
Unraveling the Role of the NLRP3 Inflammasome in Lymphoma: Implications in Pathogenesis and Therapeutic Strategies

 , , , and
, , , and
Abstract
:1. Introduction
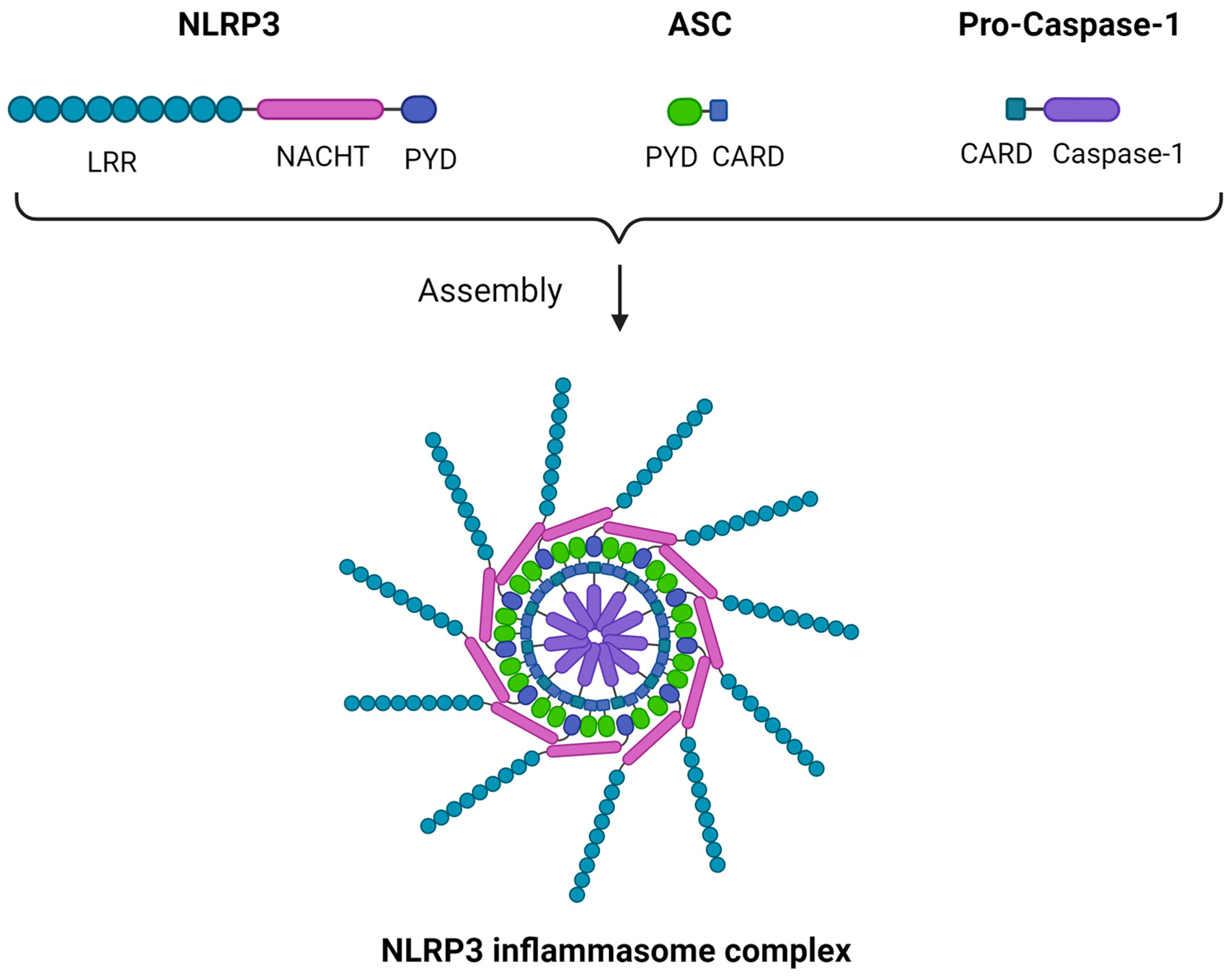
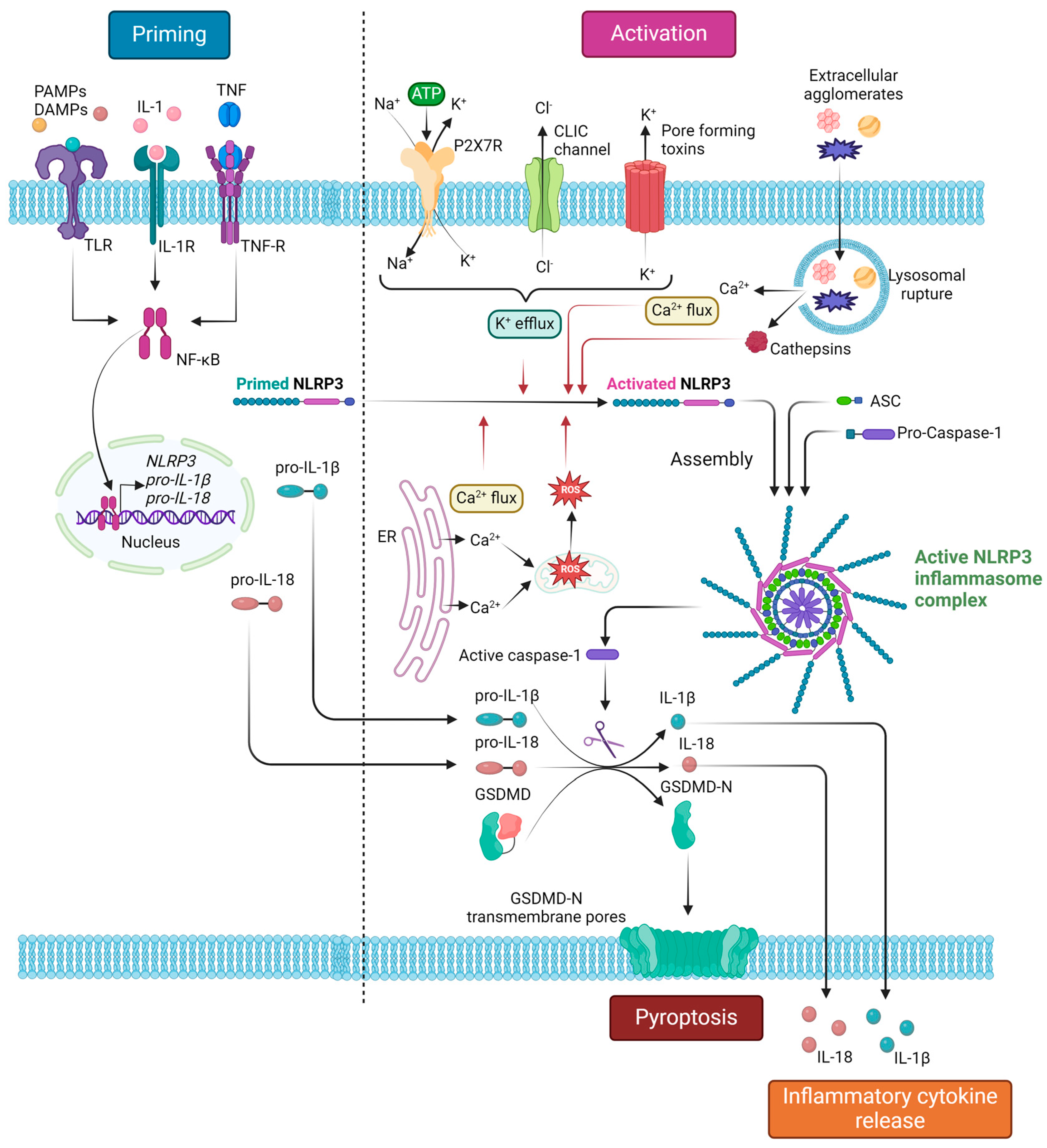
2. The NLRP3 Inflammasome: Structure, Activation, and Downstream Effects
2.1. Structure
2.2. Priming and Activation
2.3. Downstream Response
3. The NLRP3 Inflammasome in Lymphocyte Development
3.1. Normal Lymphopoiesis Overview
3.2. The NLRP3 Inflammasome in B-Cell Lymphopoiesis
3.3. The NLRP3 Inflammasome in T-Cell Lymphopoiesis
4. The NLRP3 Inflammasome in Lymphomagenesis
4.1. B-Cell NHLs
4.1.1. NLRP3 Inflammasome Activation Contributes to B-Cell NHL Development via Its Effector Cytokines
4.1.2. The Effect of NLRP3 Inflammasome Activation in the B-Cell NHL Microenvironment
4.1.3. Deciphering the Drivers of NLRP3 Inflammasome Activation in B-Cell NHL
4.2. T and NK-Cell Lymphomas
5. Exploring the Crosstalk of Tripartite Motif (TRIM) Family Proteins and NLRP3 Inflammasome Activation in Lymphoma
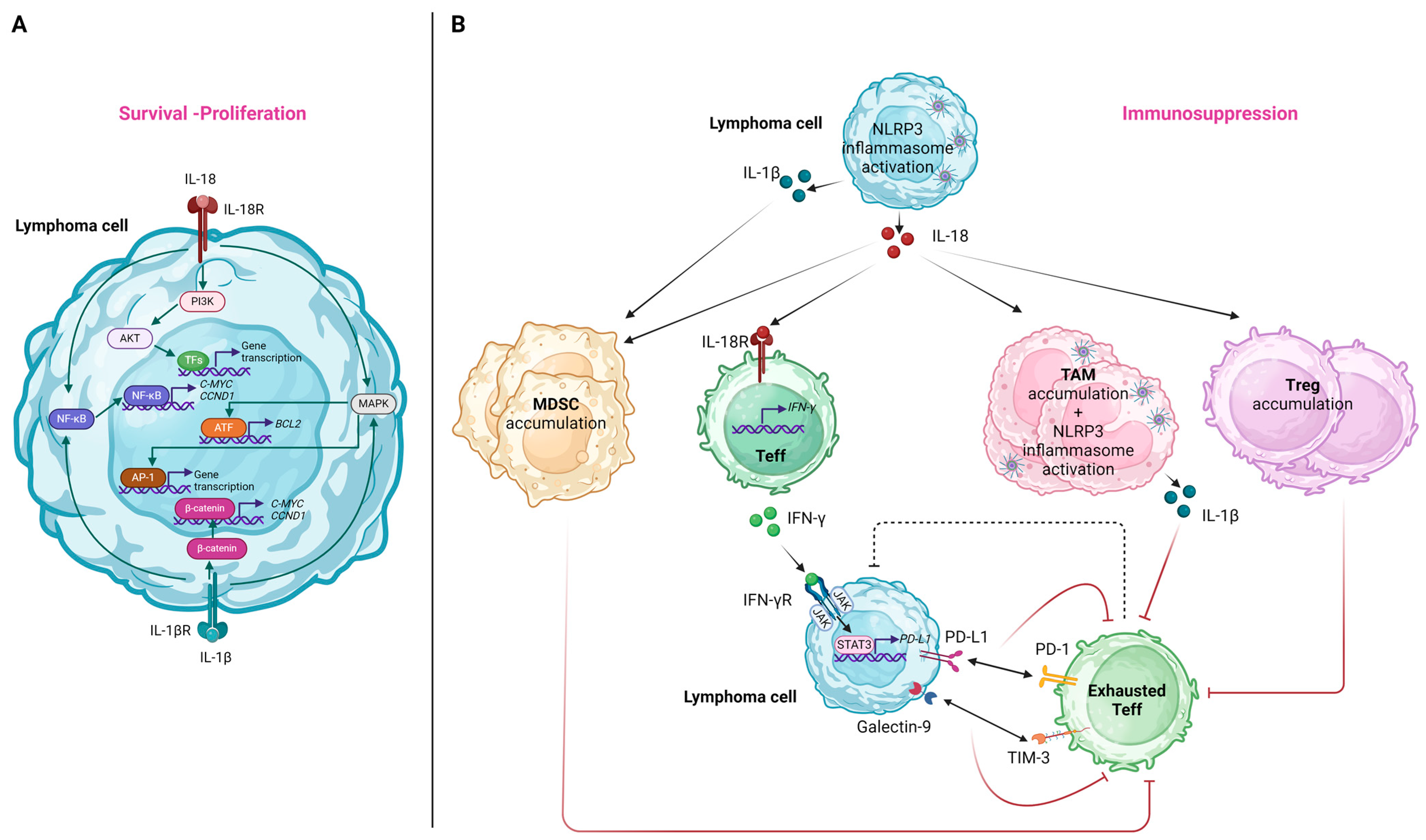
6. Molecular Pathways by Which NLRP3 Inflammasome Affects Lymphoma Cells and the Immune TME
6.1. Effects on Lymphoma Cells
6.2. Effects on the Immune TME
7. NLRP3 Inflammasome Signaling in Lymphoid Premalignant Conditions
8. Therapeutic Targeting of NLRP3 in Lymphoma
9. Discussion
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- Abdul-Sater, A.A.; Philpott, D.J. Inflammasomes. In Encyclopedia of Immunobiology; Ratcliffe, M.J.H., Ed.; Academic Press: Oxford, UK, 2016; pp. 447–453. [Google Scholar]
- Groslambert, M.; Py, B.F. Spotlight on the NLRP3 inflammasome pathway. J. Inflamm. Res. 2018, 11, 359–374. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [PubMed]
- Mortimer, L.; Moreau, F.; MacDonald, J.A.; Chadee, K. NLRP3 inflammasome inhibition is disrupted in a group of auto-inflammatory disease CAPS mutations. Nat. Immunol. 2016, 17, 1176–1186. [Google Scholar] [CrossRef] [PubMed]
- Menu, P.; Vince, J.E. The NLRP3 inflammasome in health and disease: The good, the bad and the ugly. Clin. Exp. Immunol. 2011, 166, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ungerbäck, J.; Belenki, D.; Jawad ul-Hassan, A.; Fredrikson, M.; Fransén, K.; Elander, N.; Verma, D.; Söderkvist, P. Genetic variation and alterations of genes involved in NFκB/TNFAIP3- and NLRP3-inflammasome signaling affect susceptibility and outcome of colorectal cancer. Carcinogenesis 2012, 33, 2126–2134. [Google Scholar] [CrossRef]
- Verma, D.; Bivik, C.; Farahani, E.; Synnerstad, I.; Fredrikson, M.; Enerbäck, C.; Rosdahl, I.; Söderkvist, P. Inflammasome polymorphisms confer susceptibility to sporadic malignant melanoma. Pigment Cell Melanoma Res. 2012, 25, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Miskiewicz, A.; Szparecki, G.; Durlik, M.; Rydzewska, G.; Ziobrowski, I.; Górska, R. The Q705K and F359L Single-Nucleotide Polymorphisms of NOD-Like Receptor Signaling Pathway: Association with Chronic Pancreatitis, Pancreatic Cancer, and Periodontitis. Arch. Immunol. Ther. Exp. 2015, 63, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Castaño-Rodríguez, N.; Kaakoush, N.O.; Goh, K.L.; Fock, K.M.; Mitchell, H.M. The NOD-like receptor signalling pathway in Helicobacter pylori infection and related gastric cancer: A case-control study and gene expression analyses. PLoS ONE 2015, 10, e0117870. [Google Scholar] [CrossRef]
- Chen, L.; Huang, C.F.; Li, Y.C.; Deng, W.W.; Mao, L.; Wu, L.; Zhang, W.F.; Zhang, L.; Sun, Z.J. Blockage of the NLRP3 inflammasome by MCC950 improves anti-tumor immune responses in head and neck squamous cell carcinoma. Cell. Mol. Life Sci. 2018, 75, 2045–2058. [Google Scholar] [CrossRef]
- Xue, Y.; Du, H.D.; Tang, D.; Zhang, D.; Zhou, J.; Zhai, C.W.; Yuan, C.C.; Hsueh, C.Y.; Li, S.J.; Heng, Y.; et al. Correlation between the NLRP3 Inflammasome and the Prognosis of Patients with LSCC. Front. Oncol. 2019, 9, 588. [Google Scholar] [CrossRef]
- Wu, C.S.; Chang, K.P.; OuYang, C.N.; Kao, H.K.; Hsueh, C.; Chen, L.C.; Cheng, H.Y.; Liang, Y.; Liou, W.; Liang, C.L.; et al. ASC contributes to metastasis of oral cavity squamous cell carcinoma. Oncotarget 2016, 7, 50074–50085. [Google Scholar] [CrossRef]
- Poli, G.; Brancorsini, S.; Cochetti, G.; Barillaro, F.; Egidi, M.G.; Mearini, E. Expression of inflammasome-related genes in bladder cancer and their association with cytokeratin 20 messenger RNA. Urol. Oncol. 2015, 33, e501–e507. [Google Scholar] [CrossRef] [PubMed]
- Allen, I.C.; TeKippe, E.M.; Woodford, R.M.; Uronis, J.M.; Holl, E.K.; Rogers, A.B.; Herfarth, H.H.; Jobin, C.; Ting, J.P. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J. Exp. Med. 2010, 207, 1045–1056. [Google Scholar] [CrossRef]
- Zaki, M.H.; Vogel, P.; Body-Malapel, M.; Lamkanfi, M.; Kanneganti, T.D. IL-18 production downstream of the Nlrp3 inflammasome confers protection against colorectal tumor formation. J. Immunol. 2010, 185, 4912–4920. [Google Scholar] [CrossRef]
- Wei, Q.; Mu, K.; Li, T.; Zhang, Y.; Yang, Z.; Jia, X.; Zhao, W.; Huai, W.; Guo, P.; Han, L. Deregulation of the NLRP3 inflammasome in hepatic parenchymal cells during liver cancer progression. Lab. Investig. 2014, 94, 52–62. [Google Scholar] [CrossRef]
