
Lipid Peroxidation as the Mechanism Underlying Polycyclic Aromatic Hydrocarbons and Sunlight Synergistic Toxicity in Dermal Fibroblasts

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
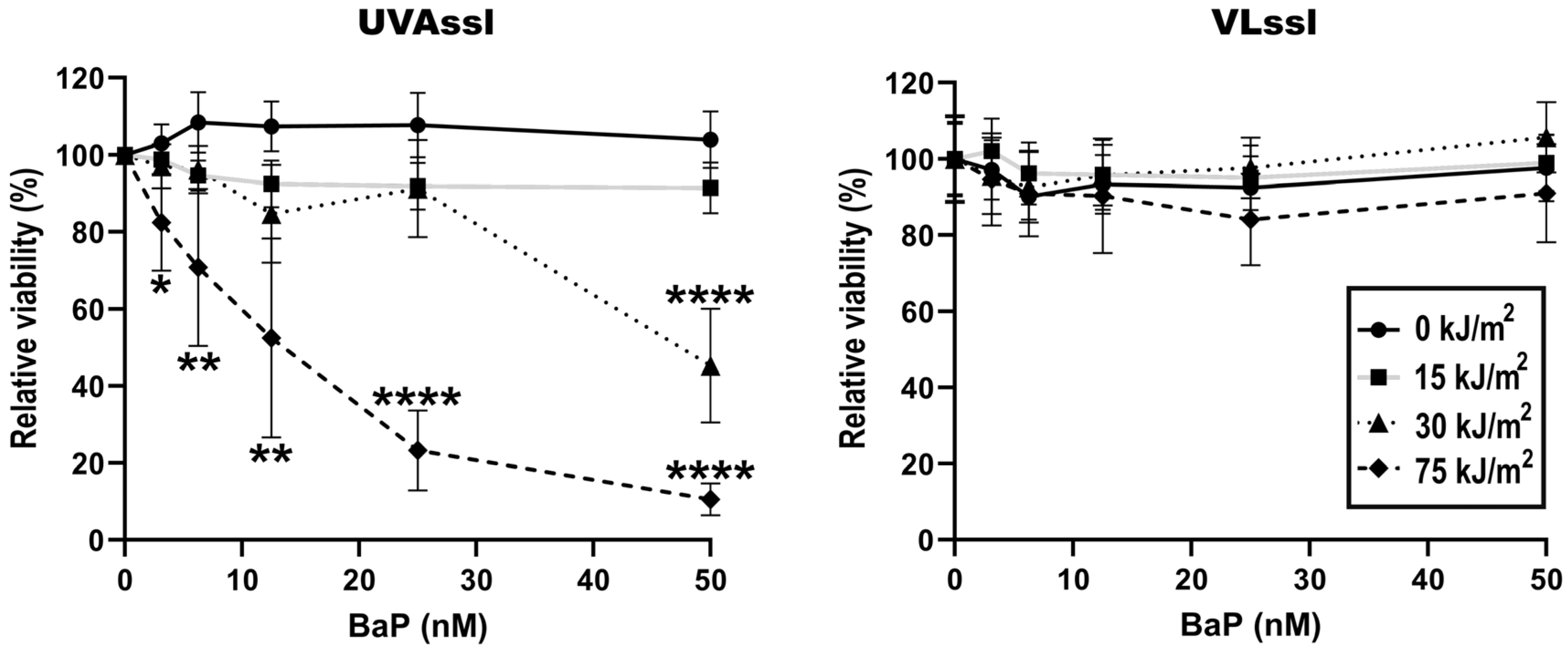
2.1. Sensitivity to UVAssl- or VLssl-Induced Cell Death in BaP-Treated Fibroblasts
2.2. Induction of Oxidative Stress in BaP-Treated Fibroblasts Exposed to UVAssl or VLssl
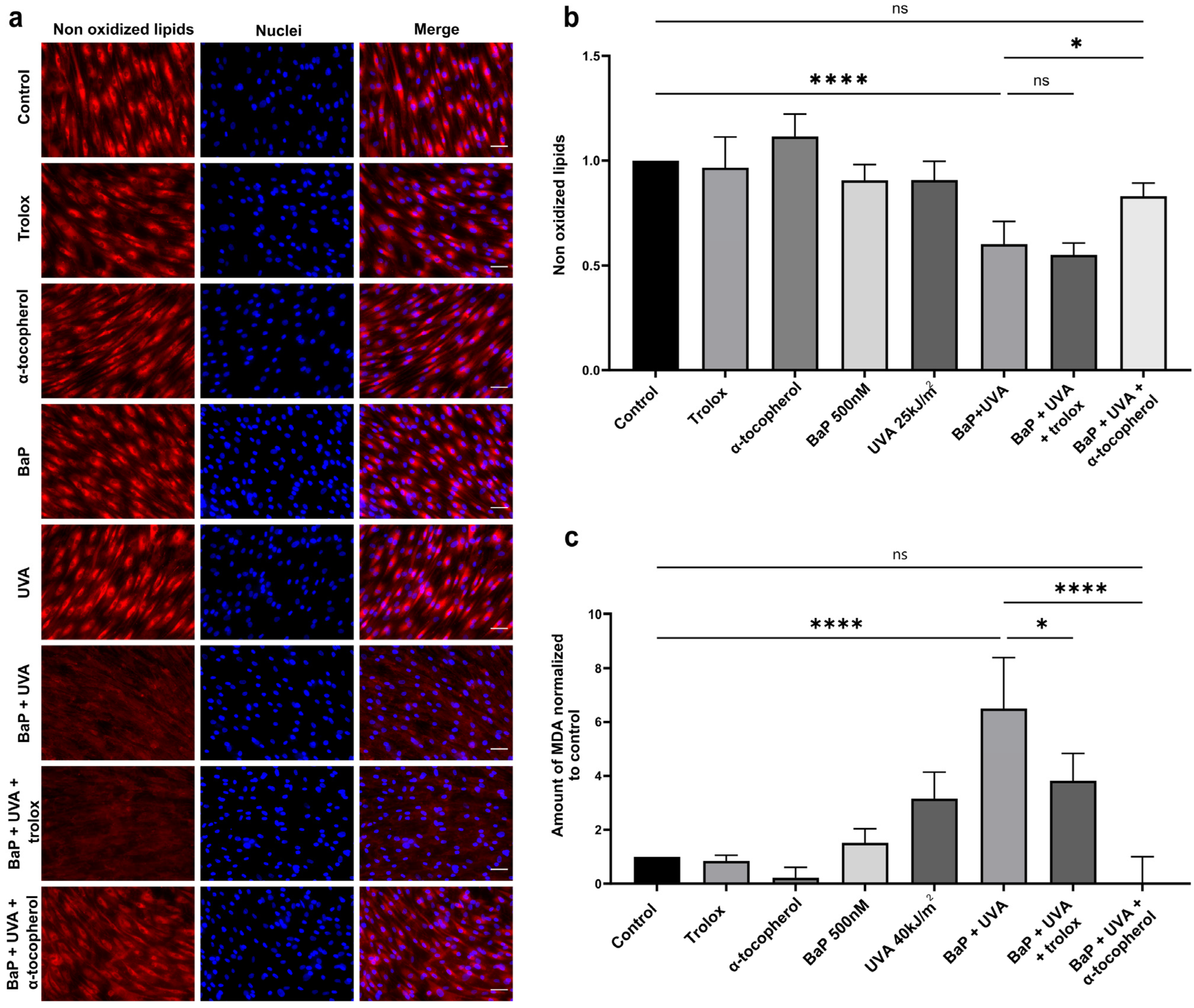
2.3. Lipid Peroxidation and MDA Formation in BaP/UVAssl-Treated Dermal Fibroblasts
2.4. Bulky DNA Adducts
3. Discussion
4. Material and Methods
4.1. Cell Culture
4.2. BaP and Light Exposure
4.3. Viability Assessment
4.4. ROS Formation Analysis
4.5. Antioxidant Treatments
4.6. Lipid Peroxidation and MDA Evaluation
4.7. Bulky DNA Adducts’ Formation
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vierkötter, A.; Schikowski, T.; Ranft, U.; Sugiri, D.; Matsui, M.; Krämer, U.; Krutmann, J. Airborne Particle Exposure and Extrinsic Skin Aging. J. Investig. Dermatol. 2010, 130, 2719–2726. [Google Scholar] [CrossRef]
- Krutmann, J.; Bouloc, A.; Sore, G.; Bernard, B.A.; Passeron, T. The Skin Aging Exposome. J. Dermatol. Sci. 2017, 85, 152–161. [Google Scholar] [CrossRef]
- Opländer, C.; Hidding, S.; Werners, F.B.; Born, M.; Pallua, N.; Suschek, C.V. Effects of Blue Light Irradiation on Human Dermal Fibroblasts. J. Photochem. Photobiol. B Biol. 2011, 103, 118–125. [Google Scholar] [CrossRef]
- Montoni, A.; George, K.M.; Soeur, J.; Tran, C.; Marrot, L.; Rochette, P.J. Chronic UVA1 Irradiation of Human Dermal Fibroblasts: Persistence of DNA Damage and Validation of a Cell Cultured–Based Model of Photoaging. J. Investig. Dermatol. 2019, 139, 1821–1824.e3. [Google Scholar] [CrossRef]
