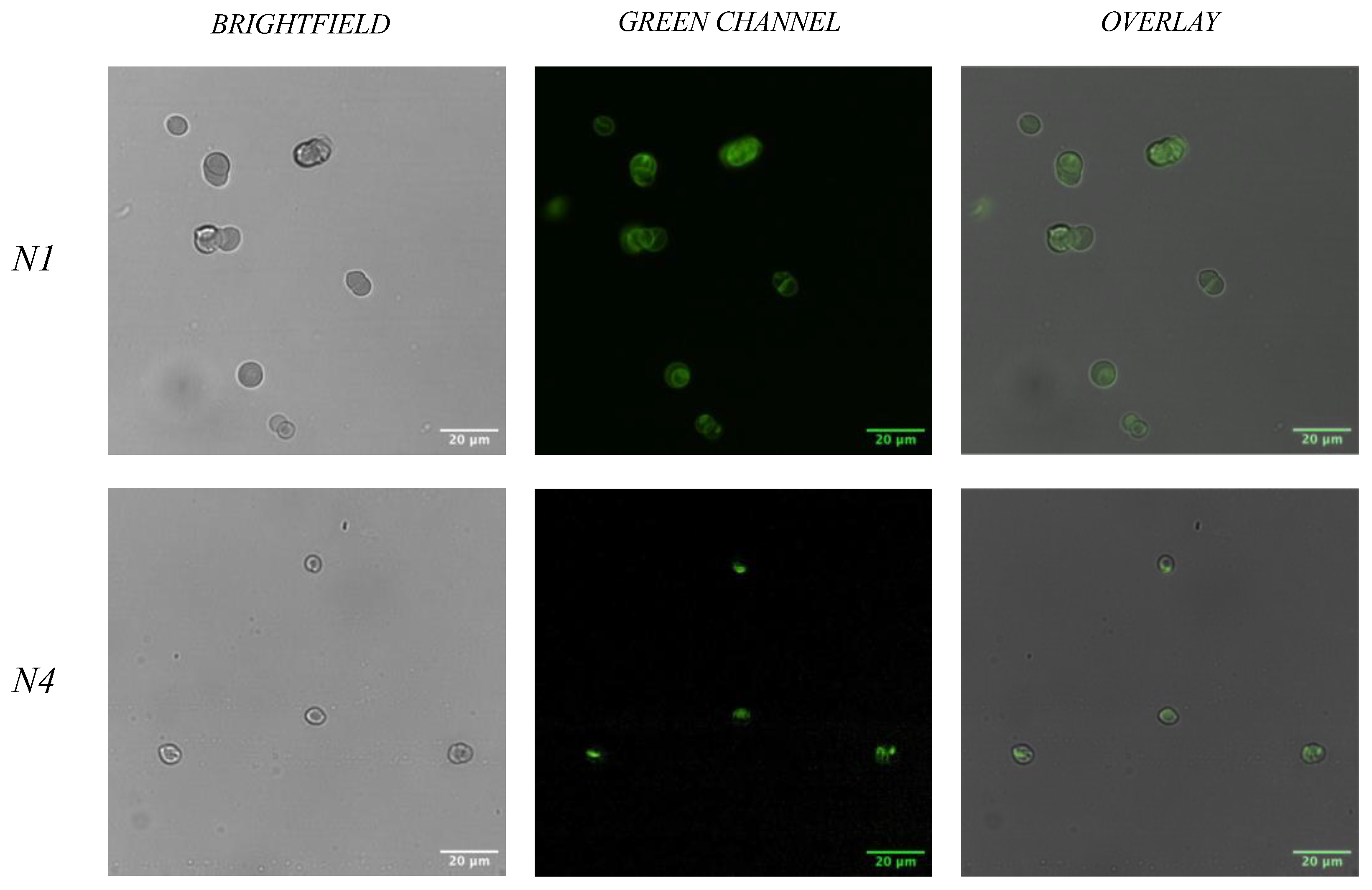
2.4.1. Fluorescent Microscopy
2.4.2. Quantitative Polymerase Chain Reaction (qPCR)
3. Materials and Methods
3.1. Reagents
3.2. Body Fluid Collection
3.3. Cell Isolation
3.4. Cell-SELEX
3.4.1. Incubation and Elution
3.4.2. PCR Amplification
3.4.3. Strand Separation
3.5. Massively Parallel Sequencing
3.6. Circular Dichroism (CD)
3.7. Fluorescent Microscopy
3.8. Quantitative Polymerase Chain Reaction (qPCR)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Williams, G.A. Body fluid identification: A case for more research and innovation. Forensic Sci. Int. Rep. 2020, 2, 100096. [Google Scholar] [CrossRef]
- Gooch, J.P. Biosensors for the Forensic Detection of Body Fluids. Ph.D. Thesis, King’s College London, London, UK, 2019. [Google Scholar]
- Dawnay, N.; Sheppard, K. From crime scene to courtroom: A review of the current bioanalytical evidence workflows used in rape and sexual assault investigations in the United Kingdom. Sci. Justice 2023, 63, 206–228. [Google Scholar] [CrossRef]
- Frascione, N.; Gooch, J.; Daniel, B. Enabling fluorescent biosensors for the forensic identification of body fluids. Analyst 2013, 138, 7279–7288. [Google Scholar] [CrossRef]
- Redhead, P.; Brown, M.K. The acid phosphatase test two minute cut-off: An insufficient time to detect some semen stains. Sci. Justice 2013, 53, 187–191. [Google Scholar] [CrossRef]
- Laporte, G. Wrongful convictions and DNA exonerations: Understanding the role of forensic science. Natl. Inst. Justice J. 2018, 279, 1–16. [Google Scholar]
- Gill, P. Analysis and implications of the miscarriages of justice of Amanda Knox and Raffaele Sollecito. Forensic Sci. Int. Genet. 2016, 23, 9–18. [Google Scholar] [CrossRef]
- Peschel, O.; Kunz, S.N.; Rothschild, M.A.; Mützel, E. Blood stain pattern analysis. Forensic Sci. Med. Pathol. 2011, 7, 257–270. [Google Scholar] [CrossRef]
- Vandewoestyne, M.; Lepez, T.; Van Hoofstat, D.; Deforce, D. Evaluation of a visualization assay for blood on forensic evidence. J. Forensic Sci. 2015, 60, 707–711. [Google Scholar] [CrossRef]
- Pollitt, E.N.; Anderson, J.C.; Scafide, K.N.; Holbrook, D.; D’Silva, G.; Sheridan, D.J. Alternate light source findings of common topical products. J. Forensic Nurs. 2016, 11, 97. [Google Scholar] [CrossRef] [PubMed]
- Kobilinsky, L.F.; Liotti, T.F.; Oeser-Sweat, J. DNA: Forensic and Legal Applications; Wiley Online Library: Hoboken, NJ, USA, 2005. [Google Scholar]
- Gooch, J.; Daniel, B.; Frascione, N. Application of fluorescent substrates to the in situ detection of prostate specific antigen. Talanta 2014, 125, 210–214. [Google Scholar] [CrossRef] [PubMed]
- Tobe, S.S.; Watson, N.; Daeid, N.N. Evaluation of six presumptive tests for blood, their specificity, sensitivity, and effect on high molecular-weight DNA. J. Forensic Sci. 2007, 52, 102–109. [Google Scholar] [CrossRef]