- van Deventer, H.W.; Burgents, J.E.; Wu, Q.P.; Woodford, R.M.; Brickey, W.J.; Allen, I.C.; McElvania-Tekippe, E.; Serody, J.S.; Ting, J.P. The inflammasome component NLRP3 impairs antitumor vaccine by enhancing the accumulation of tumor-associated myeloid-derived suppressor cells. Cancer Res. 2010, 70, 10161–10169. [Google Scholar] [CrossRef]
- Hamarsheh, S.A.; Zeiser, R. NLRP3 Inflammasome Activation in Cancer: A Double-Edged Sword. Front. Immunol. 2020, 11, 1444. [Google Scholar] [CrossRef] [PubMed]
- Tomasik, J.; Basak, G.W. Inflammasomes—New Contributors to Blood Diseases. Int. J. Mol. Sci. 2022, 23, 8129. [Google Scholar] [CrossRef] [PubMed]
- Urwanisch, L.; Luciano, M.; Horejs-Hoeck, J. The NLRP3 Inflammasome and Its Role in the Pathogenicity of Leukemia. Int. J. Mol. Sci. 2021, 22, 1271. [Google Scholar] [CrossRef]
- Ratajczak, M.Z.; Bujko, K.; Cymer, M.; Thapa, A.; Adamiak, M.; Ratajczak, J.; Abdel-Latif, A.K.; Kucia, M. The Nlrp3 inflammasome as a “rising star” in studies of normal and malignant hematopoiesis. Leukemia 2020, 34, 1512–1523. [Google Scholar] [CrossRef] [PubMed]
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.d.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th Edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- de Leval, L.; Jaffe, E.S. Lymphoma Classification. Cancer J. 2020, 26, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Eigenbrod, T.; Muñoz-Planillo, R.; Nuñez, G. The inflammasome: A caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 2009, 10, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Liszewski, M.K.; Kolev, M.; Le Friec, G.; Leung, M.; Bertram, P.G.; Fara, A.F.; Subias, M.; Pickering, M.C.; Drouet, C.; Meri, S.; et al. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity 2013, 39, 1143–1157. [Google Scholar] [CrossRef] [PubMed]
- Heeger, P.S.; Kemper, C. Novel roles of complement in T effector cell regulation. Immunobiology 2012, 217, 216–224. [Google Scholar] [CrossRef]
- Phan, T.G.; Grigorova, I.; Okada, T.; Cyster, J.G. Subcapsular encounter and complement-dependent transport of immune complexes by lymph node B cells. Nat. Immunol. 2007, 8, 992–1000. [Google Scholar] [CrossRef]
- Hsu, M.L.; Zouali, M. Inflammasome is a central player in B cell development and homing. Life Sci. Alliance 2023, 6, e202201700. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and diseases. Signal Transduct. Target. Ther. 2021, 6, 128. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Lu, L.; Li, B.; Shi, X.; Jin, H.; Hu, W. The roles of inflammasomes in cancer. Front. Immunol. 2023, 14, 1195572. [Google Scholar] [CrossRef] [PubMed]
- Blevins, H.M.; Xu, Y.; Biby, S.; Zhang, S. The NLRP3 Inflammasome Pathway: A Review of Mechanisms and Inhibitors for the Treatment of Inflammatory Diseases. Front. Aging Neurosci. 2022, 14, 879021. [Google Scholar] [CrossRef]
- Wang, L.; Sharif, H.; Vora, S.M.; Zheng, Y.; Wu, H. Structures and functions of the inflammasome engine. J. Allergy Clin. Immunol. 2021, 147, 2021–2029. [Google Scholar] [CrossRef] [PubMed]
- Fernandes-Alnemri, T.; Wu, J.; Yu, J.W.; Datta, P.; Miller, B.; Jankowski, W.; Rosenberg, S.; Zhang, J.; Alnemri, E.S. The pyroptosome: A supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 2007, 14, 1590–1604. [Google Scholar] [CrossRef]
- de Zoete, M.R.; Palm, N.W.; Zhu, S.; Flavell, R.A. Inflammasomes. Cold Spring Harb. Perspect. Biol. 2014, 6, a016287. [Google Scholar] [CrossRef]
- Song, L.; Pei, L.; Yao, S.; Wu, Y.; Shang, Y. NLRP3 Inflammasome in Neurological Diseases, from Functions to Therapies. Front. Cell Neurosci. 2017, 11, 63. [Google Scholar] [CrossRef]
- Sundaram, B.; Kanneganti, T.D. Advances in Understanding Activation and Function of the NLRC4 Inflammasome. Int. J. Mol. Sci. 2021, 22, 1048. [Google Scholar] [CrossRef]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, Z.; Xiao, T.S. Post-translational regulation of inflammasomes. Cell. Mol. Immunol. 2017, 14, 65–79. [Google Scholar] [CrossRef]
- Katsnelson, M.A.; Rucker, L.G.; Russo, H.M.; Dubyak, G.R. K+ efflux agonists induce NLRP3 inflammasome activation independently of Ca2+ signaling. J. Immunol. 2015, 194, 3937–3952. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef]
- Tang, T.; Lang, X.; Xu, C.; Wang, X.; Gong, T.; Yang, Y.; Cui, J.; Bai, L.; Wang, J.; Jiang, W.; et al. CLICs-dependent chloride efflux is an essential and proximal upstream event for NLRP3 inflammasome activation. Nat. Commun. 2017, 8, 202. [Google Scholar] [CrossRef]
- Rajamäki, K.; Lappalainen, J.; Oörni, K.; Välimäki, E.; Matikainen, S.; Kovanen, P.T.; Eklund, K.K. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: A novel link between cholesterol metabolism and inflammation. PLoS ONE 2010, 5, e11765. [Google Scholar] [CrossRef]
- Martinon, F.; Pétrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef]