- Piao, M.J.; Ahn, M.J.; Kang, K.A.; Ryu, Y.S.; Hyun, Y.J.; Shilnikova, K.; Zhen, A.X.; Jeong, J.W.; Choi, Y.H.; Kang, H.K.; et al. Particulate Matter 2.5 Damages Skin Cells by Inducing Oxidative Stress, Subcellular Organelle Dysfunction, and Apoptosis. Arch. Toxicol. 2018, 92, 2077–2091. [Google Scholar] [CrossRef]
- Zinflou, C.; Rochette, P.J. Absorption of Blue Light by Cigarette Smoke Components Is Highly Toxic for Retinal Pigmented Epithelial Cells. Arch. Toxicol. 2019, 93, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Botta, C.; Di Giorgio, C.; Sabatier, A.-S.; De Méo, M. Effects of UVA and Visible Light on the Photogenotoxicity of Benzo[a]Pyrene and Pyrene. Environ. Toxicol. 2009, 24, 492–505. [Google Scholar] [CrossRef] [PubMed]
- Soeur, J.; Belaïdi, J.-P.; Chollet, C.; Denat, L.; Dimitrov, A.; Jones, C.; Perez, P.; Zanini, M.; Zobiri, O.; Mezzache, S.; et al. Photo-Pollution Stress in Skin: Traces of Pollutants (PAH and Particulate Matter) Impair Redox Homeostasis in Keratinocytes Exposed to UVA1. J. Dermatol. Sci. 2017, 86, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Burke, K.; Wei, H. Synergistic Damage by UVA Radiation and Pollutants. Toxicol Ind Health 2009, 25, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Von Koschembahr, A.; Youssef, A.; Béal, D.; Gudimard, L.; Giot, J.-P.; Douki, T. Toxicity and DNA Repair in Normal Human Keratinocytes Co-Exposed to Benzo[a]Pyrene and Sunlight. Toxicol. Vitr. 2020, 63, 104744. [Google Scholar] [CrossRef]
- Wang, S.; Sheng, Y.; Feng, M.; Leszczynski, J.; Wang, L.; Tachikawa, H.; Yu, H. Light-Induced Cytotoxicity of 16 Polycyclic Aromatic Hydrocarbons on the US EPA Priority Pollutant List in Human Skin HaCaT Keratinocytes: Relationship between Phototoxicity and Excited State Properties. Environ. Toxicol. 2007, 22, 318–327. [Google Scholar] [CrossRef]
