
Factor H’s Control of Complement Activation Emerges as a Significant and Promising Therapeutic Target for Alzheimer’s Disease Treatment
 , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. Factor H Plays a Protective Role against Complement-Induced Lysis in Blood Endothelial Cells
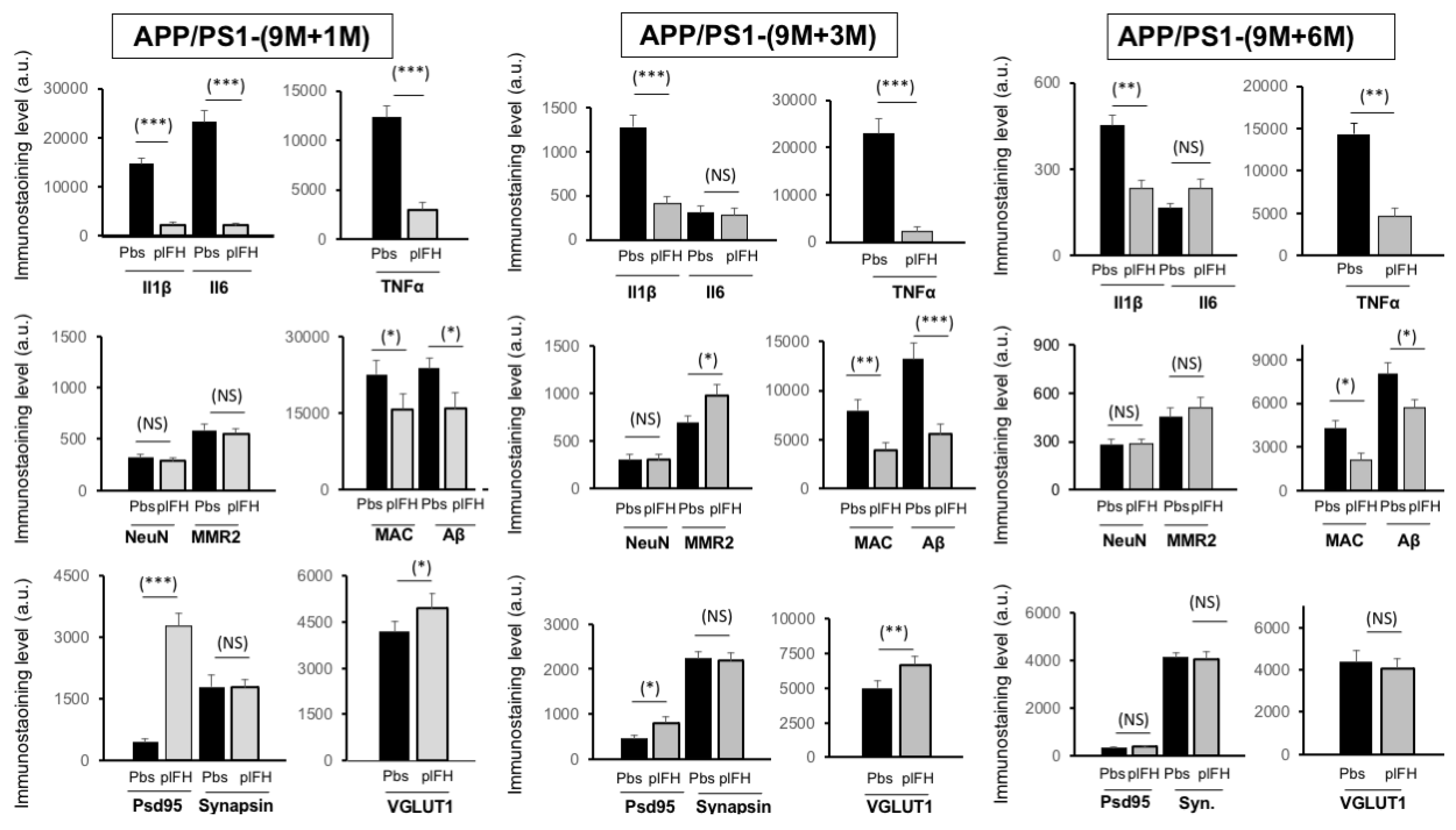
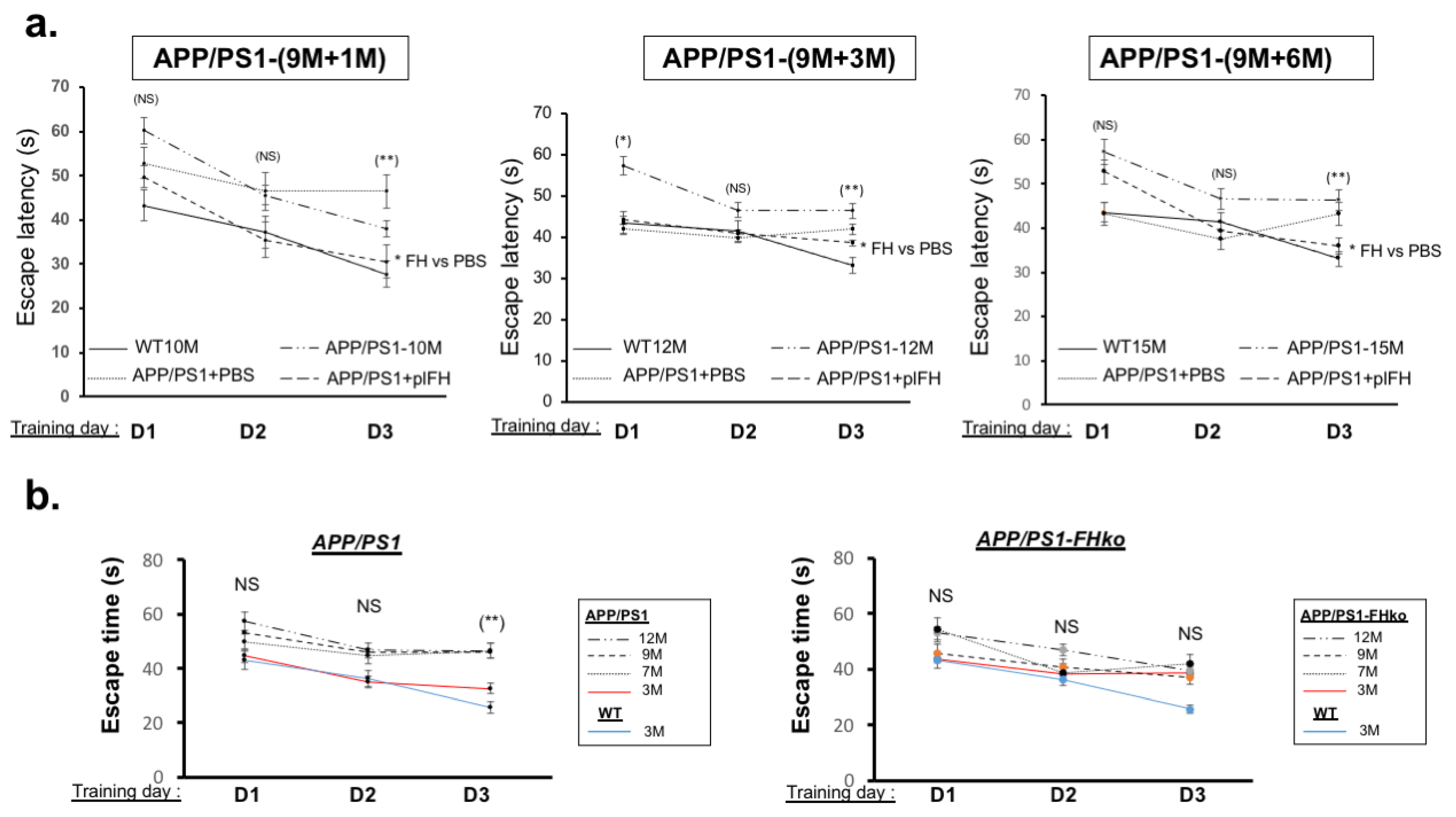
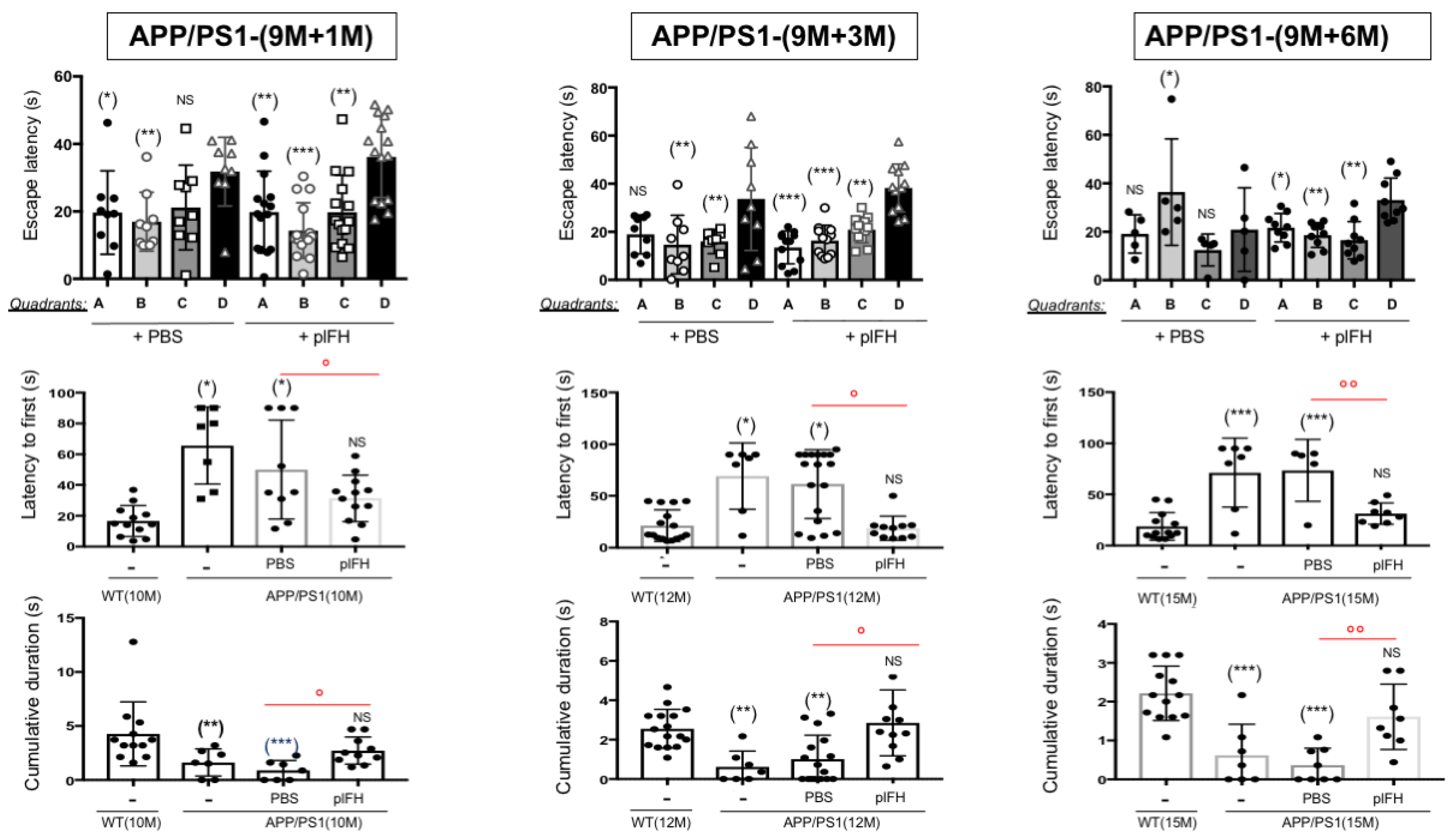
2.2. Factor H Transiently Improves Cognitive Functions in Early-Stage AD-APP/PS1 Mice
2.3. Factor H Improves the Cognitive Functions of 9-Month-Old APP/PS1 Mice up to 6 Months Post-Injection
2.4. Deletion of Factor H Expression Accelerates Loss of Cognitive Functions in APP/PS1 Mice
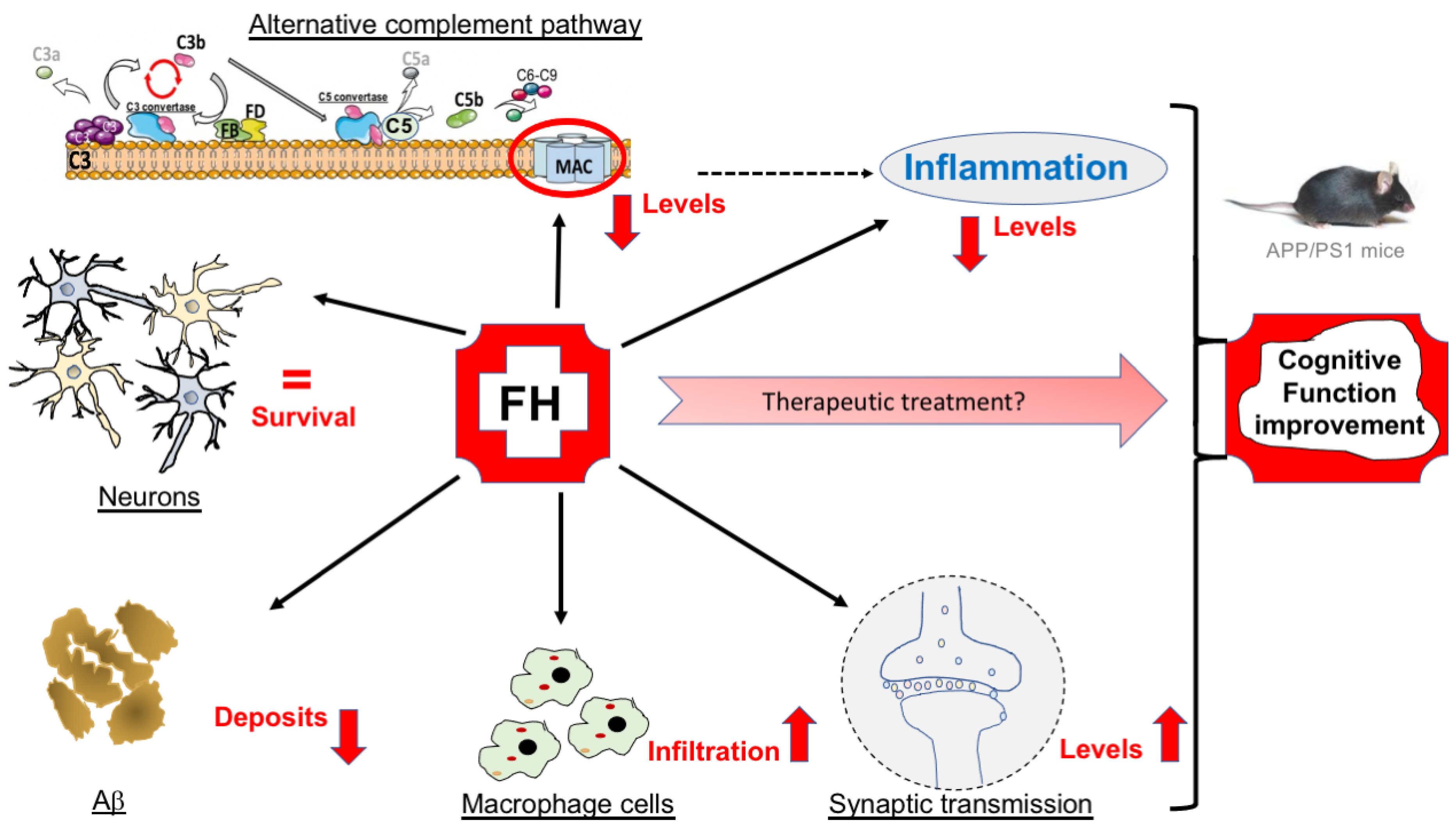
3. Discussion
4. Materials and Methods
4.1. Ethics for Animal Use
4.2. Factor H Injection in Cerebrospinal Fluid Mice
4.3. Optical Clearing:
4.4. Western Blot Analysis
4.5. Immunohistochemistry on Mice Brain
4.6. Morris Water Maze Experiments
4.7. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- PubMed. Alzheimer’s Disease. Available online: https://pubmed.ncbi.nlm.nih.gov/20107219/ (accessed on 28 April 2023).
- Scheff, S.W.; Price, D.A.; Schmitt, F.A.; Mufson, E.J. Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging 2006, 27, 1372–1384. [Google Scholar] [CrossRef]
- Reddy, P.H.; Mani, G.; Park, B.S.; Jacques, J.; Murdoch, G.; Whetsell, W., Jr.; Kaye, J.; Manczak, M. Differential loss of synaptic proteins in Alzheimer’s disease: Implications for synaptic dysfunction. J. Alzheimer’s Dis. 2005, 7, 103–117. [Google Scholar] [CrossRef]
- Gong, Y.; Lippa, C.F.; Zhu, J.; Lin, Q.; Rosso, A.L. Disruption of glutamate receptors at Shank-postsynaptic platform in Alzheimer’s disease. Brain Res. 2009, 1292, 191–198. [Google Scholar] [CrossRef]
- Briggs, R.; Kennelly, S.P.; O’Neill, D. Drug treatments in Alzheimer’s disease. Clin. Med. 2016, 16, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Schenk, D.; Basi, G.S.; Pangalos, M.N. Treatment Strategies Targeting Amyloid β-Protein. Cold Spring Harb. Perspect. Med. 2012, 2, a006387. [Google Scholar] [CrossRef] [PubMed]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef]