- Dostert, C.; Pétrilli, V.; Van Bruggen, R.; Steele, C.; Mossman, B.T.; Tschopp, J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 2008, 320, 674–677. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Huang, Y.; Chen, M.; Yang, Y.; Li, X.; Zhang, W. Mitochondrial DNA in NLRP3 inflammasome activation. Int. Immunopharmacol. 2022, 108, 108719. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.S.; He, Q.; Janczy, J.R.; Elliott, E.I.; Zhong, Z.; Olivier, A.K.; Sadler, J.J.; Knepper-Adrian, V.; Han, R.; Qiao, L.; et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 2013, 39, 311–323. [Google Scholar] [CrossRef]
- Chen, J.; Chen, Z.J. PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature 2018, 564, 71–76. [Google Scholar] [CrossRef]
- Krumm, B.; Xiang, Y.; Deng, J. Structural biology of the IL-1 superfamily: Key cytokines in the regulation of immune and inflammatory responses. Protein Sci. 2014, 23, 526–538. [Google Scholar] [CrossRef]
- Wyble, C.W.; Hynes, K.L.; Kuchibhotla, J.; Marcus, B.C.; Hallahan, D.; Gewertz, B.L. TNF-α and IL-1 upregulate membrane-bound and soluble E-selectin through a common pathway. J. Surg. Res. 1997, 73, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Bochner, B.S.; Luscinskas, F.W.; Gimbrone, M.A., Jr.; Newman, W.; Sterbinsky, S.A.; Derse-Anthony, C.P.; Klunk, D.; Schleimer, R.P. Adhesion of human basophils, eosinophils, and neutrophils to interleukin 1-activated human vascular endothelial cells: Contributions of endothelial cell adhesion molecules. J. Exp. Med. 1991, 173, 1553–1557. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.D.; Fan, F.; McConkey, D.J.; Jean, M.E.; Liu, W.; Reinmuth, N.; Stoeltzing, O.; Ahmad, S.A.; Parikh, A.A.; Mukaida, N.; et al. Role of P38 MAPK, AP-1, and NF-κB in interleukin-1β-induced IL-8 expression in human vascular smooth muscle cells. Cytokine 2002, 18, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Wang, H.; Chen, W.; Meng, G. Regulation of adaptive immunity by the NLRP3 inflammasome. Int. Immunopharmacol. 2011, 11, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Novick, D.; Kim, S.; Kaplanski, G.; Dinarello, C.A. Interleukin-18, more than a Th1 cytokine. Semin. Immunol. 2013, 25, 439–448. [Google Scholar] [CrossRef]
- Zhang, H.; Gao, J.; Tang, Y.; Jin, T.; Tao, J. Inflammasomes cross-talk with lymphocytes to connect the innate and adaptive immune response. J. Adv. Res. 2023, 54, 181–193. [Google Scholar] [CrossRef]
- Deets, K.A.; Vance, R.E. Inflammasomes and adaptive immune responses. Nat. Immunol. 2021, 22, 412–422. [Google Scholar] [CrossRef]
- Lenkiewicz, A.M.; Adamiak, M.; Thapa, A.; Bujko, K.; Pedziwiatr, D.; Abdel-Latif, A.K.; Kucia, M.; Ratajczak, J.; Ratajczak, M.Z. The Nlrp3 Inflammasome Orchestrates Mobilization of Bone Marrow-Residing Stem Cells into Peripheral Blood. Stem Cell Rev. Rep. 2019, 15, 391–403. [Google Scholar] [CrossRef]
- Yang, L.; Hu, M.; Lu, Y.; Han, S.; Wang, J. Inflammasomes and the Maintenance of Hematopoietic Homeostasis: New Perspectives and Opportunities. Molecules 2021, 26, 309. [Google Scholar] [CrossRef]
- Dorshkind, K. Multilineage development from adult bone marrow cells. Nat. Immunol. 2002, 3, 311–313. [Google Scholar] [CrossRef]
- Månsson, R.; Hultquist, A.; Luc, S.; Yang, L.; Anderson, K.; Kharazi, S.; Al-Hashmi, S.; Liuba, K.; Thorén, L.; Adolfsson, J.; et al. Molecular evidence for hierarchical transcriptional lineage priming in fetal and adult stem cells and multipotent progenitors. Immunity 2007, 26, 407–419. [Google Scholar] [CrossRef] [PubMed]
- LeBien, T.W.; Tedder, T.F. B lymphocytes: How they develop and function. Blood 2008, 112, 1570–1580. [Google Scholar] [CrossRef] [PubMed]
- Pieper, K.; Grimbacher, B.; Eibel, H. B-cell biology and development. J. Allergy Clin. Immunol. 2013, 131, 959–971. [Google Scholar] [CrossRef] [PubMed]
- Janeway, C.; Travers, P.; Walport, M.; Shlomchik, M.J. Immunobiology: The Immune System in Health and Disease, 5th ed.; Taylor & Francis, Inc.: Abingdon, UK, 1994. [Google Scholar]
- Koch, U.; Radtke, F. Mechanisms of T cell development and transformation. Annu. Rev. Cell Dev. Biol. 2011, 27, 539–562. [Google Scholar] [CrossRef]
- Miller, J.P.; Allman, D. Linking age-related defects in B lymphopoiesis to the aging of hematopoietic stem cells. Semin. Immunol. 2005, 17, 321–329. [Google Scholar] [CrossRef]
- Morrison, S.J.; Wandycz, A.M.; Akashi, K.; Globerson, A.; Weissman, I.L. The aging of hematopoietic stem cells. Nat. Med. 1996, 2, 1011–1016. [Google Scholar] [CrossRef] [PubMed]
- Tuljapurkar, S.R.; McGuire, T.R.; Brusnahan, S.K.; Jackson, J.D.; Garvin, K.L.; Kessinger, M.A.; Lane, J.T.; O’Kane, B.J.; Sharp, J.G. Changes in human bone marrow fat content associated with changes in hematopoietic stem cell numbers and cytokine levels with aging. J. Anat. 2011, 219, 574–581. [Google Scholar] [CrossRef]
- Frasca, D.; Blomberg, B.B. Inflammaging decreases adaptive and innate immune responses in mice and humans. Biogerontology 2016, 17, 7–19. [Google Scholar] [CrossRef]
- Kennedy, D.E.; Knight, K.L. Inhibition of B Lymphopoiesis by Adipocytes and IL-1-Producing Myeloid-Derived Suppressor Cells. J. Immunol. 2015, 195, 2666–2674. [Google Scholar] [CrossRef]
- Kennedy, D.E.; Knight, K.L. Inflammatory Changes in Bone Marrow Microenvironment Associated with Declining B Lymphopoiesis. J. Immunol. 2017, 198, 3471–3479. [Google Scholar] [CrossRef]