- Foote, C.S. Mechanisms of Photosensitized Oxidation: There Are Several Different Types of Photosensitized Oxidation Which May Be Important in Biological Systems. Science 1968, 162, 963–970. [Google Scholar] [CrossRef]
- Armstrong, B.K.; Kricker, A. The Epidemiology of UV Induced Skin Cancer. J. Photochem. Photobiol. B Biol. 2001, 63, 8–18. [Google Scholar] [CrossRef]
- Brash, D.E. UV Signature Mutations. Photochem. Photobiol. 2015, 91, 15–26. [Google Scholar] [CrossRef] [PubMed]
- El Ghissassi, F.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; Bouvard, V.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A Review of Human Carcinogens—Part D: Radiation. Lancet Oncol. 2009, 10, 751–752. [Google Scholar] [CrossRef] [PubMed]
- Tornaletti, S.; Pfeifer, G.P. UV Damage and Repair Mechanisms in Mammalian Cells. Bioessays 1996, 18, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Douki, T.; Reynaud-Angelin, A.; Cadet, J.; Sage, E. Bipyrimidine Photoproducts Rather than Oxidative Lesions Are the Main Type of DNA Damage Involved in the Genotoxic Effect of Solar UVA Radiation. Biochemistry 2003, 42, 9221–9226. [Google Scholar] [CrossRef] [PubMed]
- Rochette, P.J. UVA-Induced Cyclobutane Pyrimidine Dimers Form Predominantly at Thymine-Thymine Dipyrimidines and Correlate with the Mutation Spectrum in Rodent Cells. Nucleic Acids Res. 2003, 31, 2786–2794. [Google Scholar] [CrossRef] [PubMed]
- Mouret, S.; Baudouin, C.; Charveron, M.; Favier, A.; Cadet, J.; Douki, T. Cyclobutane Pyrimidine Dimers Are Predominant DNA Lesions in Whole Human Skin Exposed to UVA Radiation. Proc. Natl. Acad. Sci. USA 2006, 103, 13765–13770. [Google Scholar] [CrossRef] [PubMed]