- Gueorguieva, I.; Willis, B.A.; Chua, L.; Chow, K.; Ernest, C.S.; Shcherbinin, S.; Ardayfio, P.; Mullins, G.R.; Sims, J.R. Donanemab Population Pharmacokinetics, Amyloid Plaque Reduction, and Safety in Participants with Alzheimer’s Disease. Clin. Pharmacol. Ther. 2023, 113, 1258–1267. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Sánchez-Rodríguez, I.; Djebari, S.; Temprano-Carazo, S.; Vega-Avelaira, D.; Jiménez-Herrera, R.; Iborra-Lázaro, G.; Yajeya, J.; Jiménez-Díaz, L.; Navarro-López, J.D. Hippocampal long-term synaptic depression and memory deficits induced in early amyloidopathy are prevented by enhancing G-protein-gated inwardly rectifying potassium channel activity. J. Neurochem. 2019, 153, 362–376. [Google Scholar] [CrossRef]
- Kim, H.Y.; Lee, D.K.; Chung, B.-R.; Kim, H.V.; Kim, Y. Intracerebroventricular Injection of Amyloid-β Peptides in Normal Mice to Acutely Induce Alzheimer-like Cognitive Deficits. J. Vis. Exp. 2016, 109, e53308. [Google Scholar] [CrossRef]
- Beason-Held, L.L.; Goh, J.O.; An, Y.; Kraut, M.A.; O’Brien, R.J.; Ferrucci, L.; Resnick, S.M. Changes in Brain Function Occur Years before the Onset of Cognitive Impairment. J. Neurosci. 2013, 33, 18008–18014. [Google Scholar] [CrossRef]
- Aisen, P.S.; Cummings, J.; Jack, C.R., Jr.; Morris, J.C.; Sperling, R.; Frölich, L.; Jones, R.W.; Dowsett, S.A.; Matthews, B.R.; Raskin, J.; et al. On the path to 2025: Understanding the Alzheimer’s disease continuum. Alzheimer’s Res. Ther. 2017, 9, 60. [Google Scholar] [CrossRef] [PubMed]
- Eikelenboom, P.; Stam, F.C. Immunoglobulins and complement factors in senile plaques. An immunoperoxidase study. Acta Neuropathol. 1982, 57, 239–242. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G. The inflammatory response system of brain: Implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res. Rev. 1995, 21, 195–218. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Rogers, J.; McGeer, E.G. Inflammation, Antiinflammatory Agents, and Alzheimer’s Disease: The Last 22 Years. J. Alzheimer’s Dis. 2016, 54, 853–857. [Google Scholar] [CrossRef] [PubMed]
- Kolev, M.; Ruseva, M.; Harris, C.; Morgan, B.; Donev, R. Implication of Complement System and its Regulators in Alzheimers Disease. Curr. Neuropharmacol. 2009, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Yasojima, K.; Schwab, C.; McGeer, E.G.; McGeer, P.L. Up-Regulated Production and Activation of the Complement System in Alzheimer’s Disease Brain. Am. J. Pathol. 1999, 154, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Bellenguez, C.; Küçükali, F.; Jansen, I.; Andrade, V.; Moreno-Grau, S.; Amin, N.; Naj, A.C.; Grenier-Boley, B.; Campos-Martin, R.; Holmans, P.A.; et al. New Insights on the Genetic Etiology of Alzheimer’s and Related Dementia. Nat. Genet. 2020, 54, 412–436. [Google Scholar] [CrossRef] [PubMed]