- Xu, H.; Chaudhri, V.K.; Wu, Z.; Biliouris, K.; Dienger-Stambaugh, K.; Rochman, Y.; Singh, H. Regulation of bifurcating B cell trajectories by mutual antagonism between transcription factors IRF4 and IRF8. Nat. Immunol. 2015, 16, 1274–1281. [Google Scholar] [CrossRef]
- Pathak, S.; Ma, S.; Trinh, L.; Lu, R. A role for interferon regulatory factor 4 in receptor editing. Mol. Cell. Biol. 2008, 28, 2815–2824. [Google Scholar] [CrossRef]
- Bruchard, M.; Rebé, C.; Derangère, V.; Togbé, D.; Ryffel, B.; Boidot, R.; Humblin, E.; Hamman, A.; Chalmin, F.; Berger, H.; et al. The receptor NLRP3 is a transcriptional regulator of TH2 differentiation. Nat. Immunol. 2015, 16, 859–870. [Google Scholar] [CrossRef]
- Cyster, J.G. Chemokines and cell migration in secondary lymphoid organs. Science 1999, 286, 2098–2102. [Google Scholar] [CrossRef]
- Ali, M.F.; Dasari, H.; Van Keulen, V.P.; Carmona, E.M. Canonical Stimulation of the NLRP3 Inflammasome by Fungal Antigens Links Innate and Adaptive B-Lymphocyte Responses by Modulating IL-1β and IgM Production. Front. Immunol. 2017, 8, 1504. [Google Scholar] [CrossRef] [PubMed]
- Chidgey, A.; Dudakov, J.; Seach, N.; Boyd, R. Impact of niche aging on thymic regeneration and immune reconstitution. Semin. Immunol. 2007, 19, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Youm, Y.H.; Kanneganti, T.D.; Vandanmagsar, B.; Zhu, X.; Ravussin, A.; Adijiang, A.; Owen, J.S.; Thomas, M.J.; Francis, J.; Parks, J.S.; et al. The Nlrp3 inflammasome promotes age-related thymic demise and immunosenescence. Cell Rep. 2012, 1, 56–68. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, C.; Hua, M.; Wang, R.; Zhong, C.; Yu, J.; Han, F.; He, N.; Zhao, Y.; Liu, G.; et al. NLRP3 inflammasome activation plays a carcinogenic role through effector cytokine IL-18 in lymphoma. Oncotarget 2017, 8, 108571–108583. [Google Scholar] [CrossRef]
- Takubo, T.; Kumura, T.; Nakao, T.; Nakamae, H.; Aoyama, Y.; Nishiki, S.; Kinoshita, Y.; Koh, K.-R.; Ohta, K.; Yamane, T.; et al. Comparative Study on Complete Remission Rate and Overall Survival in Three Groups Classified Based on the Serum Interleukin-18 Level in Non-Hodgkin’s Lymphoma Patients. Acta Haematol. 2001, 104, 220–222. [Google Scholar] [CrossRef]
- Khaled, H.M.; Abdelhamid, T.M.; Abu-Taleb, F.M.; El-Hifnawi, N.M.; Waley, A.B. Impact of serum levels of IL-18 and soluble IL-2 receptor on the clinical outcome of patients with diffuse large B-cell lymphoma treated with R-CHOP regimen. Future Sci. OA 2019, 5, Fso414. [Google Scholar] [CrossRef]
- Lu, F.; Zhao, Y.; Pang, Y.; Ji, M.; Sun, Y.; Wang, H.; Zou, J.; Wang, Y.; Li, G.; Sun, T.; et al. NLRP3 inflammasome upregulates PD-L1 expression and contributes to immune suppression in lymphoma. Cancer Lett. 2021, 497, 178–189. [Google Scholar] [CrossRef]
- Liu, Y.; Barta, S.K. Diffuse large B-cell lymphoma: 2019 update on diagnosis, risk stratification, and treatment. Am. J. Hematol. 2019, 94, 604–616. [Google Scholar] [CrossRef]
- Soydinc, H.O.; Guney, N.; Basaran, M.; Duranyildiz, D.; Yasasever, V. Clinical significance of interleukin-4 and interleukin-18 levels in aggressive non-Hodgkin’s lymphoma patients. Genet. Mol. Res. 2016, 15, gmr.15038590. [Google Scholar] [CrossRef] [PubMed]
- Baldini, C.; Santini, E.; Rossi, C.; Donati, V.; Solini, A. The P2X7 receptor-NLRP3 inflammasome complex predicts the development of non-Hodgkin’s lymphoma in Sjogren’s syndrome: A prospective, observational, single-centre study. J. Intern. Med. 2017, 282, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Ek, S.; Högerkorp, C.M.; Dictor, M.; Ehinger, M.; Borrebaeck, C.A. Mantle cell lymphomas express a distinct genetic signature affecting lymphocyte trafficking and growth regulation as compared with subpopulations of normal human B cells. Cancer Res. 2002, 62, 4398–4405. [Google Scholar]
- Singh, V.V.; Kerur, N.; Bottero, V.; Dutta, S.; Chakraborty, S.; Ansari, M.A.; Paudel, N.; Chikoti, L.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus latency in endothelial and B cells activates gamma interferon-inducible protein 16-mediated inflammasomes. J. Virol. 2013, 87, 4417–4431. [Google Scholar] [CrossRef]
- Salaro, E.; Rambaldi, A.; Falzoni, S.; Amoroso, F.S.; Franceschini, A.; Sarti, A.C.; Bonora, M.; Cavazzini, F.; Rigolin, G.M.; Ciccone, M.; et al. Involvement of the P2X7-NLRP3 axis in leukemic cell proliferation and death. Sci. Rep. 2016, 6, 26280. [Google Scholar] [CrossRef] [PubMed]
- Reinhart, N.M.; Akinyemi, I.A.; Frey, T.R.; Xu, H.; Agudelo, C.; Brathwaite, J.; Burton, E.M.; Burgula, S.; McIntosh, M.T.; Bhaduri-McIntosh, S. The danger molecule HMGB1 cooperates with the NLRP3 inflammasome to sustain expression of the EBV lytic switch protein in Burkitt lymphoma cells. Virology 2022, 566, 136–142. [Google Scholar] [CrossRef]
- Ihim, S.A.; Abubakar, S.D.; Zian, Z.; Sasaki, T.; Saffarioun, M.; Maleknia, S.; Azizi, G. Interleukin-18 cytokine in immunity, inflammation, and autoimmunity: Biological role in induction, regulation, and treatment. Front. Immunol. 2022, 13, 919973. [Google Scholar] [CrossRef] [PubMed]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Marzec, M.; Zhang, Q.; Goradia, A.; Raghunath, P.N.; Liu, X.; Paessler, M.; Wang, H.Y.; Wysocka, M.; Cheng, M.; Ruggeri, B.A.; et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc. Natl. Acad. Sci. USA 2008, 105, 20852–20857. [Google Scholar] [CrossRef] [PubMed]