- Tewari, A.; Sarkany, R.P.; Young, A.R. UVA1 Induces Cyclobutane Pyrimidine Dimers but Not 6-4 Photoproducts in Human Skin In Vivo. J. Investig. Dermatol. 2012, 132, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Baier, J.; Maisch, T.; Maier, M.; Engel, E.; Landthaler, M.; Bäumler, W. Singlet Oxygen Generation by UVA Light Exposure of Endogenous Photosensitizers. Biophys. J. 2006, 91, 1452–1459. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Douki, T.; Ravanat, J.-L.; Di Mascio, P. Sensitized Formation of Oxidatively Generated Damage to Cellular DNA by UVA Radiation. Photochem. Photobiol. Sci. 2009, 8, 903–911. [Google Scholar] [CrossRef]
- Cadet, J.; Douki, T.; Ravanat, J.-L. Oxidatively Generated Damage to Cellular DNA by UVB and UVA Radiation. Photochem. Photobiol. 2015, 91, 140–155. [Google Scholar] [CrossRef]
- Di Mascio, P.; Martinez, G.R.; Miyamoto, S.; Ronsein, G.E.; Medeiros, M.H.G.; Cadet, J. Singlet Molecular Oxygen Reactions with Nucleic Acids, Lipids, and Proteins. Chem. Rev. 2019, 119, 2043–2086. [Google Scholar] [CrossRef] [PubMed]
- D’Orazio, J.; Jarrett, S.; Amaro-Ortiz, A.; Scott, T. UV Radiation and the Skin. Int. J. Mol. Sci. 2013, 14, 12222–12248. [Google Scholar] [CrossRef]
- Kwon, H.-S.; Ryu, M.H.; Carlsten, C. Ultrafine Particles: Unique Physicochemical Properties Relevant to Health and Disease. Exp. Mol. Med. 2020, 52, 318–328. [Google Scholar] [CrossRef]
- Dabestani, R.; Ivanov, I.N. A Compilation of Physical, Spectroscopic and Photophysical Properties of Polycyclic Aromatic Hydrocarbons. Photochem. Photobiol. 1999, 70, 10–34. [Google Scholar] [CrossRef]
- Outdoor Air Pollution; International Agency for Research on Cancer, World Health Organization: Lyon, France, 2016.