- Torvell, M.; Carpanini, S.M.; Daskoulidou, N.; Byrne, R.A.J.; Sims, R.; Morgan, B.P. Genetic Insights into the Impact of Complement in Alzheimer’s Disease. Genes 2021, 12, 1990. [Google Scholar] [CrossRef]
- Zhang, D.-F.; Li, J.; Wu, H.; Cui, Y.; Bi, R.; Zhou, H.-J.; Wang, H.-Z.; Zhang, C.; Wang, D.; Alzheimer’s Disease Neuroimaging Initiative (ADNI); et al. CFH Variants Affect Structural and Functional Brain Changes and Genetic Risk of Alzheimer’s Disease. Neuropsychopharmacology 2015, 41, 1034–1045. [Google Scholar] [CrossRef]
- Zetterberg, M.; Landgren, S.; Andersson, M.E.; Palmér, M.S.; Gustafson, D.R.; Skoog, I.; Minthon, L.; Thelle, D.S.; Wallin, A.; Bogdanovic, N.; et al. Association of complement factor H Y402H gene polymorphism with Alzheimer’s disease. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2008, 147B, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Le Fur, I.; Laumet, G.; Richard, F.; Fievet, N.; Berr, C.; Rouaud, O.; Delcourt, C.; Amouyel, P.; Lambert, J.-C. Association study of the CFH Y402H polymorphism with Alzheimer’s disease. Neurobiol. Aging 2010, 31, 165–166. [Google Scholar] [CrossRef] [PubMed]
- Borras, C.; Canonica, J.; Jorieux, S.; Abache, T.; El Sanharawi, M.; Klein, C.; Delaunay, K.; Jonet, L.; Salvodelli, M.; Naud, M.-C.; et al. CFH exerts anti-oxidant effects on retinal pigment epithelial cells independently from protecting against membrane attack complex. Sci. Rep. 2019, 9, 13873. [Google Scholar] [CrossRef]
- Borras, C.; Delaunay, K.; Slaoui, Y.; Abache, T.; Jorieux, S.; Naud, M.-C.; El Sanharawi, M.; Gelize, E.; Lassiaz, P.; An, N.; et al. Mechanisms of FH Protection Against Neovascular AMD. Front. Immunol. 2020, 11, 443. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Dejanovic, B.; Gandham, V.D.; Gogineni, A.; Edmonds, R.; Schauer, S.; Srinivasan, K.; Huntley, M.A.; Wang, Y.; Wang, T.-M.; et al. Complement C3 Is Activated in Human AD Brain and Is Required for Neurodegeneration in Mouse Models of Amyloidosis and Tauopathy. Cell Rep. 2019, 28, 2111–2123.e6. [Google Scholar] [CrossRef]