- Virkler, K.; Lednev, I.K. Analysis of body fluids for forensic purposes: From laboratory testing to non-destructive rapid confirmatory identification at a crime scene. Forensic Sci. Int. 2009, 188, 1–17. [Google Scholar] [CrossRef]
- Harris, H.A.; Lee, H.C. Introduction to Forensic Science and Criminalistics; CRC Press: Boca Raton, FL, USA, 2019. [Google Scholar]
- Tombelli, S.; Minunni, M.; Mascini, M. Analytical applications of aptamers. Biosens. Bioelectron. 2005, 20, 2424–2434. [Google Scholar] [CrossRef]
- Gooch, J.; Daniel, B.; Parkin, M.; Frascione, N. Developing aptasensors for forensic analysis. TrAC Trends Anal. Chem. 2017, 94, 150–160. [Google Scholar] [CrossRef]
- Arshavsky-Graham, S.; Heuer, C.; Jiang, X.; Segal, E. Aptasensors versus immunosensors—Which will prevail? Eng. Life Sci. 2022, 22, 319–333. [Google Scholar] [CrossRef]
- Roxo, C.; Kotkowiak, W.; Pasternak, A. G-quadruplex-forming aptamers—Characteristics, applications, and perspectives. Molecules 2019, 24, 3781. [Google Scholar] [CrossRef] [PubMed]
- Gooch, J.; Morillas, A.V.; Frascione, N. Bioanalytical Advancements in the Reliable Visualization and Discrimination of Bodily Fluids. In Emerging Technologies for the Analysis of Forensic Traces; Springer: Berlin/Heidelberg, Germany, 2019; pp. 75–102. [Google Scholar]
- Van Dorst, B.; Mehta, J.; Bekaert, K.; Rouah-Martin, E.; De Coen, W.; Dubruel, P.; Blust, R.; Robbens, J. Recent advances in recognition elements of food and environmental biosensors: A review. Biosens. Bioelectron. 2010, 26, 1178–1194. [Google Scholar] [CrossRef] [PubMed]
- Sefah, K.; Shangguan, D.; Xiong, X.; O’donoghue, M.B.; Tan, W. Development of DNA aptamers using Cell-SELEX. Nat. Protoc. 2010, 5, 1169–1185. [Google Scholar] [CrossRef] [PubMed]
- The Galaxy Community. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2022 Update. Nucleic Acids Res. 2022, 50, W345–W351. [Google Scholar] [CrossRef]
- Hoinka, J.; Backofen, R.; Przytycka, T.M. AptaSUITE: A full-featured bioinformatics framework for the comprehensive analysis of aptamers from HT-SELEX experiments. Mol. Ther. Nucleic Acids 2018, 11, 515–517. [Google Scholar] [CrossRef]
- Gooch, J.; Tungsirisurp, S.; Costanzo, H.; Napier, R.; Frascione, N. Generating aptamers towards human sperm cells using massively parallel sequencing. Anal. Bioanal. Chem. 2021, 413, 5821–5834. [Google Scholar] [CrossRef]
- Hanson, M.S.; Stephenson, A.H.; Bowles, E.A.; Sridharan, M.; Adderley, S.; Sprague, R.S. Phosphodiesterase 3 is present in rabbit and human erythrocytes and its inhibition potentiates iloprost-induced increases in cAMP. Am. J. Physiol.-Heart Circ. Physiol. 2008, 295, H786–H793. [Google Scholar] [CrossRef] [PubMed]
- Higgins, J.M. Red blood cell population dynamics. Clin. Lab. Med. 2015, 35, 43–57. [Google Scholar] [CrossRef]
- Dean, L.; Dean, L. Blood Groups and Red Cell Antigens; NCBI: Bethesda, MD, USA, 2005; Volume 2. [Google Scholar]