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Some Non-Heterocyclic Polycyclic Aromatic Hydrocarbons and Some Related Exposures. IARC Monogr. Eval. Carcinog. Risks Hum. 2010, 92, 1–853. [Google Scholar]
- Ding, Y.S.; Ashley, D.L.; Watson, C.H. Determination of 10 Carcinogenic Polycyclic Aromatic Hydrocarbons in Mainstream Cigarette Smoke. J. Agric. Food Chem. 2007, 55, 5966–5973. [Google Scholar] [CrossRef]
- Hoffmann, D.; Hoffmann, I.; El-Bayoumy, K. The Less Harmful Cigarette: A Controversial Issue. A Tribute to Ernst L. Wynder. Chem. Res. Toxicol. 2001, 14, 767–790. [Google Scholar] [CrossRef]
- Yang, L.; Li, C.; Tang, X. The Impact of PM2.5 on the Host Defense of Respiratory System. Front. Cell Dev. Biol. 2020, 8, 91. [Google Scholar] [CrossRef] [PubMed]
- Parrado, C.; Mercado-Saenz, S.; Perez-Davo, A.; Gilaberte, Y.; Gonzalez, S.; Juarranz, A. Environmental Stressors on Skin Aging. Mechanistic Insights. Front. Pharmacol. 2019, 10, 759. [Google Scholar] [CrossRef] [PubMed]
- Song, X.F.; Chen, Z.Y.; Zang, Z.J.; Zhang, Y.N.; Zeng, F.; Peng, Y.P.; Yang, C. Investigation of Polycyclic Aromatic Hydrocarbon Level in Blood and Semen Quality for Residents in Pearl River Delta Region in China. Environ. Int. 2013, 60, 97–105. [Google Scholar] [CrossRef]
- Palazzi, P.; Mezzache, S.; Bourokba, N.; Hardy, E.M.; Schritz, A.; Bastien, P.; Emond, C.; Li, J.; Soeur, J.; Appenzeller, B.M.R. Exposure to Polycyclic Aromatic Hydrocarbons in Women Living in the Chinese Cities of BaoDing and Dalian Revealed by Hair Analysis. Environ. Int. 2018, 121, 1341–1354. [Google Scholar] [CrossRef]
- Yin, L.; Morita, A.; Tsuji, T. Alterations of Extracellular Matrix Induced by Tobacco Smoke Extract. Arch. Dermatol. Res. 2000, 292, 188–194. [Google Scholar] [CrossRef]
- Shin, K.-O.; Uchida, Y.; Park, K. Diesel Particulate Extract Accelerates Premature Skin Aging in Human Fibroblasts via Ceramide-1-Phosphate-Mediated Signaling Pathway. Int. J. Mol. Sci. 2022, 23, 2691. [Google Scholar] [CrossRef]
- Park, S.-Y.; Byun, E.; Lee, J.; Kim, S.; Kim, H. Air Pollution, Autophagy, and Skin Aging: Impact of Particulate Matter (PM10) on Human Dermal Fibroblasts. Int. J. Mol. Sci. 2018, 19, 2727. [Google Scholar] [CrossRef] [PubMed]
- Yu, H. Environmental Carcinogenic Polycyclic Aromatic Hydrocarbons: Photochemistry and Phototoxicity. J. Environ. Sci. Health Part C 2002, 20, 149–183. [Google Scholar] [CrossRef]
- Marrot, L. Pollution and Sun Exposure: A Deleterious Synergy. Mechanisms and Opportunities for Skin Protection. CMC 2019, 25, 5469–5486. [Google Scholar] [CrossRef]
- Kadunce, D.P. Cigarette Smoking: Risk Factor for Premature Facial Wrinkling. Ann. Intern. Med. 1991, 114, 840. [Google Scholar] [CrossRef]
- Yin, L.; Morita, A.; Tsuji, T. Skin Aging Induced by Ultraviolet Exposure and Tobacco Smoking: Evidence from Epidemiological and Molecular Studies: Skin Aging Induced by UV and Tobacco Smoking. Photodermatol. Photoimmunol. Photomed. 2001, 17, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Grenier, A.; Morissette, M.C.; Rochette, P.J.; Pouliot, R. The Combination of Cigarette Smoke and Solar Rays Causes Effects Similar to Skin Aging in a Bilayer Skin Model. Sci. Rep. 2023, 13, 17969. [Google Scholar] [CrossRef] [PubMed]
- Ibuki, Y.; Warashina, T.; Noro, T.; Goto, R. Coexposure to Benzo[a]Pyrene plus Ultraviolet A Induces 8-Oxo-7,8-Dihydro-2′-Deoxyguanosine Formation in Human Skin Fibroblasts: Preventive Effects of Anti-Oxidant Agents. Environ. Toxicol. Pharmacol. 2002, 12, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Xia, Q.; Chiang, H.-M.; Yin, J.-J.; Chen, S.; Cai, L.; Yu, H.; Fu, P.P. UVA Photoirradiation of Benzo[a]Pyrene Metabolites: Induction of Cytotoxicity, Reactive Oxygen Species, and Lipid Peroxidation. Toxicol. Ind. Health 2015, 31, 898–910. [Google Scholar] [CrossRef]
