
Epidermal Growth Factor Receptor Inhibitors in Glioblastoma: Current Status and Future Possibilities


Abstract
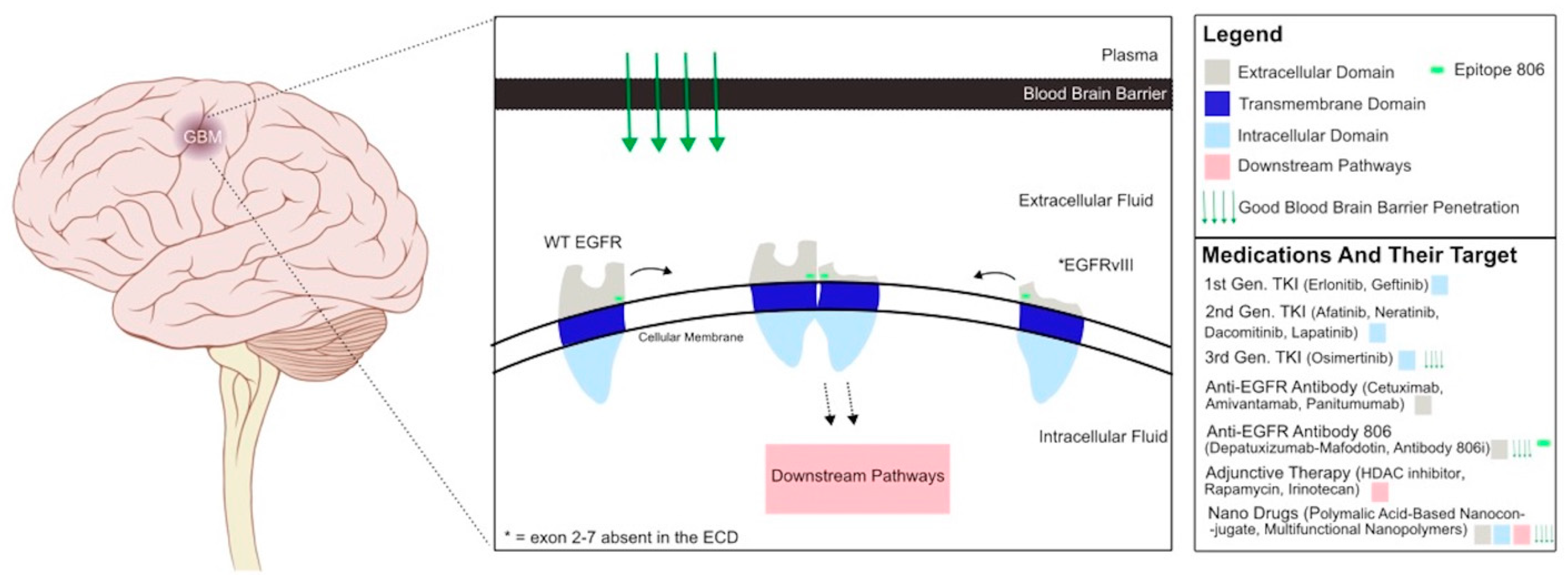
:1. Introduction
2. EGFR Receptors: Structure and Signaling Pathways
3. EGFR Mutations in Glioblastoma
4. Current EGFR Inhibitors for Cancer Therapy
5. EGFR Inhibitors in Glioblastoma: Preclinical Studies
6. Clinical Trials with EGFR Inhibitors in Glioblastoma
7. Challenges and Potential Strategies
8. Future Perspectives and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gilard, V.; Tebani, A.; Dabaj, I.; Laquerrière, A.; Fontanilles, M.; Derrey, S.; Marret, S.; Bekri, S. Diagnosis and Management of Glioblastoma: A Comprehensive Perspective. J. Pers. Med. 2021, 11, 258. [Google Scholar] [CrossRef]
- Sareen, H.; Ma, Y.; Becker, T.M.; Roberts, T.L.; de Souza, P.; Powter, B. Molecular Biomarkers in Glioblastoma: A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2022, 23, 8835. [Google Scholar] [CrossRef]
- Lin, B.; Ziebro, J.; Smithberger, E.; Skinner, K.R.; Zhao, E.; Cloughesy, T.F.; Binder, Z.A.; O’Rourke, D.M.; Nathanson, D.A.; Furnari, F.B.; et al. EGFR, the Lazarus target for precision oncology in glioblastoma. Neuro-Oncology 2022, 24, 2035–2062. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Mohammed, S.; Dinesan, M.; Ajayakumar, T. Survival and quality of life analysis in glioblastoma multiforme with adjuvant chemoradiotherapy: A retrospective study. Rep. Pract. Oncol. Radiother. 2022, 27, 1026–1036. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Hynes, N.E.; Lane, H.A. ERBB receptors and cancer: The complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef]
- Zhu, H.; Acquaviva, J.; Ramachandran, P.; Boskovitz, A.; Woolfenden, S.; Pfannl, R.; Bronson, R.T.; Chen, J.W.; Weissleder, R.; Housman, D.E.; et al. Oncogenic EGFR signaling cooperates with loss of tumor suppressor gene functions in gliomagenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 2712–2716. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.H.; Xu, A.M.; White, F.M. Oncogenic EGFR signaling networks in glioma. Sci. Signal. 2009, 2, re6. [Google Scholar] [CrossRef]
- Darmanis, S.; Sloan, S.A.; Croote, D.; Mignardi, M.; Chernikova, S.; Samghababi, P.; Zhang, Y.; Neff, N.; Kowarsky, M.; Caneda, C.; et al. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep. 2017, 21, 1399–1410. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zeng, F.; Forrester, S.J.; Eguchi, S.; Zhang, M.-Z.; Harris, R.C. Expression and Function of the Epidermal Growth Factor Receptor in Physiology and Disease. Physiol. Rev. 2016, 96, 1025–1069. [Google Scholar] [CrossRef]
- Singh, S.; Barik, D.; Lawrie, K.; Mohapatra, I.; Prasad, S.; Naqvi, A.R.; Singh, A.; Singh, G. Unveiling Novel Avenues in mTOR-Targeted Therapeutics: Advancements in Glioblastoma Treatment. Int. J. Mol. Sci. 2023, 24, 14960. [Google Scholar] [CrossRef] [PubMed]