- Stefanidou, M.; Alevisopoulos, G.; Spiliopoulou, C. Fundamental Issues in Forensic Semen Detection. West Indian Med. J. 2010, 59, 280. [Google Scholar]
- Garvin, A.M.; Fischer, A.; Schnee-Griese, J.; Jelinski, A.; Bottinelli, M.; Soldati, G.; Tubio, M.; Castella, V.; Monney, N.; Malik, N.; et al. Isolating DNA from sexual assault cases: A comparison of standard methods with a nuclease-based approach. Investig. Genet. 2012, 3, 25. [Google Scholar] [CrossRef]
- Tucker, T.; Marra, M.; Friedman, J.M. Massively parallel sequencing: The next big thing in genetic medicine. Am. J. Hum. Genet. 2009, 85, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Ballard, D.; Winkler-Galicki, J.; Wesoły, J. Massive parallel sequencing in forensics: Advantages, issues, technicalities, and prospects. Int. J. Leg. Med. 2020, 134, 1291–1303. [Google Scholar] [CrossRef]
- Carratto, T.M.T.; Moraes, V.M.S.; Recalde, T.S.F.; Oliveira, M.L.G.D.; Teixeira Mendes-Junior, C. Applications of massively parallel sequencing in forensic genetics. Genet. Mol. Biol. 2022, 45 (Suppl. 1), e20220077. [Google Scholar] [CrossRef]
- Lee, S.J.; Cho, J.; Lee, B.H.; Hwang, D.; Park, J.W. Design and Prediction of Aptamers Assisted by In Silico Methods. Biomedicines 2023, 11, 356. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Zadeh, J.N.; Steenberg, C.D.; Bois, J.S.; Wolfe, B.R.; Pierce, M.B.; Khan, A.R.; Dirks, R.M.; Pierce, N.A. NUPACK: Analysis and design of nucleic acid systems. J. Comput. Chem. 2011, 32, 170–173. [Google Scholar] [CrossRef]
- Abreu, R.D.S.; Antunes, D.; Moreira, A.D.S.; Passetti, F.; Mendonça, J.B.; de Araújo, N.S.; Sassaro, T.F.; Alberto, A.V.P.; Carrossini, N.; Fernandes, P.V.; et al. Next Generation of Ovarian Cancer Detection Using Aptamers. Int. J. Mol. Sci. 2023, 24, 6315. [Google Scholar] [CrossRef]
- Ilgu, M.; Yan, S.; Khounlo, R.M.; Lamm, M.H.; Nilsen-Hamilton, M. Common secondary and tertiary structural features of aptamer–ligand interaction shared by RNA aptamers with different primary sequences. Molecules 2019, 24, 4535. [Google Scholar] [CrossRef]
- Kikin, O.; D’Antonio, L.; Bagga, P.S. QGRS Mapper: A web-based server for predicting G-quadruplexes in nucleotide sequences. Nucleic Acids Res. 2006, 34 (Suppl. 2), W676–W682. [Google Scholar] [CrossRef]
- Burge, S.; Parkinson, G.N.; Hazel, P.; Todd, A.K.; Neidle, S. Quadruplex DNA: Sequence, topology and structure. Nucleic Acids Res. 2006, 34, 5402–5415. [Google Scholar] [CrossRef] [PubMed]
- del Villar-Guerra, R.; Gray, R.D.; Chaires, J.B. Quadruplex DNA structure characterization by circular dichroism. Curr. Protoc. Nucleic Acid Chem. 2017, 68, 17–18. [Google Scholar] [CrossRef] [PubMed]
- Yoshitomi, T.; Hayashi, M.; Oguro, T.; Kimura, K.; Wayama, F.; Furusho, H.; Yoshimoto, K. Binding and structural properties of DNA aptamers with VEGF-A-mimic activity. Mol. Ther. Nucleic Acids 2020, 19, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Kypr, J.; Kejnovská, I.; Renčiuk, D.; Vorlíčková, M. Circular dichroism and conformational polymorphism of DNA. Nucleic Acids Res. 2009, 37, 1713–1725. [Google Scholar] [CrossRef]