- Crallan, R.A.; Ingham, E.; Routledge, M.N. Wavelength Dependent Responses of Primary Human Keratinocytes to Combined Treatment with Benzo[a]Pyrene and UV Light. Mutagenesis 2005, 20, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Saladi, R.; Austin, L.; Gao, D.; Lu, Y.; Phelps, R.; Lebwohl, M.; Wei, H. The Combination of Benzo[a]Pyrene and Ultraviolet A Causes an in Vivo Time-Related Accumulation of DNA Damage in Mouse Skin. Photochem. Photobiol. 2003, 77, 413–419. [Google Scholar] [CrossRef]
- Marnett, L.J. Lipid Peroxidation—DNA Damage by Malondialdehyde. Mutat. Res./Fundam. Mol. Mech. Mutagen. 1999, 424, 83–95. [Google Scholar] [CrossRef]
- Voulgaridou, G.-P.; Anestopoulos, I.; Franco, R.; Panayiotidis, M.I.; Pappa, A. DNA Damage Induced by Endogenous Aldehydes: Current State of Knowledge. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2011, 711, 13–27. [Google Scholar] [CrossRef]
- Yoon, S.; Lim, C.; Chung, H.-J.; Kim, J.-H.; Huh, Y.; Park, K.; Jeong, S. Autophagy Activation by Crepidiastrum Denticulatum Extract Attenuates Environmental Pollutant-Induced Damage in Dermal Fibroblasts. Int. J. Mol. Sci. 2019, 20, 517. [Google Scholar] [CrossRef]
- Poon, P.Y.; Kwok, H.H.; Yue, P.Y.K.; Yang, M.S.M.; Mak, N.K.; Wong, C.K.C.; Wong, R.N.S. Cytoprotective Effect of 20( S )-Rg3 on Benzo[ a ]Pyrene-Induced DNA Damage. Drug Metab. Dispos. 2012, 40, 120–129. [Google Scholar] [CrossRef]
- Kim, K.-H.; Jahan, S.A.; Kabir, E.; Brown, R.J.C. A Review of Airborne Polycyclic Aromatic Hydrocarbons (PAHs) and Their Human Health Effects. Environ. Int. 2013, 60, 71–80. [Google Scholar] [CrossRef]
- Takamura-Enya, T.; Suzuki, H.; Hisamatsu, Y. Mutagenic Activities and Physicochemical Properties of Selected Nitrobenzanthrones. Mutagenesis 2006, 21, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Neal, M.S.; Zhu, J.; Foster, W.G. Quantification of Benzo[a]Pyrene and Other PAHs in the Serum and Follicular Fluid of Smokers versus Non-Smokers. Reprod. Toxicol. 2008, 25, 100–106. [Google Scholar] [CrossRef]
- Kuluncsics, Z.; Perdiz, D.; Brulay, E.; Muel, B.; Sage, E. Wavelength Dependence of Ultraviolet-Induced DNA Damage Distribution: Involvement of Direct or Indirect Mechanisms and Possible Artefacts. J. Photochem. Photobiol. B Biol. 1999, 49, 71–80. [Google Scholar] [CrossRef] [PubMed]
- VanderVeen, L.A.; Hashim, M.F.; Shyr, Y.; Marnett, L.J. Induction of Frameshift and Base Pair Substitution Mutations by the Major DNA Adduct of the Endogenous Carcinogen Malondialdehyde. Proc. Natl. Acad. Sci. USA 2003, 100, 14247–14252. [Google Scholar] [CrossRef] [PubMed]
- Henkler, F.; Stolpmann, K.; Luch, A. Exposure to Polycyclic Aromatic Hydrocarbons: Bulky DNA Adducts and Cellular Responses. In Molecular, Clinical and Environmental Toxicology; Experientia Supplementum; Luch, A., Ed.; Springer: Basel, Switzerland, 2012; Volume 101, pp. 107–131. [Google Scholar] [CrossRef]
- Marquis, O.; Miaud, C.; Ficetola, G.F.; Boscher, A.; Mouchet, F.; Guittonneau, S.; Devaux, A. Variation in Genotoxic Stress Tolerance among Frog Populations Exposed to UV and Pollutant Gradients. Aquat. Toxicol. 2009, 95, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Toyooka, T.; Ohnuki, G.; Ibuki, Y. Solar-Simulated Light-Exposed Benzo[a]Pyrene Induces Phosphorylation of Histone H2AX. Mutat. Res. 2008, 650, 132–139. [Google Scholar] [CrossRef]