- Shinojima, N.; Tada, K.; Shiraishi, S.; Kamiryo, T.; Kochi, M.; Nakamura, H.; Makino, K.; Saya, H.; Hirano, H.; Kuratsu, J.-I.; et al. Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Res. 2003, 63, 6962–6970. [Google Scholar] [PubMed]
- Orellana, L.; Thorne, A.H.; Lema, R.; Gustavsson, J.; Parisian, A.D.; Hospital, A.; Cordeiro, T.N.; Bernadó, P.; Scott, A.M.; Brun-Heath, I.; et al. Oncogenic mutations at the EGFR ectodomain structurally converge to remove a steric hindrance on a kinase-coupled cryptic epitope. Proc. Natl. Acad. Sci. USA 2019, 116, 10009–10018. [Google Scholar] [CrossRef] [PubMed]
- Francis, J.M.; Zhang, C.-Z.; Maire, C.L.; Jung, J.; Manzo, V.E.; Adalsteinsson, V.A.; Homer, H.; Haidar, S.; Blumenstiel, B.; Pedamallu, C.S.; et al. EGFR variant heterogeneity in glioblastoma resolved through single-nucleus sequencing. Cancer Discov. 2014, 4, 956–971. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Binder, Z.A.; Thorne, A.H.; Bakas, S.; Wileyto, E.P.; Bilello, M.; Akbari, H.; Rathore, S.; Ha, S.M.; Zhang, L.; Ferguson, C.J.; et al. Epidermal Growth Factor Receptor Extracellular Domain Mutations in Glioblastoma Present Opportunities for Clinical Imaging and Therapeutic Development. Cancer Cell 2018, 34, 163–177.e7. [Google Scholar] [CrossRef]
- Huang, W.; Li, J.; Zhu, H.; Qin, X.; Chen, C.; Wang, B.; Wei, J.; Song, Y.; Lu, X.; Li, Z.; et al. A novel EGFR variant EGFRx maintains glioblastoma stem cells through STAT5. Neuro-Oncology 2023, 26, 85–99. [Google Scholar] [CrossRef]
- Pines, G.; Huang, P.H.; Zwang, Y.; White, F.M.; Yarden, Y. EGFRvIV: A previously uncharacterized oncogenic mutant reveals a kinase autoinhibitory mechanism. Oncogene 2010, 29, 5850–5860. [Google Scholar] [CrossRef] [PubMed]
- Vivanco, I.; Robins, H.I.; Rohle, D.; Campos, C.; Grommes, C.; Nghiemphu, P.L.; Kubek, S.; Oldrini, B.; Chheda, M.G.; Yannuzzi, N.; et al. Differential sensitivity of glioma- versus lung cancer-specific EGFR mutations to EGFR kinase inhibitors. Cancer Discov. 2012, 2, 458–471. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Orellana, L. Convergence of EGFR glioblastoma mutations: Evolution and allostery rationalizing targeted therapy. Mol. Cell. Oncol. 2019, 6, e1630798. [Google Scholar] [CrossRef] [PubMed]
- Johns, T.G.; Mellman, I.; Cartwright, G.A.; Ritter, G.; Old, L.J.; Burgess, A.W.; Scott, A.M. The antitumor monoclonal antibody 806 recognizes a high-mannose form of the EGF receptor that reaches the cell surface when cells over-express the receptor. FASEB J. 2005, 19, 780–782. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef]
- Yang, S.-R.; Schultheis, A.M.; Yu, H.; Mandelker, D.; Ladanyi, M.; Büttner, R. Precision medicine in non-small cell lung cancer: Current applications and future directions. Semin. Cancer Biol. 2022, 84, 184–198. [Google Scholar] [CrossRef]
- Suda, K.; Onozato, R.; Yatabe, Y.; Mitsudomi, T. EGFR T790M mutation: A double role in lung cancer cell survival? J. Thorac. Oncol. 2009, 4, 1–4. [Google Scholar] [CrossRef]