- Doi, T.; Ishikawa, T.; Okayama, T.; Oka, K.; Mizushima, K.; Yasuda, T.; Sakamoto, N.; Katada, K.; Kamada, K.; Uchiyama, K.; et al. The JAK/STAT pathway is involved in the upregulation of PD-L1 expression in pancreatic cancer cell lines. Oncol. Rep. 2017, 37, 1545–1554. [Google Scholar] [CrossRef]
- Atsaves, V.; Tsesmetzis, N.; Chioureas, D.; Kis, L.; Leventaki, V.; Drakos, E.; Panaretakis, T.; Grander, D.; Medeiros, L.J.; Young, K.H.; et al. PD-L1 is commonly expressed and transcriptionally regulated by STAT3 and MYC in ALK-negative anaplastic large-cell lymphoma. Leukemia 2017, 31, 1633–1637. [Google Scholar] [CrossRef]
- Sasidharan Nair, V.; Toor, S.M.; Ali, B.R.; Elkord, E. Dual inhibition of STAT1 and STAT3 activation downregulates expression of PD-L1 in human breast cancer cells. Expert Opin. Ther. Targets 2018, 22, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Serna, L.; Azcoaga, P.; Brahmachary, M.; Caffarel, M.M.; Braza, M.S. Diffuse large B-cell lymphoma microenvironment displays a predominant macrophage infiltrate marked by a strong inflammatory signature. Front. Immunol. 2023, 14, 1048567. [Google Scholar] [CrossRef]
- Liu, Z.H.; Zhang, L.; Jing, F.J.; Xiao, S.X.; Gao, Y.; Bian, H.Y.; Zhao, X. Genetic Polymorphisms in NLRP3 Inflammasome-Associated Genes in Patients with B-Cell Non-Hodgkin’s Lymphoma. J. Inflamm. Res. 2021, 14, 5687–5697. [Google Scholar] [CrossRef]
- Wang, W.; Xu, S.W.; Teng, Y.; Zhu, M.; Guo, Q.Y.; Wang, Y.W.; Mao, X.L.; Li, S.W.; Luo, W.D. The Dark Side of Pyroptosis of Diffuse Large B-Cell Lymphoma in B-Cell Non-Hodgkin Lymphoma: Mediating the Specific Inflammatory Microenvironment. Front. Cell Dev. Biol. 2021, 9, 779123. [Google Scholar] [CrossRef]
- Ma, J.; Wang, W.; Ma, J.; Xu, Z. A Novel and Validated 8-Pyroptosis-Related Genes Based Risk Prediction Model for Diffuse Large B Cell Lymphoma. Biomolecules 2022, 12, 1835. [Google Scholar] [CrossRef]
- Mackay, F.; Schneider, P. Cracking the BAFF code. Nat. Rev. Immunol. 2009, 9, 491–502. [Google Scholar] [CrossRef]
- Schiemann, B.; Gommerman, J.L.; Vora, K.; Cachero, T.G.; Shulga-Morskaya, S.; Dobles, M.; Frew, E.; Scott, M.L. An essential role for BAFF in the normal development of B cells through a BCMA-independent pathway. Science 2001, 293, 2111–2114. [Google Scholar] [CrossRef]
- Briones, J.; Timmerman, J.M.; Hilbert, D.M.; Levy, R. BLyS and BLyS receptor expression in non-Hodgkin’s lymphoma. Exp. Hematol. 2002, 30, 135–141. [Google Scholar] [CrossRef]
- Novak, A.J.; Grote, D.M.; Stenson, M.; Ziesmer, S.C.; Witzig, T.E.; Habermann, T.M.; Harder, B.; Ristow, K.M.; Bram, R.J.; Jelinek, D.F.; et al. Expression of BLyS and its receptors in B-cell non-Hodgkin lymphoma: Correlation with disease activity and patient outcome. Blood 2004, 104, 2247–2253. [Google Scholar] [CrossRef]
- Novak, A.J.; Bram, R.J.; Kay, N.E.; Jelinek, D.F. Aberrant expression of B-lymphocyte stimulator by B chronic lymphocytic leukemia cells: A mechanism for survival. Blood 2002, 100, 2973–2979. [Google Scholar] [CrossRef]
- Haiat, S.; Billard, C.; Quiney, C.; Ajchenbaum-Cymbalista, F.; Kolb, J.P. Role of BAFF and APRIL in human B-cell chronic lymphocytic leukaemia. Immunology 2006, 118, 281–292. [Google Scholar] [CrossRef]
- Novak, A.J.; Darce, J.R.; Arendt, B.K.; Harder, B.; Henderson, K.; Kindsvogel, W.; Gross, J.A.; Greipp, P.R.; Jelinek, D.F. Expression of BCMA, TACI, and BAFF-R in multiple myeloma: A mechanism for growth and survival. Blood 2004, 103, 689–694. [Google Scholar] [CrossRef]
- He, B.; Chadburn, A.; Jou, E.; Schattner, E.J.; Knowles, D.M.; Cerutti, A. Lymphoma B cells evade apoptosis through the TNF family members BAFF/BLyS and APRIL. J. Immunol. 2004, 172, 3268–3279. [Google Scholar] [CrossRef]
- Yang, S.; Li, J.Y.; Xu, W. Role of BAFF/BAFF-R axis in B-cell non-Hodgkin lymphoma. Crit. Rev. Oncol. Hematol. 2014, 91, 113–122. [Google Scholar] [CrossRef]
- Lim, K.H.; Chen, L.C.; Hsu, K.; Chang, C.C.; Chang, C.Y.; Kao, C.W.; Chang, Y.F.; Chang, M.C.; Chen, C.G. BAFF-driven NLRP3 inflammasome activation in B cells. Cell Death Dis. 2020, 11, 820. [Google Scholar] [CrossRef]
- Quartuccio, L.; Salvin, S.; Fabris, M.; Maset, M.; Pontarini, E.; Isola, M.; De Vita, S. BLyS upregulation in Sjogren’s syndrome associated with lymphoproliferative disorders, higher ESSDAI score and B-cell clonal expansion in the salivary glands. Rheumatology 2013, 52, 276–281. [Google Scholar] [CrossRef]
- Fragoulis, G.E.; Vakrakou, A.G.; Papadopoulou, A.; Germenis, A.; Kanavakis, E.; Moutsopoulos, H.M.; Manoussakis, M.N. Impaired degradation and aberrant phagocytosis of necrotic cell debris in the peripheral blood of patients with primary Sjögren’s syndrome. J. Autoimmun. 2015, 56, 12–22. [Google Scholar] [CrossRef]
- Manoussakis, M.N.; Fragoulis, G.E.; Vakrakou, A.G.; Moutsopoulos, H.M. Impaired clearance of early apoptotic cells mediated by inhibitory IgG antibodies in patients with primary Sjögren’s syndrome. PLoS ONE 2014, 9, e112100. [Google Scholar] [CrossRef]