- Zinflou, C.; Rochette, P.J. Indenopyrene and Blue-Light Co-Exposure Impairs the Tightly Controlled Activation of Xenobiotic Metabolism in Retinal Pigment Epithelial Cells: A Mechanism for Synergistic Toxicity. Int. J. Mol. Sci. 2023, 24, 17385. [Google Scholar] [CrossRef]
- Al-Baker, E.A.; Oshin, M.; Hutchison, C.J.; Kill, I.R. Analysis of UV-Induced Damage and Repair in Young and Senescent Human Dermal Fibroblasts Using the Comet Assay. Mech. Ageing Dev. 2005, 126, 664–672. [Google Scholar] [CrossRef]
- Sauvaigo, S.; Bonnet-Duquennoy, M.; Odin, F.; Hazane-Puch, F.; Lachmann, N.; Bonté, F.; Kurfürst, R.; Favier, A. DNA Repair Capacities of Cutaneous Fibroblasts: Effect of Sun Exposure, Age and Smoking on Response to an Acute Oxidative Stress. Br. J. Dermatol. 2007, 157, 26–32. [Google Scholar] [CrossRef]
- Moriwaki, S.; Takahashi, Y. Photoaging and DNA Repair. J. Dermatol. Sci. 2008, 50, 169–176. [Google Scholar] [CrossRef]
- Hazane, F.; Sauvaigo, S.; Douki, T.; Favier, A.; Beani, J.-C. Age-Dependent DNA Repair and Cell Cycle Distribution of Human Skin Fibroblasts in Response to UVA Irradiation. J. Photochem. Photobiol. B Biol. 2006, 82, 214–223. [Google Scholar] [CrossRef]
- Ali, R.; Trump, S.; Lehmann, I.; Hanke, T. Live Cell Imaging of the Intracellular Compartmentalization of the Contaminate Benzo[a]Pyrene: Imaging B[a]P Compartmentalization. J. Biophoton 2015, 8, 361–371. [Google Scholar] [CrossRef]
- Ayala-Torres, S.; Chen, Y.; Svoboda, T.; Rosenblatt, J.; Van Houten, B. Analysis of Gene-Specific DNA Damage and Repair Using Quantitative Polymerase Chain Reaction. Methods 2000, 22, 135–147. [Google Scholar] [CrossRef]
- Jung, D.; Cho, Y.; Meyer, J.N.; Di Giulio, R.T. The Long Amplicon Quantitative PCR for DNA Damage Assay as a Sensitive Method of Assessing DNA Damage in the Environmental Model, Atlantic Killifish (Fundulus Heteroclitus). Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2009, 149, 182–186. [Google Scholar] [CrossRef]
- Ponti, M.; Forrow, S.M.; Souhami, R.L.; D’Incalci, M.; Hartley, J.A. Measurement of the Sequence Specificity of Covalent DNA Modification by Antineoplastic Agents Using Taq DNA Polymerase. Nucleic Acids Res. 1991, 19, 2929–2933. [Google Scholar] [CrossRef] [PubMed]
- Furda, A.; Santos, J.H.; Meyer, J.N.; Van Houten, B. Quantitative PCR-Based Measurement of Nuclear and Mitochondrial DNA Damage and Repair in Mammalian Cells. Methods Mol. Biol. 2014, 1105, 419–437. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A Tool to Design Target-Specific Primers for Polymerase Chain Reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Larnac, E.; Montoni, A.; Haydont, V.; Marrot, L.; Rochette, P.J. Lipid Peroxidation as the Mechanism Underlying Polycyclic Aromatic Hydrocarbons and Sunlight Synergistic Toxicity in Dermal Fibroblasts. Int. J. Mol. Sci. 2024, 25, 1905. https://doi.org/10.3390/ijms25031905
Larnac E, Montoni A, Haydont V, Marrot L, Rochette PJ. Lipid Peroxidation as the Mechanism Underlying Polycyclic Aromatic Hydrocarbons and Sunlight Synergistic Toxicity in Dermal Fibroblasts. International Journal of Molecular Sciences. 2024; 25(3):1905. https://doi.org/10.3390/ijms25031905
Chicago/Turabian StyleLarnac, Eloïse, Alicia Montoni, Valérie Haydont, Laurent Marrot, and Patrick J. Rochette. 2024. "Lipid Peroxidation as the Mechanism Underlying Polycyclic Aromatic Hydrocarbons and Sunlight Synergistic Toxicity in Dermal Fibroblasts" International Journal of Molecular Sciences 25, no. 3: 1905. https://doi.org/10.3390/ijms25031905