- Wang, X.; Goldstein, D.; Crowe, P.J.; Yang, J.-L. Next-generation EGFR/HER tyrosine kinase inhibitors for the treatment of patients with non-small-cell lung cancer harboring EGFR mutations: A review of the evidence. OncoTargets Ther. 2016, 9, 5461–5473. [Google Scholar] [CrossRef]
- Opdam, F.L.; Guchelaar, H.-J.; Beijnen, J.H.; Schellens, J.H.M. Lapatinib for Advanced or Metastatic Breast Cancer. Oncologist 2012, 17, 536–542. [Google Scholar] [CrossRef] [PubMed]
- InformedHealth.org. Neratinib (Nerlynx) for the Treatment of Breast Cancer: Overview; Institute for Quality and Efficiency in Health Care (IQWiG): Cologne, Germany, 2020. Available online: https://www.ncbi.nlm.nih.gov/books/NBK556938/ (accessed on 7 September 2023).
- Park, K.; Tan, E.-H.; O’Byrne, K.; Zhang, L.; Boyer, M.; Mok, T.; Hirsh, V.; Yang, J.C.-H.; Lee, K.H.; Lu, S.; et al. Afatinib versus gefitinib as first-line treatment of patients with EGFR mutation-positive non-small-cell lung cancer (LUX-Lung 7): A phase 2B, open-label, randomised controlled trial. Lancet Oncol. 2016, 17, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.; Wu, Y.-L.; Ahn, M.-J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.M.E.; et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef]
- Wu, S.-G.; Shih, J.-Y. Management of acquired resistance to EGFR TKI-targeted therapy in advanced non-small cell lung cancer. Mol. Cancer 2018, 17, 38. [Google Scholar] [CrossRef] [PubMed]
- Goss, G.; Tsai, C.-M.; Shepherd, F.A.; Ahn, M.-J.; Bazhenova, L.; Crinò, L.; de Marinis, F.; Felip, E.; Morabito, A.; Hodge, R.; et al. CNS response to osimertinib in patients with T790M-positive advanced NSCLC: Pooled data from two phase II trials. Ann. Oncol. 2018, 29, 687–693. [Google Scholar] [CrossRef]
- Campos, S.; Davey, P.; Hird, A.; Pressnail, B.; Bilbao, J.; Aviv, R.I.; Symons, S.; Pirouzmand, F.; Sinclair, E.; Culleton, S.; et al. Brain metastasis from an unknown primary, or primary brain tumour? A diagnostic dilemma. Curr. Oncol. 2009, 16, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Lagerwaard, F.; Levendag, P.; Nowak, P.C.M.; Eijkenboom, W.H.; Hanssens, P.J.; Schmitz, P.M. Identification of prognostic factors in patients with brain metastases: A review of 1292 patients. Int. J. Radiat. Oncol. Biol. Phys. 1999, 43, 795–803. [Google Scholar] [CrossRef]
- Ceresoli, G.L.; Cappuzzo, F.; Gregorc, V.; Bartolini, S.; Crinò, L.; Villa, E. Gefitinib in patients with brain metastases from non-small-cell lung cancer: A prospective trial. Ann. Oncol. 2004, 15, 1042–1047. [Google Scholar] [CrossRef]
- Iuchi, T.; Shingyoji, M.; Sakaida, T.; Hatano, K.; Nagano, O.; Itakura, M.; Kageyama, H.; Yokoi, S.; Hasegawa, Y.; Kawasaki, K.; et al. Phase II trial of gefitinib alone without radiation therapy for Japanese patients with brain metastases from EGFR-mutant lung adenocarcinoma. Lung Cancer 2013, 82, 282–287. [Google Scholar] [CrossRef]
- Schuler, M.; Wu, Y.-L.; Hirsh, V.; O’Byrne, K.; Yamamoto, N.; Mok, T.; Popat, S.; Sequist, L.V.; Massey, D.; Zazulina, V.; et al. First-Line Afatinib versus Chemotherapy in Patients with Non-Small Cell Lung Cancer and Common Epidermal Growth Factor Receptor Gene Mutations and Brain Metastases. J. Thorac. Oncol. 2016, 11, 380–390. [Google Scholar] [CrossRef]