- Małgowska, M.; Gudanis, D.; Teubert, A.; Dominiak, G.; Gdaniec, Z. How to study G-quadruplex structures. Biotechnologia 2012, 93, 381–390. [Google Scholar] [CrossRef]
- Kratschmer, C.; Levy, M. Effect of chemical modifications on aptamer stability in serum. Nucleic Acid Ther. 2017, 27, 335–344. [Google Scholar] [CrossRef]
- Zhu, Q.; Liu, G.; Kai, M. DNA aptamers in the diagnosis and treatment of human diseases. Molecules 2015, 20, 20979–20997. [Google Scholar] [CrossRef]
- Lakhin, A.; Tarantul, V.; Gening, L. Aptamers: Problems, solutions and prospects. Acta Naturae 2013, 5, 34–43. [Google Scholar] [CrossRef]
- Ding, F.; Guo, S.; Xie, M.; Luo, W.; Yuan, C.; Huang, W.; Zhou, Y.; Zhang, X.L.; Zhou, X. Diagnostic applications of gastric carcinoma cell aptamers in vitro and in vivo. Talanta 2015, 134, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Zeng, H. Aptamer-based ELISA assay for highly specific and sensitive detection of Zika NS1 protein. Anal. Chem. 2017, 89, 12743–12748. [Google Scholar] [CrossRef]
- Shi, H.; Tang, Z.; Kim, Y.; Nie, H.; Huang, Y.F.; He, X.; Deng, K.; Wang, K.; Tan, W. In vivo fluorescence imaging of tumors using molecular aptamers generated by cell-SELEX. Chem. Asian J. 2010, 5, 2209–2213. [Google Scholar] [CrossRef]
- Aljohani, M.M.; Cialla-May, D.; Popp, J.; Chinnappan, R.; Al-Kattan, K.; Zourob, M. Aptamers: Potential diagnostic and therapeutic agents for blood diseases. Molecules 2022, 27, 383. [Google Scholar] [CrossRef]
- Wang, R.E.; Wu, H.; Niu, Y.; Cai, J. Improving the stability of aptamers by chemical modification. Curr. Med. Chem. 2011, 18, 4126–4138. [Google Scholar] [CrossRef]
- Siddiqui, S.; Yuan, J. Binding Characteristics Study of DNA based Aptamers for E. coli O157:H7. Molecules 2021, 26, 204. [Google Scholar] [CrossRef]
- de Melo, M.I.A.; da Silva Cunha, P.; Ferreira, I.M.; de Andrade, A.S.R. DNA aptamers selection for Staphylococcus aureus cells by SELEX and Cell-SELEX. Mol. Biol. Rep. 2023, 50, 157–165. [Google Scholar] [CrossRef]
- Dos Santos, V.C.; Almeida, N.B.; de Sousa, T.A.; Araujo, E.N.; de Andrade, A.S.; Plentz, F. Real-time PCR for direct aptamer quantification on functionalized graphene surfaces. Sci. Rep. 2019, 9, 19311. [Google Scholar] [CrossRef]
- El-Husseini, D.M.; Sayour, A.E.; Melzer, F.; Mohamed, M.F.; Neubauer, H.; Tammam, R.H. Generation and selection of specific aptamers targeting Brucella species through an enhanced cell-SELEX methodology. Int. J. Mol. Sci. 2022, 23, 6131. [Google Scholar] [CrossRef]
- World Heath Organisation (WHO). WHO Laboratory Manual for the Examination and Processing of Human Semen, 5th ed.; World Health Organisation (WHO): Geneva, Switzerland, 2010.