- Shi, Q.; Chowdhury, S.; Ma, R.; Le, K.X.; Hong, S.; Caldarone, B.J.; Stevens, B.; Lemere, C.A. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci. Transl. Med. 2017, 9, eaaf6295. [Google Scholar] [CrossRef]
- An, X.-Q.; Xi, W.; Gu, C.-Y.; Huang, X. Complement protein C5a enhances the β-amyloid-induced neuro-inflammatory response in microglia in Alzheimer’s disease. Med. Sci. 2018, 34, 116–120. [Google Scholar] [CrossRef]
- Mukherjee, P.; Pasinetti, G.M. The role of complement anaphylatoxin C5a in neurodegeneration: Implications in Alzheimer’s disease. J. Neuroimmunol. 2000, 105, 124–130. [Google Scholar] [CrossRef]
- Panayiotou, E.; Fella, E.; Andreou, S.; Papacharalambous, R.; Gerasimou, P.; Costeas, P.; Angeli, S.; Kousiappa, I.; Papacostas, S.; Kyriakides, T. C5aR agonist enhances phagocytosis of fibrillar and non-fibrillar Aβ amyloid and preserves memory in a mouse model of familial Alzheimer’s disease. PLoS ONE 2019, 14, e0225417. [Google Scholar] [CrossRef]
- Ordoñez-Gutierrez, L.; Fernandez-Perez, I.; Herrera, J.L.; Anton, M.; Benito-Cuesta, I.; Wandosell, F. AβPP/PS1 Transgenic Mice Show Sex Differences in the Cerebellum Associated with Aging. J. Alzheimer’s Dis. 2016, 54, 645–656. [Google Scholar] [CrossRef]
- Wang, J.; Tanila, H.; Puoliväli, J.; Kadish, I.; van Groen, T. Gender differences in the amount and deposition of amyloidβ in APPswe and PS1 double transgenic mice. Neurobiol. Dis. 2003, 14, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Vinters, H.V. Cerebral amyloid angiopathy. A critical review. Stroke 1987, 18, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Charidimou, A.; Gang, Q.; Werring, D.J. Sporadic cerebral amyloid angiopathy revisited: Recent insights into pathophysiology and clinical spectrum. J. Neurol. Neurosurg. Psychiatry 2011, 83, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Krstic, D.; Knuesel, I. Deciphering the mechanism underlying late-onset Alzheimer disease. Nat. Rev. Neurol. 2012, 9, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Cavanagh, C.; Colby-Milley, J.; Bouvier, D.; Farso, M.; Chabot, J.-G.; Quirion, R.; Krantic, S. βCTF-Correlated Burst of Hippocampal TNFα Occurs at a Very Early, Pre-Plaque Stage in the TgCRND8 Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2013, 36, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Cavanagh, C.; Tse, Y.C.; Nguyen, H.