- Reungwetwattana, T.; Nakagawa, K.; Cho, B.C.; Cobo, M.; Cho, E.K.; Bertolini, A.; Bohnet, S.; Zhou, C.; Lee, K.H.; Nogami, N.; et al. CNS Response to Osimertinib Versus Standard Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Patients with Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 3290–3297. [Google Scholar] [CrossRef] [PubMed]
- Douez, E.; D’Atri, V.; Guillarme, D.; Antier, D.; Guerriaud, M.; Beck, A.; Watier, H.; Foucault-Fruchard, L. Why is there no biosimilar of Erbitux®? J. Pharm. Biomed. Anal. 2023, 234, 115544. [Google Scholar] [CrossRef] [PubMed]
- Chidharla, A.; Parsi, M.; Kasi, A. Cetuximab. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: http://www.ncbi.nlm.nih.gov/books/NBK459293/ (accessed on 3 September 2023).
- Gemmete, J.J.; Mukherji, S.K. Panitumumab (vectibix). AJNR Am. J. Neuroradiol. 2011, 32, 1002–1003. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Haura, E.B.; Leighl, N.B.; Mitchell, P.; Shu, C.A.; Girard, N.; Viteri, S.; Han, J.-Y.; Kim, S.-W.; Lee, C.K.; et al. Amivantamab in EGFR Exon 20 Insertion-Mutated Non-Small-Cell Lung Cancer Progressing on Platinum Chemotherapy: Initial Results From the CHRYSALIS Phase I Study. J. Clin. Oncol. 2021, 39, 3391–3402. [Google Scholar] [CrossRef] [PubMed]
- Mukasa, A.; Wykosky, J.; Ligon, K.L.; Chin, L.; Cavenee, W.K.; Furnari, F. Mutant EGFR is required for maintenance of glioma growth in vivo, and its ablation leads to escape from receptor dependence. Proc. Natl. Acad. Sci. USA 2010, 107, 2616–2621. [Google Scholar] [CrossRef] [PubMed]
- Mishima, K.; Johns, T.G.; Luwor, R.B.; Scott, A.M.; Stockert, E.; Jungbluth, A.A.; Ji, X.D.; Suvarna, P.; Voland, J.R.; Old, L.J.; et al. Growth suppression of intracranial xenografted glioblastomas overexpressing mutant epidermal growth factor receptors by systemic administration of monoclonal antibody (mAb) 806, a novel monoclonal antibody directed to the receptor. Cancer Res. 2001, 61, 5349–5354. [Google Scholar]
- Johns, T.G.; Luwor, R.B.; Murone, C.; Walker, F.; Weinstock, J.; Vitali, A.A.; Perera, R.M.; Jungbluth, A.A.; Stockert, E.; Old, L.J.; et al. Antitumor efficacy of cytotoxic drugs and the monoclonal antibody 806 is enhanced by the EGF receptor inhibitor AG1478. Proc. Natl. Acad. Sci. USA 2003, 100, 15871–15876. [Google Scholar] [CrossRef]
- Luwor, R.B.; Johns, T.G.; Murone, C.; Huang, H.J.; Cavenee, W.K.; Ritter, G.; Old, L.J.; Burgess, A.W.; Scott, A.M. Monoclonal antibody 806 inhibits the growth of tumor xenografts expressing either the de2–7 or amplified epidermal growth factor receptor (EGFR) but not wild-type EGFR. Cancer Res. 2001, 61, 5355–5361. [Google Scholar]
- Eller, J.L.; Longo, S.L.; Kyle, M.M.; Bassano, D.; Hicklin, D.J.; Canute, G.W. Anti-epidermal growth factor receptor monoclonal antibody cetuximab augments radiation effects in glioblastoma multiforme in vitro and in vivo. Neurosurgery 2005, 56, 155–162, discussion 162. [Google Scholar] [CrossRef]