- Kono, H.; Kimura, Y.; Latz, E. Inflammasome activation in response to dead cells and their metabolites. Curr. Opin. Immunol. 2014, 30, 91–98. [Google Scholar] [CrossRef]
- Vakrakou, A.G.; Boiu, S.; Ziakas, P.D.; Xingi, E.; Boleti, H.; Manoussakis, M.N. Systemic activation of NLRP3 inflammasome in patients with severe primary Sjögren’s syndrome fueled by inflammagenic DNA accumulations. J. Autoimmun. 2018, 91, 23–33. [Google Scholar] [CrossRef]
- Patil, K.; Kuttikrishnan, S.; Khan, A.Q.; Ahmad, F.; Alam, M.; Buddenkotte, J.; Ahmad, A.; Steinhoff, M.; Uddin, S. Molecular pathogenesis of Cutaneous T cell Lymphoma: Role of chemokines, cytokines, and dysregulated signaling pathways. Semin. Cancer Biol. 2022, 86, 382–399. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, K.; Clark, R.; Dowgiert, R.; Hurwitz, D.; Shibata, M.; Rich, B.E.; Hirahara, K.; Jones, D.A.; Eapen, S.; Mizutani, H.; et al. Expression of interleukin-18 and caspase-1 in cutaneous T-cell lymphoma. Clin. Cancer Res. 2006, 12, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Okamura, H.; Tsutsi, H.; Komatsu, T.; Yutsudo, M.; Hakura, A.; Tanimoto, T.; Torigoe, K.; Okura, T.; Nukada, Y.; Hattori, K.; et al. Cloning of a new cytokine that induces IFN-γ production by T cells. Nature 1995, 378, 88–91. [Google Scholar] [CrossRef]
- Yoshimoto, T.; Mizutani, H.; Tsutsui, H.; Noben-Trauth, N.; Yamanaka, K.; Tanaka, M.; Izumi, S.; Okamura, H.; Paul, W.E.; Nakanishi, K. IL-18 induction of IgE: Dependence on CD4+ T cells, IL-4 and STAT6. Nat. Immunol. 2000, 1, 132–137. [Google Scholar] [CrossRef]
- Yoshimoto, T.; Tsutsui, H.; Tominaga, K.; Hoshino, K.; Okamura, H.; Akira, S.; Paul, W.E.; Nakanishi, K. IL-18, although antiallergic when administered with IL-12, stimulates IL-4 and histamine release by basophils. Proc. Natl. Acad. Sci. USA 1999, 96, 13962–13966. [Google Scholar] [CrossRef]
- Huanosta-Murillo, E.; Alcántara-Hernández, M.; Hernández-Rico, B.; Victoria-Acosta, G.; Miranda-Cruz, P.; Domínguez-Gómez, M.A.; Jurado-Santacruz, F.; Patiño-López, G.; Pérez-Koldenkova, V.; Palma-Guzmán, A.; et al. NLRP3 Regulates IL-4 Expression in TOX+ CD4+ T Cells of Cutaneous T Cell Lymphoma to Potentially Promote Disease Progression. Front. Immunol. 2021, 12, 668369. [Google Scholar] [CrossRef]
- Willemze, R.; Cerroni, L.; Kempf, W.; Berti, E.; Facchetti, F.; Swerdlow, S.H.; Jaffe, E.S. The 2018 update of the WHO-EORTC classification for primary cutaneous lymphomas. Blood 2019, 133, 1703–1714. [Google Scholar] [CrossRef]
- Campbell, J.; Clark, R.A.; Kupper, T.S. Sezary Syndrome Is a Malignancy of Central Memory T Cells with Skin Homing Properties, While Mycosis Fungoides Is a Malignancy of Skin Homing Effector Memory T Cells. Blood 2009, 114, 3954. [Google Scholar] [CrossRef]
- Manfrere, K.C.G.; Torrealba, M.P.; Ferreira, F.M.; de Sousa, E.S.A.; Miyashiro, D.; Teixeira, F.M.E.; Custódio, R.W.A.; Nakaya, H.I.; Ramos, Y.A.L.; Sotto, M.N.; et al. Imbalanced IL-1B and IL-18 Expression in Sézary Syndrome. Int. J. Mol. Sci. 2023, 24, 4674. [Google Scholar] [CrossRef]
- Lim, S.W.; Ryu, K.J.; Lee, H.; Ko, Y.H.; Kim, W.S.; Kim, S.J. Serum IL18 is associated with hemophagocytosis and poor survival in extranodal natural killer/T-cell lymphoma. Leuk. Lymphoma 2019, 60, 317–325. [Google Scholar] [CrossRef]
- Chen, Q.; Liu, S.; Zhang, K.; Yu, B.; Zhang, W.; Zhang, H.; Chen, X. Hsa-miR-372-5p regulates the NIMA related kinase 7 and IL-1β release in NK/T-cell lymphoma. Leuk. Lymphoma 2021, 62, 2648–2656. [Google Scholar] [CrossRef]
- Popovic, D.; Vucic, D.; Dikic, I. Ubiquitination in disease pathogenesis and treatment. Nat. Med. 2014, 20, 1242–1253. [Google Scholar] [CrossRef]
- Ciechanover, A.; Stanhill, A. The complexity of recognition of ubiquitinated substrates by the 26S proteasome. Biochim. Biophys. Acta 2014, 1843, 86–96. [Google Scholar] [CrossRef]
- Yau, R.; Rape, M. The increasing complexity of the ubiquitin code. Nat. Cell Biol. 2016, 18, 579–586. [Google Scholar] [CrossRef]
- Caldeira, M.V.; Salazar, I.L.; Curcio, M.; Canzoniero, L.M.; Duarte, C.B. Role of the ubiquitin-proteasome system in brain ischemia: Friend or foe? Prog. Neurobiol. 2014, 112, 50–69. [Google Scholar] [CrossRef]
- Deng, N.H.; Zhou, Z.X.; Liu, H.T.; Tian, Z.; Wu, Z.F.; Liu, X.Y.; Xiong, W.H.; Wang, Z.; Jiang, Z.S. TRIMs: Generalists Regulating the NLRP3 Inflammasome Signaling Pathway. DNA Cell Biol. 2022, 41, 262–275. [Google Scholar] [CrossRef]
- Crawford, L.J.; Johnston, C.K.; Irvine, A.E. TRIM proteins in blood cancers. J. Cell Commun. Signal. 2018, 12, 21–29. [Google Scholar] [CrossRef]
- Xie, X.; Wang, F.; Li, X. Inhibition of TRIM14 protects cerebral ischemia/reperfusion injury through regulating NF-κB/NLRP3 pathway-mediated inflammation and apoptosis. J. Recept. Signal Transduct. Res. 2022, 42, 197–205. [Google Scholar] [CrossRef]
- Munding, C.; Keller, M.; Niklaus, G.; Papin, S.; Tschopp, J.; Werner, S.; Beer, H.D. The estrogen-responsive B box protein: A novel enhancer of interleukin-1β secretion. Cell Death Differ. 2006, 13, 1938–1949. [Google Scholar] [CrossRef]