-B.; Krantic, S.; Breitner, J.C.; Quirion, R.; Wong, T.P. Inhibiting tumor necrosis factor-α before amyloidosis prevents synaptic deficits in an Alzheimer’s disease model. Neurobiol. Aging 2016, 47, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Dinet, V.; Arouche-Delaperche, L.; Dégardin, J.; Naud, M.-C.; Picaud, S.; Krantic, S. Concomitant Retinal Alterations in Neuronal Activity and TNFα Pathway Are Detectable during the Pre-Symptomatic Stage in a Mouse Model of Alzheimer’s Disease. Cells 2022, 11, 1650. [Google Scholar] [CrossRef]
- Doméné, A.; Cavanagh, C.; Page, G.; Bodard, S.; Klein, C.; Delarasse, C.; Chalon, S.; Krantic, S. Expression of Phenotypic Astrocyte Marker Is Increased in a Transgenic Mouse Model of Alzheimer’s Disease versus Age-Matched Controls: A Presymptomatic Stage Study. Int. J. Alzheimer’s Dis. 2016, 2016, 5696241. [Google Scholar] [CrossRef]
- Decourt, B.; Lahiri, D.K.; Sabbagh, M.N. Targeting Tumor Necrosis Factor Alpha for Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 412–425. [Google Scholar] [CrossRef]
- Liu, J.; Tan, Y.; Zhang, J.; Zou, L.; Deng, G.; Xu, X.; Wang, F.; Ma, Z.; Zhang, J.; Zhao, T.; et al. C5aR, TNF-α, and FGL2 contribute to coagulation and complement activation in virus-induced fulminant hepatitis. J. Hepatol. 2015, 62, 354–362. [Google Scholar] [CrossRef]
- Forloni, G.; Demicheli, F.; Giorgi, S.; Bendotti, C.; Angeretti, N. Expression of amyloid precursor protein mRNAs in endothelial, neuronal and glial cells: Modulation by interleukin-1. Mol. Brain Res. 1992, 16, 128–134. [Google Scholar] [CrossRef]
- Li, Y.; Liu, L.; Barger, S.W.; Griffin, W.S.T. Interleukin-1 Mediates Pathological Effects of Microglia on Tau Phosphorylation and on Synaptophysin Synthesis in Cortical Neurons through a p38-MAPK Pathway. J. Neurosci. 2003, 23, 1605–1611. [Google Scholar] [CrossRef]
- Kitazawa, M.; Cheng, D.; Tsukamoto, M.R.; Koike, M.A.; Wes, P.D.; Vasilevko, V.; Cribbs, D.H.; LaFerla, F.M. Blocking IL-1 Signaling Rescues Cognition, Attenuates Tau Pathology, and Restores Neuronal β-Catenin Pathway Function in an Alzheimer’s Disease Model. J. Immunol. 2011, 187, 6539–6549. [Google Scholar] [CrossRef]
- Huell, M.; Strauss, S.; Volk, B.; Berger, M.; Bauer, J. Interleukin-6 is present in early stages of plaque formation and is restricted to the brains of Alzheimer’s disease patients. Acta Neuropathol. 1995, 89, 544–551. [Google Scholar] [CrossRef]