- Buendia Duque, M.; de Vargas Pinheiro, K.; Thomaz, A.; da Silva, C.A.; Freire, N.H.; Brunetto, A.T.; Schwartsmann, G.; Jaeger, M.; de Farias, C.B.; Roesler, R. Combined Inhibition of HDAC and EGFR Reduces Viability and Proliferation and Enhances STAT3 mRNA Expression in Glioblastoma Cells. J. Mol. Neurosci. 2019, 68, 49–57. [Google Scholar] [CrossRef]
- Xu, W.; Bi, Y.; Kong, J.; Zhang, J.; Wang, B.; Li, K.; Tian, M.; Pan, X.; Shi, B.; Gu, J.; et al. Combination of an anti-EGFRvIII antibody CH12 with Rapamycin synergistically inhibits the growth of EGFRvIII+PTEN− glioblastoma in vivo. Oncotarget 2016, 7, 24752–24765. [Google Scholar] [CrossRef] [PubMed]
- Hohwieler Schloss, M.; Freidberg, S.R.; Heatley, G.J.; Lo, T.C. Glucocorticoid dependency as a prognostic factor in radiotherapy for cerebral gliomas. Acta Oncol. 1989, 28, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Shields, L.B.E.; Shelton, B.J.; Shearer, A.J.; Chen, L.; Sun, D.A.; Parsons, S.; Bourne, T.D.; LaRocca, R.; Spalding, A.C. Dexamethasone administration during definitive radiation and temozolomide renders a poor prognosis in a retrospective analysis of newly diagnosed glioblastoma patients. Radiat. Oncol. 2015, 10, 222. [Google Scholar] [CrossRef] [PubMed]
- Watne, K.; Hannisdal, E.; Nome, O.; Hager, B.; Hirschberg, H. Prognostic factors in malignant gliomas with special reference to intra-arterial chemotherapy. Acta Oncol. 1993, 32, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Aldaz, P.; Auzmendi-Iriarte, J.; Durántez, M.; Lasheras-Otero, I.; Carrasco-Garcia, E.; Zelaya, M.V.; Bragado, L.; Olías-Arjona, A.; Egaña, L.; Samprón, N.; et al. Identification of a Dexamethasone Mediated Radioprotection Mechanism Reveals New Therapeutic Vulnerabilities in Glioblastoma. Cancers 2021, 13, 361. [Google Scholar] [CrossRef] [PubMed]
- Raizer, J.J.; Giglio, P.; Hu, J.; Groves, M.; Merrell, R.; Conrad, C.; Phuphanich, S.; Puduvalli, V.K.; Loghin, M.; Paleologos, N.; et al. A phase II study of bevacizumab and erlotinib after radiation and temozolomide in MGMT unmethylated GBM patients. J. Neurooncol. 2016, 126, 185–192. [Google Scholar] [CrossRef]
- Peereboom, D.M.; Ahluwalia, M.S.; Ye, X.; Supko, J.G.; Hilderbrand, S.L.; Phuphanich, S.; Nabors, L.B.; Rosenfeld, M.R.; Mikkelsen, T.; Grossman, S.A.; et al. NABTT 0502: A phase II and pharmacokinetic study of erlotinib and sorafenib for patients with progressive or recurrent glioblastoma multiforme. Neuro-Oncology 2013, 15, 490–496. [Google Scholar] [CrossRef]
- Sathornsumetee, S.; Desjardins, A.; Vredenburgh, J.J.; McLendon, R.E.; Marcello, J.; Herndon, J.E.; Mathe, A.; Hamilton, M.; Rich, J.N.; Norfleet, J.A.; et al. Phase II trial of bevacizumab and erlotinib in patients with recurrent malignant glioma. Neuro-Oncology 2010, 12, 1300–1310. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.-C.; Bady, P.; Kamoshima, Y.; Kouwenhoven, M.C.M.; Delorenzi, M.; Lambiv, W.L.; Hamou, M.-F.; Matter, M.S.; Koch, A.; et al. Pathway analysis of glioblastoma tissue after preoperative treatment with the EGFR tyrosine kinase inhibitor gefitinib—A phase II trial. Mol. Cancer Ther. 2011, 10, 1102–1112. [Google Scholar] [CrossRef]