- Jena, K.K.; Kolapalli, S.P.; Mehto, S.; Nath, P.; Das, B.; Sahoo, P.K.; Ahad, A.; Syed, G.H.; Raghav, S.K.; Senapati, S.; et al. TRIM16 controls assembly and degradation of protein aggregates by modulating the p62-NRF2 axis and autophagy. EMBO J. 2018, 37, e98358. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Jain, A.; Choi, S.W.; Mandell, M.A.; Schroder, K.; Johansen, T.; Deretic, V. TRIM-mediated precision autophagy targets cytoplasmic regulators of innate immunity. J. Cell Biol. 2015, 210, 973–989. [Google Scholar] [CrossRef] [PubMed]
- Papin, S.; Cuenin, S.; Agostini, L.; Martinon, F.; Werner, S.; Beer, H.D.; Grütter, C.; Grütter, M.; Tschopp, J. The SPRY domain of Pyrin, mutated in familial Mediterranean fever patients, interacts with inflammasome components and inhibits proIL-1β processing. Cell Death Differ. 2007, 14, 1457–1466. [Google Scholar] [CrossRef] [PubMed]
- Vajjhala, P.R.; Kaiser, S.; Smith, S.J.; Ong, Q.R.; Soh, S.L.; Stacey, K.J.; Hill, J.M. Identification of multifaceted binding modes for pyrin and ASC pyrin domains gives insights into pyrin inflammasome assembly. J. Biol. Chem. 2014, 289, 23504–23519. [Google Scholar] [CrossRef]
- Gao, W.; Li, Y.; Liu, X.; Wang, S.; Mei, P.; Chen, Z.; Liu, K.; Li, S.; Xu, X.W.; Gan, J.; et al. TRIM21 regulates pyroptotic cell death by promoting Gasdermin D oligomerization. Cell Death Differ. 2022, 29, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Lu, Z.; Zhu, G.; Chen, Y.; Wu, Y. Knockdown of TRIM22 Relieves Oxygen-Glucose Deprivation/Reoxygenation-Induced Apoptosis and Inflammation through Inhibition of NF-κB/NLRP3 Axis. Cell. Mol. Neurobiol. 2021, 41, 341–351. [Google Scholar] [CrossRef]
- Hang, Y.; Tan, L.; Chen, Q.; Liu, Q.; Jin, Y. E3 ubiquitin ligase TRIM24 deficiency promotes NLRP3/caspase-1/IL-1β-mediated pyroptosis in endometriosis. Cell Biol. Int. 2021, 45, 1561–1570. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tao, S.; Liao, L.; Li, Y.; Li, H.; Li, Z.; Lin, L.; Wan, X.; Yang, X.; Chen, L. TRIM25 promotes the cell survival and growth of hepatocellular carcinoma through targeting Keap1-Nrf2 pathway. Nat. Commun. 2020, 11, 348. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Li, Q.; Liang, W.; Yan, R.; Tong, L.; Jia, M.; Zhao, C.; Zhao, W. TRIM28 SUMOylates and stabilizes NLRP3 to facilitate inflammasome activation. Nat. Commun. 2021, 12, 4794. [Google Scholar] [CrossRef]
- Hu, Y.; Mao, K.; Zeng, Y.; Chen, S.; Tao, Z.; Yang, C.; Sun, S.; Wu, X.; Meng, G.; Sun, B. Tripartite-motif protein 30 negatively regulates NLRP3 inflammasome activation by modulating reactive oxygen species production. J. Immunol. 2010, 185, 7699–7705. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Zhao, Z.; Zheng, S.; Wu, M.; Song, X.; Li, Y.; Zheng, Y.; Liu, B.; Chen, L.; Gao, C.; et al. The E3 ubiquitin ligase TRIM31 is involved in cerebral ischemic injury by promoting degradation of TIGAR. Redox Biol. 2021, 45, 102058. [Google Scholar] [CrossRef]
- Song, H.; Liu, B.; Huai, W.; Yu, Z.; Wang, W.; Zhao, J.; Han, L.; Jiang, G.; Zhang, L.; Gao, C.; et al. The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nat. Commun. 2016, 7, 13727. [Google Scholar] [CrossRef]
- Weng, L.; Mitoma, H.; Trichot, C.; Bao, M.; Liu, Y.; Zhang, Z.; Liu, Y.J. The E3 ubiquitin ligase tripartite motif 33 is essential for cytosolic RNA-induced NLRP3 inflammasome activation. J. Immunol. 2014, 193, 3676–3682. [Google Scholar] [CrossRef]
- Mitoma, H.; Hanabuchi, S.; Kim, T.; Bao, M.; Zhang, Z.; Sugimoto, N.; Liu, Y.J. The DHX33 RNA helicase senses cytosolic RNA and activates the NLRP3 inflammasome. Immunity 2013, 39, 123–135. [Google Scholar] [CrossRef]
- Shen, J.; Wu, Q.; Liang, T.; Zhang, J.; Bai, J.; Yuan, M.; Shen, P. TRIM40 inhibits IgA1-induced proliferation of glomerular mesangial cells by inactivating NLRP3 inflammasome through ubiquitination. Mol. Immunol. 2021, 140, 225–232. [Google Scholar] [CrossRef]
- Huttlin, E.L.; Bruckner, R.J.; Paulo, J.A.; Cannon, J.R.; Ting, L.; Baltier, K.; Colby, G.; Gebreab, F.; Gygi, M.P.; Parzen, H.; et al. Architecture of the human interactome defines protein communities and disease networks. Nature 2017, 545, 505–509. [Google Scholar] [CrossRef]
- Liu, X.; Lei, Q. TRIM62 knockout protects against cerebral ischemic injury in mice by suppressing NLRP3-regulated neuroinflammation. Biochem. Biophys. Res. Commun. 2020, 529, 140–147. [Google Scholar] [CrossRef]
- Cao, Z.; Conway, K.L.; Heath, R.J.; Rush, J.S.; Leshchiner, E.S.; Ramirez-Ortiz, Z.G.; Nedelsky, N.B.; Huang, H.; Ng, A.; Gardet, A.; et al. Ubiquitin Ligase TRIM62 Regulates CARD9-Mediated Anti-Fungal Immunity and Intestinal Inflammation. Immunity 2015, 43, 715–726. [Google Scholar] [CrossRef]
- Tang, T.; Li, P.; Zhou, X.; Wang, R.; Fan, X.; Yang, M.; Qi, K. The E3 Ubiquitin Ligase TRIM65 Negatively Regulates Inflammasome Activation through Promoting Ubiquitination of NLRP3. Front. Immunol. 2021, 12, 741839. [Google Scholar] [CrossRef]
- Bernardi, R.; Pandolfi, P.P. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat. Rev. Mol. Cell Biol. 2007, 8, 1006–1016. [Google Scholar] [CrossRef] [PubMed]