- Walsh, K.P.; Minamide, L.S.; Kane, S.J.; Shaw, A.E.; Brown, D.R.; Pulford, B.; Zabel, M.D.; Lambeth, J.D.; Kuhn, T.B.; Bamburg, J.R. Amyloid-β and Proinflammatory Cytokines Utilize a Prion Protein-Dependent Pathway to Activate NADPH Oxidase and Induce Cofilin-Actin Rods in Hippocampal Neurons. PLoS ONE 2014, 9, e95995. [Google Scholar] [CrossRef] [PubMed]
- Heyser, C.J.; Masliah, E.; Samimi, A.; Campbell, I.L.; Gold, L.H. Progressive decline in avoidance learning paralleled by inflammatory neurodegeneration in transgenic mice expressing interleukin 6 in the brain. Proc. Natl. Acad. Sci. USA 1997, 94, 1500–1505. [Google Scholar] [CrossRef]
- e Silva, N.M.L.; Gonçalves, R.A.; Pascoal, T.A.; Lima-Filho, R.A.S.; Resende, E.d.P.F.; Vieira, E.L.M.; Teixeira, A.L.; de Souza, L.C.; Peny, J.A.; Fortuna, J.T.S.; et al. Pro-inflammatory interleukin-6 signaling links cognitive impairments and peripheral metabolic alterations in Alzheimer’s disease. Transl. Psychiatry 2021, 11, 251. [Google Scholar] [CrossRef] [PubMed]
- Swardfager, W.; Lanctôt, K.; Rothenburg, L.; Wong, A.; Cappell, J.; Herrmann, N. A Meta-Analysis of Cytokines in Alzheimer’s Disease. Biol. Psychiatry 2010, 68, 930–941. [Google Scholar] [CrossRef]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen, L.A.; Katzman, R. Physical basis of cognitive alterations in alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991, 30, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Fayed, N.; Modrego, P.J.; Rojas-Salinas, G.; Aguilar, K. Brain Glutamate Levels Are Decreased in Alzheimer’s Disease. Am. J. Alzheimer’s Dis. Other Dementiasr 2011, 26, 450–456. [Google Scholar] [CrossRef]
- Johnson, J.; Sherry, D.M.; Liu, X.; Fremeau, R.T.; Seal, R.P.; Edwards, R.H.; Copenhagen, D.R. Vesicular glutamate transporter 3 expression identifies glutamatergic amacrine cells in the rodent retina. J. Comp. Neurol. 2004, 477, 386–398. [Google Scholar] [CrossRef] [PubMed]