- Choi, S.W.; Jung, H.A.; Cho, H.; Kim, T.M.; Park, C.; Nam, D.; Lee, S. A multicenter, phase II trial of GC1118, a novel anti-EGFR antibody, for recurrent glioblastoma patients with EGFR amplification. Cancer Med. 2023, 12, 15788–15796. [Google Scholar] [CrossRef]
- Lassman, A.B.; Pugh, S.L.; Wang, T.J.C.; Aldape, K.; Gan, H.K.; Preusser, M.; Vogelbaum, M.A.; Sulman, E.P.; Won, M.; Zhang, P.; et al. Depatuxizumab mafodotin in EGFR-amplified newly diagnosed glioblastoma: A phase III randomized clinical trial. Neuro-Oncology 2022, 25, 339–350. [Google Scholar] [CrossRef]
- Hasselbalch, B.; Lassen, U.; Hansen, S.; Holmberg, M.; Sørensen, M.; Kosteljanetz, M.; Broholm, H.; Stockhausen, M.-T.; Poulsen, H.S. Cetuximab, bevacizumab, and irinotecan for patients with primary glioblastoma and progression after radiation therapy and temozolomide: A phase II trial. Neuro-Oncology 2010, 12, 508–516. [Google Scholar] [CrossRef] [PubMed]
- McCrea, H.J.; Ivanidze, J.; O’Connor, A.; Hersh, E.H.; Boockvar, J.A.; Gobin, Y.P.; Knopman, J.; Greenfield, J.P. Intraarterial delivery of bevacizumab and cetuximab utilizing blood-brain barrier disruption in children with high-grade glioma and diffuse intrinsic pontine glioma: Results of a phase I trial. J. Neurosurg. Pediatr. 2021, 28, 371–379. [Google Scholar] [CrossRef]
- Pérez-Soler, R.; Delord, J.P.; Halpern, A.; Kelly, K.; Krueger, J.; Sureda, B.M.; von Pawel, J.; Temel, J.; Siena, S.; Soulières, D.; et al. HER1/EGFR inhibitor-associated rash: Future directions for management and investigation outcomes from the HER1/EGFR inhibitor rash management forum. Oncologist 2005, 10, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M. Cetuximab: Adverse event profile and recommendations for toxicity management. Clin. J. Oncol. Nurs. 2005, 9, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Sepúlveda-Sánchez, J.M.; Vaz, M.Á.; Balañá, C.; Gil-Gil, M.; Reynés, G.; Gallego, Ó.; Martínez-García, M.; Vicente, E.; Quindós, M.; Luque, R.; et al. Phase II trial of dacomitinib, a pan-human EGFR tyrosine kinase inhibitor, in recurrent glioblastoma patients with EGFR amplification. Neuro-Oncology 2017, 19, 1522–1531. [Google Scholar] [CrossRef]
- Lv, S.; Teugels, E.; Sadones, J.; De Brakeleer, S.; Duerinck, J.; Du Four, S.; Michotte, A.; De Grève, J.; Neyns, B. Correlation of EGFR, IDH1 and PTEN status with the outcome of patients with recurrent glioblastoma treated in a phase II clinical trial with the EGFR-blocking monoclonal antibody cetuximab. Int. J. Oncol. 2012, 41, 1029–1035. [Google Scholar] [CrossRef]
- Teleanu, D.M.; Chircov, C.; Grumezescu, A.M.; Volceanov, A.; Teleanu, R.I. Blood-Brain Delivery Methods Using Nanotechnology. Pharmaceutics 2018, 10, 269. [Google Scholar] [CrossRef]