- Zhang, Y.; Xu, H.; Wang, X.; Wang, L.; Liu, R.; Li, L.; Zhou, H. Single-strained DNA aptamers mask RhD antigenic epitopes on human RhD+ red blood cells to escape alloanti-RhD immunological recognition. Mol. Med. Rep. 2020, 21, 1841–1848. [Google Scholar] [CrossRef]
- Hosseini, S.M.H.; Bassami, M.R.; Haghparast, A.; Sankian, M.; Tabar, G.H. Identification of Aptamers that Specifically Bind to A1 Antigen by Performing Cell-on Human Erythrocytes. Galen Med. J. 2020, 9, e1657. [Google Scholar] [CrossRef]
- Li, X.; Wang, T.; Xie, T.; Dai, J.; Zhang, Y.; Ling, N.; Guo, J.; Li, C.; Sun, X.; Zhang, X.; et al. Aptamer-Mediated Enrichment of Rare Circulating Fetal Nucleated Red Blood Cells for Noninvasive Prenatal Diagnosis. Anal. Chem. 2023, 95, 5419–5427. [Google Scholar] [CrossRef]
- Birch, C.M.; Hou, H.W.; Han, J.; Niles, J.C. Identification of malaria parasite-infected red blood cell surface aptamers by inertial microfluidic SELEX (I-SELEX). Sci. Rep. 2015, 5, 11347. [Google Scholar] [CrossRef]
- Bushnik, E. Partially Robotic Selection of Aptamers to Red Blood Cell Protein Glycophorin A. Master’s Thesis, Université d’Ottawa/University of Ottawa, Ottawa, ON, Canada, 2018. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Round | Donor | RBC Count | Incubation Time | Washes | Negative Selection |
|---|---|---|---|---|---|
| 1 | 1 | 1 × 107 | 1 h | 3 | No |
| 2 | 2 | 5 × 106 | 1 h | 3 | No |
| 3 | 3 | 4 × 106 | 1 h | 3 | No |
| 4 | 4 | 3 × 106 | 1 h | 3 | No |
| 5 | 5 | 2 × 106 | 1 h | 3 | No |
| 6 | 1 | 1 × 106 | 1 h | 3 | No |
| 7 | 2 | 1 × 106 | 30 min | 3 | Yes |
| 8 | 3 | 1 × 106 | 30 min | 4 | Yes |
| 9 | 4 | 1 × 106 | 30 min | 5 | Yes |
| 10 | 5 | 1 × 106 | 30 min | 6 | Yes |
| Sequence (5′–3′) | |
|---|---|
| N1 | ATCCAGAGTGACGCAGCACGGGTTGGGGCTGGTTGTGTGTTGTTTTT TTGGCTGTATGTGGACACGGTGGCTTAGT |
| N2 | ATCCAGAGTGACGCAGCATGTGTGAGTTGGATTGAGTGTGGATTGGG GGGATGATGGGTGGACACGGTGGCTTAGT |
| N3 | ATCCAGAGTGACGCAGCAGGATGGATGTGGTTGGGGATCGGCTGGG GGATTTAGTTGGTGGACACGGTGGCTTAGT |
| N4 | ATCCAGAGTGACGCAGCATGCGGGGAGAGGAGTGTGGGATGGGTTT GTTTGTTTAGGGTGGACACGGTGGCTTAGT |
| N5 | ATCCAGAGTGACGCAGCAATTTGGAGGCGGGATGATATGCGTAGGGG TTTGAGGGGGGTGGACACGGTGGCTTAGT |
| N6 | ATCCAGAGTGACGCAGCAGCAGGGTGTGGGGTAAGTGTGTGGGGTG TTGTTGTTTGGGTGGACACGGTGGCTTAGT |
| N7 | ATCCAGAGTGACGCAGCACGTGGTGTTTTGGGGGTGTTTGATTGGGT TTCTGTTCGGATGGACACGGTGGCTTAGT |
| N8 | ATCCAGAGTGACGCAGCAGGAGAGTTGATTTGGGTGTTGGTGGCTG GGATAGTAAGGGTGGACACGGTGGCTTAGT |
| N9 | ATCCAGAGTGACGCAGCATTATGTGGGGGGTGGTGTACTTGTTTGGG GATCGGTTGGTTGGACACGGTGGCTTAGT |