- Kashani, A.; Lepicard, E.; Poirel, O.; Videau, C.; David, J.P.; Fallet-Bianco, C.; Simon, A.; Delacourte, A.; Giros, B.; Epelbaum, J.; et al. Loss of VGLUT1 and VGLUT2 in the prefrontal cortex is correlated with cognitive decline in Alzheimer disease. Neurobiol. Aging 2008, 29, 1619–1630. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Perdigon, M.; Tordera, R.M.; Gil-Bea, F.J.; Gerenu, G.; Ramirez, M.J.; Solas, M. Down-regulation of glutamatergic terminals (VGLUT1) driven by Aβ in Alzheimer’s disease. Hippocampus 2016, 26, 1303–1312. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Perdigon, M.; Solas, M.; Ramirez, M.J. JNK: A Putative Link between Insulin Signaling and VGLUT1 in Alzheimer’s Disease. J. Alzheimer’s Dis. 2016, 50, 963–967. [Google Scholar] [CrossRef] [PubMed]
- Proctor, D.T.; Coulson, E.J.; Dodd, P.R. Reduction in Post-Synaptic Scaffolding PSD-95 and SAP-102 Protein Levels in the Alzheimer Inferior Temporal Cortex is Correlated with Disease Pathology. J. Alzheimer’s Dis. 2010, 21, 795–811. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Colodner, K.J.; Matousek, S.B.; Merry, K.; Hong, S.; Kenison, J.E.; Frost, J.L.; Le, K.X.; Li, S.; Dodart, J.-C.; et al. ComplementC3-Deficient Mice Fail to Display Age-Related Hippocampal Decline. J. Neurosci. 2015, 35, 13029–13042. [Google Scholar] [CrossRef]
- Laskowski, J.; Renner, B.; Pickering, M.C.; Serkova, N.J.; Smith-Jones, P.M.; Clambey, E.T.; Nemenoff, R.A.; Thurman, J.M. Complement factor H–deficient mice develop spontaneous hepatic tumors. J. Clin. Investig. 2020, 130, 4039–4054. [Google Scholar] [CrossRef]
- Podcasy, J.L.; Epperson, C.N. Considering sex and gender in Alzheimer disease and other dementias. Dialog. Clin. Neurosci. 2016, 18, 437–446. [Google Scholar] [CrossRef]
- Renier, N.; Adams, E.L.; Kirst, C.; Wu, Z.; Azevedo, R.; Kohl, J.; Autry, A.E.; Kadiri, L.; Venkataraju, K.U.; Zhou, Y.; et al. Mapping of Brain Activity by Automated Volume Analysis of Immediate Early Genes. Cell 2016, 165, 1789–1802. [Google Scholar] [CrossRef]
- Nicolas, N.; Roux, E. 3D Imaging and Quantitative Characterization of Mouse Capillary Coronary Network Architecture. Biology 2021, 10, 306. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stage (Months M) | WT | APP/PS1 | +PBS | +FH |
|---|---|---|---|---|
| 6 M + 1 M | 2.61 | 0.77 | 1.1 | 3.03 |
| ↓70% | ↓60% | = | ||
| 6 M + 3 M | 4.27 | 1.623 | 1.615 | 1.265 |
| ↓62% | ↓63% | ↓70% | ||
| 9 M + 1 M | 4.267 | 1.29 | 0.88 | 2.721 |
| ↓70% | ↓75% | ↓36% | ||
| 9 M + 3 M | 2.55 | 0.61 | 1.011 | 2.851 |
| ↓72% | ↓61% | = | ||
| 9 M + 6 M | 2.211 | 0.62 | 0.64 | 1.573 |
| ↓73% | ↓72% | ↓30% | ||
| Cumulative duration (s) | ||||