- Chou, S.-T.; Patil, R.; Galstyan, A.; Gangalum, P.R.; Cavenee, W.K.; Furnari, F.B.; Ljubimov, V.A.; Chesnokova, A.; Kramerov, A.A.; Ding, H.; et al. Simultaneous blockade of interacting CK2 and EGFR pathways by tumor-targeting nanobioconjugates increases therapeutic efficacy against glioblastoma multiforme. J. Control. Release 2016, 244, 14–23. [Google Scholar] [CrossRef]
- Liu, C.; Zhao, Z.; Gao, H.; Rostami, I.; You, Q.; Jia, X.; Wang, C.; Zhu, L.; Yang, Y. Enhanced blood-brain-barrier penetrability and tumor-targeting efficiency by peptide-functionalized poly(amidoamine) dendrimer for the therapy of gliomas. Nanotheranostics 2019, 3, 311–330. [Google Scholar] [CrossRef]
- Patil, R.; Sun, T.; Rashid, M.H.; Israel, L.L.; Ramesh, A.; Davani, S.; Black, K.L.; Ljubimov, A.V.; Holler, E.; Ljubimova, J.Y. Multifunctional Nanopolymers for Blood-Brain Barrier Delivery and Inhibition of Glioblastoma Growth through EGFR/EGFRvIII, c-Myc, and PD-1. Nanomaterials 2021, 11, 2892. [Google Scholar] [CrossRef]
- D’Amico, R.S.; Aghi, M.K.; Vogelbaum, M.A.; Bruce, J.N. Convection-enhanced drug delivery for glioblastoma: A review. J. Neurooncol. 2021, 151, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Kunwar, S.; Chang, S.; Westphal, M.; Vogelbaum, M.; Sampson, J.; Barnett, G.; Shaffrey, M.; Ram, Z.; Piepmeier, J.; Prados, M.; et al. Phase III randomized trial of CED of IL13-PE38QQR vs Gliadel wafers for recurrent glioblastoma. Neuro-Oncology 2010, 12, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.K.; Burge, M.; Solomon, B.; Lee, S.T.; Holen, K.D.; Zhang, Y.; Ciprotti, M.; Lee, F.T.; Munasinghe, W.; Fischer, J.; et al. A Phase 1 and Biodistribution Study of ABT-806i, an 111In-Radiolabeled Conjugate of the Tumor-Specific Anti-EGFR Antibody ABT-806. J. Nucl. Med. 2021, 62, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Nadeem Abbas, M.; Kausar, S.; Wang, F.; Zhao, Y.; Cui, H. Advances in Targeting the Epidermal Growth Factor Receptor Pathway by Synthetic Products and Its Regulation by Epigenetic Modulators as a Therapy for Glioblastoma. Cells 2019, 8, 350. [Google Scholar] [CrossRef]

{kind=link}
| Trial | Regimen | Target Dose | Side Effects | Prospective Trials in Glioblastoma | Summary of Results |
|---|---|---|---|---|---|
| A Phase II Study Of Bevacizumab And Erlotinib After Radiation And Temozolomide In MGMT Unmethylated Gbm Patients. | Bevacizumab And Erlotinib | Bevacizumab (intramuscular) at 10 mg/kg every 2 weeks Erlotinib (oral) 150 mg/day. Temozolomide 75 mg/m2/day | Lymphopenia, rash, hypertension, fatigue | No active clinical trials | No improvement in OS |
| Phase II trial of bevacizumab and erlotinib in patients with recurrent malignant glioma | Bevacizumab And Erlotinib | Bevacizumab (intravenously) (10 mg/kg) every 2 weeks Erlotinib (oral) 200 mg/day | Rash, mucositis, diarrhea, and fatigue | No active clinical trials | Drug combination showed activity but resulted in no improved PFS or radiaographic response |