| Position | Length | QGRS | G-Score | |
|---|---|---|---|---|
| N1 | 21 | 12 | GGTTGGGGCTGG | 19 |
| 50 | 21 | GGCTGTATGTGGACACGGTGG | 14 | |
| N4 | 22 | 22 | GGGGAGAGGAGTGTGGGATGGG | 20 |
| 57 | 14 | GGTGGACACGGTGG | 18 |
| Amplification Step | Temperature (°C) | Time (s) |
|---|---|---|
| Denature | 95.0 | 120 |
| Denature * | 95.0 | 30 |
| Annealing * | 56.3 | 30 |
| Extension * | 72.0 | 30 |
| Extension | 92.0 | 180 |
| Hold | 4.0 | ∞ |
| Reagents | Preliminary PCR (μL) | Trial PCR (μL) | Preparative PCR (μL) |
|---|---|---|---|
| X10 PCR Buffer | 100.00 | 35.00 | 400.00 |
| dNTP Mixture (10 mM) | 20.00 | 7.00 | 80.00 |
| MgCl2 (50 mM) | 40.00 | 14.00 | 160.00 |
| Forward Primer (100 μM) | 5.00 | 1.75 | 20.00 |
| Reverse Primer (100 μM) | 5.00 | 1.75 | 20.00 |
| DNAse-Free Water | 327.00 | 254.45 | 2908.00 |
| Platinum Taq Polymerase | 3.00 | 1.05 | 12.00 |
| Eluted ssDNA Pool | 500.00 | - | 400.00 |
| Amplification Step | Temperature (°C) | Time (s) |
|---|---|---|
| Denature | 98.0 | 30 |
| Denature * | 98.0 | 10 |
| Annealing * | 65.0 | 75 |
| Extension * | 65.0 | 300 |
| Hold | 4.0 | ∞ |
| Reagents | Per Sample (μL) | Total (μL) |
|---|---|---|
| PCR Grade Water | 4.0 | 240.0 |
| KAPA SYBR® Fast Green Master Mix (2×) Universal | 10.0 | 600.0 |
| Forward Primer (10 μM) | 0.4 | 24.0 |
| Reverse Primer (10 μM) | 0.4 | 24.0 |
| 50× Low ROX | 0.2 | 12.0 |
| Template ssDNA | 5.0 | - |
| Amplification Step | Temperature (°C) | Time (s) |
|---|---|---|
| Enzyme Activation | 95.0 | 180 |
| Denature * | 95.0 | 10 |
| Annealing and Extension * | 65.0 | 30 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costanzo, H.; Gooch, J.; Tungsirisurp, S.; Frascione, N. The Development and Characterisation of ssDNA Aptamers via a Modified Cell-SELEX Methodology for the Detection of Human Red Blood Cells. Int. J. Mol. Sci. 2024, 25, 1814. https://doi.org/10.3390/ijms25031814
Costanzo H, Gooch J, Tungsirisurp S, Frascione N. The Development and Characterisation of ssDNA Aptamers via a Modified Cell-SELEX Methodology for the Detection of Human Red Blood Cells. International Journal of Molecular Sciences. 2024; 25(3):1814. https://doi.org/10.3390/ijms25031814
Chicago/Turabian StyleCostanzo, Hayley, James Gooch, Sireethorn Tungsirisurp, and Nunzianda Frascione. 2024. "The Development and Characterisation of ssDNA Aptamers via a Modified Cell-SELEX Methodology for the Detection of Human Red Blood Cells" International Journal of Molecular Sciences 25, no. 3: 1814. https://doi.org/10.3390/ijms25031814