| 6M + 1M | 16.47 | 58.5 | 51.37 | 22.81 |
| ↑3.5% | ↑3.1% | ↑1.3% | ||
| 6M + 3M | 16.59 | 46.29 | 39.71 | 34.94 |
| ↑2.8% | ↑2.4% | ↑2.1% | ||
| 9M + 1M | 18.68 | 71.33 | 88.46 | 29.45 |
| ↑3.8% | ↑4.7% | ↑1.6% | ||
| 9M + 3M | 21.3 | 69.19 | 61.5 | 18.37 |
| ↑3.2% | ↑2.8% | = (0.8) | ||
| 9M + 6M | 16.5 | 65.61 | 50.07 | 31.35 |
| ↑3.7% | ↑3% | ↑2% | ||
| Latency to first (s) | ||||
| Western Blot | Host | Dilution | Manufacturer |
|---|---|---|---|
| C3/C3b fragments | Rat | 1/1000 | Hycult |
| FH | Goat | 1/1500 | Quidel |
| Actin-β | Rabbit | 1/1500 | Santa Cruz |
| Immunostaining | Host | Dilution | Manufacturer |
| C3 | Rabbit | 1/500 | Abcam |
| C3a | Rat | 1/400 | DB Pharmingen |
| C5b-9-FITC | - | 1/500 | Santa Cruz |
| FH | Goat | 1/600 | Quidel |
| Albumin | Sheep | 1/600 | Abcam |
| Cd31 | Mouse | 1/700 | Chemicon |
| VGLUT1 | Mouse | 1/500 | Santa Cruz |
| Psd95-FITC | - | 1/400 | Santa Cruz |
| Synapsin | Mouse | 1/400 | Santa Cruz |
| NeuN | Rabbit | 1/500 | Millipore |
| IL6 | Goat | 1/500 | R&D Systems |
| IL1β | Goat | 1/500 | R&D Systems |
| TNF-α | Rat | 1/500 | Biolegend |
| MMR2 | Mouse | 1/600 | Millipore |
| Aβ(6E10) | Mouse | 1/400 | Biolegend |
| Injection | - | Quantity | Manufacturer |
| Human plFH | - | 50 µg/5 µL | Sigma-Aldrich |
| FH-His Tag | - | 50 µg/5 µL | Sigma-Aldrich |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasantari, I.; Nicolas, N.; Alzieu, P.; Leval, L.; Shalabi, A.; Grolleau, S.; Dinet, V. Factor H’s Control of Complement Activation Emerges as a Significant and Promising Therapeutic Target for Alzheimer’s Disease Treatment. Int. J. Mol. Sci. 2024, 25, 2272. https://doi.org/10.3390/ijms25042272
Hasantari I, Nicolas N, Alzieu P, Leval L, Shalabi A, Grolleau S, Dinet V. Factor H’s Control of Complement Activation Emerges as a Significant and Promising Therapeutic Target for Alzheimer’s Disease Treatment. International Journal of Molecular Sciences. 2024; 25(4):2272. https://doi.org/10.3390/ijms25042272
Chicago/Turabian StyleHasantari, Iris, Nabil Nicolas, Philippe Alzieu, Léa Leval, Andree Shalabi, Sylvain Grolleau, and Virginie Dinet. 2024. "Factor H’s Control of Complement Activation Emerges as a Significant and Promising Therapeutic Target for Alzheimer’s Disease Treatment" International Journal of Molecular Sciences 25, no. 4: 2272. https://doi.org/10.3390/ijms25042272