| A Phase II And Pharmacokinetic Study Of Erlotinib And Sorafenib For Patients With Progressive Or Recurrent Glioblastoma Multiforme. | Erlotinib And Sorafenib | Erlotinib (oral) 150 mg/day sorafenib (oral) 400 mg twice daily for 28 days | Fatigue, diarrhea, hypophosphatemia, acneiform rash | 1 active trial for sorafenib | Erlotinib did not improve OS, even when combined with temozolomide, radiation, and other synergistic drugs |
| Analysis Of Glioblastoma Tissue After Preoperative Treatment With The EGFR Tyrosine Kinase Inhibitor Gefitinib–A Phase II Trial. | Gefitinib | 500 mg gefitinib (oral) 5 days prior to surgery | Not reported | no active clinical trials | Gefitinib alone was not enough to stop tumor growth signaling |
| Phase II Trial Of Dacomitinib In Recurrent Glioblastome Patients With EGFR Amplification. | Dacomitinib | Dacomitinib (oral) 45 mg/day | Rash, diarrhea, asthenia, nausuea/vomitting | no active clinical trials | No improvement in OS |
| Phase II Trial Of GC1118, A Novel Anti-EGFR Antibody, For Recurrent Glioblastoma Patients With EGFR Amplification. | GC1118 | 4 mg/kg weekly on Days 1, 8, 15, and 22 of a 28-day cycle x6 | Rash, acneiform, mucositis, diarrhea | no active clinical trials | No improvement in progression free survival (PFS) |
| Depatuxizumab Mafodotin In EGFR-Amplified Newly Diagnosed Glioblastoma: A Phase III Randomized Clinical Trial. | Depatuxizumab-Mafodotin | Dosed at 2.0 mg/kg during radiation therapy, then 1.25 mg/kg thereafter on days 1 and 15/28 | Corneal epitheliopathy | no active clinical trials | No improvement in OS, PFS improved in all patients and more so in patients with the EGFRvIII variant |
| Cetuximab, Bevacizumab, And Irinotecan For Patients With Primary Glioblastoma And Progression After Radiation Therapy And Temozolomide: A Phase II Trial. | Cetuximab, Bevacizumab, And Irinotecan | Patients received bevacizumab (10 mg/kg) (intravenous) Irinotecan (125 mg/m2) (Intravenous) and Cetuximab (400 mg/m2 as the loading dose on day 1 followed by 250 mg/m2 weekly) every 2 weeks for up to 6 months | Nausua, vomitting, diarrhea, stomatisits | 4 active trials for cetuximab | Improved median OS, with skin toxicities |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ezzati, S.; Salib, S.; Balasubramaniam, M.; Aboud, O. Epidermal Growth Factor Receptor Inhibitors in Glioblastoma: Current Status and Future Possibilities. Int. J. Mol. Sci. 2024, 25, 2316. https://doi.org/10.3390/ijms25042316
Ezzati S, Salib S, Balasubramaniam M, Aboud O. Epidermal Growth Factor Receptor Inhibitors in Glioblastoma: Current Status and Future Possibilities. International Journal of Molecular Sciences. 2024; 25(4):2316. https://doi.org/10.3390/ijms25042316
Chicago/Turabian StyleEzzati, Shawyon, Samuel Salib, Meenakshisundaram Balasubramaniam, and Orwa Aboud. 2024. "Epidermal Growth Factor Receptor Inhibitors in Glioblastoma: Current Status and Future Possibilities" International Journal of Molecular Sciences 25, no. 4: 2316. https://doi.org/10.3390/ijms25042316