- de Thé, H.; Lavau, C.; Marchio, A.; Chomienne, C.; Degos, L.; Dejean, A. The PML-RARα fusion mRNA generated by the t(15;17) translocation in acute promyelocytic leukemia encodes a functionally altered RAR. Cell 1991, 66, 675–684. [Google Scholar] [CrossRef]
- Gurrieri, C.; Capodieci, P.; Bernardi, R.; Scaglioni, P.P.; Nafa, K.; Rush, L.J.; Verbel, D.A.; Cordon-Cardo, C.; Pandolfi, P.P. Loss of the tumor suppressor PML in human cancers of multiple histologic origins. J. Natl. Cancer Inst. 2004, 96, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Louria-Hayon, I.; Alsheich-Bartok, O.; Levav-Cohen, Y.; Silberman, I.; Berger, M.; Grossman, T.; Matentzoglu, K.; Jiang, Y.H.; Muller, S.; Scheffner, M.; et al. E6AP promotes the degradation of the PML tumor suppressor. Cell Death Differ. 2009, 16, 1156–1166. [Google Scholar] [CrossRef]
- Dong, L.; Zhang, H.; Zan, T.; Han, J.; Xue, Q.; Sun, Y. LncRNA LUADT1 Is Upregulated in Mantle Cell Lymphoma and Modulates TRIM11 by Sponging miR-24-3p to Inhibit Cell Apoptosis. Crit. Rev. Eukaryot. Gene Expr. 2021, 31, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Ding, M.; Wang, C.; Yang, X.; Ye, T.; Yu, H. TRIM11 promotes lymphomas by activating the β-catenin signaling and Axin1 ubiquitination degradation. Exp. Cell Res. 2020, 387, 111750. [Google Scholar] [CrossRef]
- Kapanadze, B.; Makeeva, N.; Corcoran, M.; Jareborg, N.; Hammarsund, M.; Baranova, A.; Zabarovsky, E.; Vorontsova, O.; Merup, M.; Gahrton, G.; et al. Comparative sequence analysis of a region on human chromosome 13q14, frequently deleted in B-cell chronic lymphocytic leukemia, and its homologous region on mouse chromosome 14. Genomics 2000, 70, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Rondeau, G.; Moreau, I.; Bézieau, S.; Cadoret, E.; Moisan, J.P.; Devilder, M.C. Exclusion of Leu1 and Leu2 genes as tumor suppressor genes in 13q14.3-deleted B-CLL. Leukemia 1999, 13, 1630–1632. [Google Scholar] [CrossRef] [PubMed]
- Bullrich, F.; Fujii, H.; Calin, G.; Mabuchi, H.; Negrini, M.; Pekarsky, Y.; Rassenti, L.; Alder, H.; Reed, J.C.; Keating, M.J.; et al. Characterization of the 13q14 tumor suppressor locus in CLL: Identification of ALT1, an alternative splice variant of the LEU2 gene. Cancer Res. 2001, 61, 6640–6648. [Google Scholar]
- Baranova, A.; Hammarsund, M.; Ivanov, D.; Skoblov, M.; Sangfelt, O.; Corcoran, M.; Borodina, T.; Makeeva, N.; Pestova, A.; Tyazhelova, T.; et al. Distinct organization of the candidate tumor suppressor gene RFP2 in human and mouse: Multiple mRNA isoforms in both species- and human-specific antisense transcript RFP2OS. Gene 2003, 321, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Litvinov, I.V.; Netchiporouk, E.; Cordeiro, B.; Zargham, H.; Pehr, K.; Gilbert, M.; Zhou, Y.; Moreau, L.; Woetmann, A.; Ødum, N.; et al. Ectopic expression of embryonic stem cell and other developmental genes in cutaneous T-cell lymphoma. Oncoimmunology 2014, 3, e970025. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.P.; Ding, D.Z.; Shi, B.; Zhang, S.Q.; Gu, L.L.; Wang, Y.C.; Cheng, C. Expression of TRIM28 correlates with proliferation and Bortezomib-induced apoptosis in B-cell non-Hodgkin lymphoma. Leuk. Lymphoma 2018, 59, 2639–2649. [Google Scholar] [CrossRef] [PubMed]
- Slezak-Prochazka, I.; Kluiver, J.; de Jong, D.; Smigielska-Czepiel, K.; Kortman, G.; Winkle, M.; Rutgers, B.; Koerts, J.; Visser, L.; Diepstra, A.; et al. Inhibition of the miR-155 target NIAM phenocopies the growth promoting effect of miR-155 in B-cell lymphoma. Oncotarget 2016, 7, 2391–2400. [Google Scholar] [CrossRef]
- Tan, X.; Cao, F.; Tang, F.; Lu, C.; Yu, Q.; Feng, S.; Yang, Z.; Chen, S.; He, X.; He, J.; et al. Suppression of DLBCL Progression by the E3 Ligase Trim35 Is Mediated by CLOCK Degradation and NK Cell Infiltration. J. Immunol. Res. 2021, 2021, 9995869. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liang, X.; Yu, T.; Xu, Y.L.; Xu, L.H.; Zhang, X.J.; Ma, J.; Wang, Y.R.; He, S.L. TRIM65 is a potential oncogenic protein via ERK1/2 on Jurkat and Raji cells: A therapeutic target in human lymphoma malignancies. Cell Biol. Int. 2018, 42, 1503–1510. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, X.; Wang, Y.; Wei, Y.; Wei, X. Involvement of inflammasomes in tumor microenvironment and tumor therapies. J. Hematol. Oncol. 2023, 16, 24. [Google Scholar] [CrossRef]
- Karki, R.; Kanneganti, T.D. Diverging inflammasome signals in tumorigenesis and potential targeting. Nat. Rev. Cancer 2019, 19, 197–214. [Google Scholar] [CrossRef] [PubMed]
- Tomura, M.; Maruo, S.; Mu, J.; Zhou, X.Y.; Ahn, H.J.; Hamaoka, T.; Okamura, H.; Nakanishi, K.; Clark, S.; Kurimoto, M.; et al. Differential capacities of CD4+, CD8+, and CD4− CD8− T cell subsets to express IL-18 receptor and produce IFN-γ in response to IL-18. J. Immunol. 1998, 160, 3759–3765. [Google Scholar] [CrossRef] [PubMed]
- Airoldi, I.; Raffaghello, L.; Cocco, C.; Guglielmino, R.; Roncella, S.; Fedeli, F.; Gambini, C.; Pistoia, V. Heterogeneous expression of interleukin-18 and its receptor in B-cell lymphoproliferative disorders deriving from naive, germinal center, and memory B lymphocytes. Clin. Cancer Res. 2004, 10, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Jost, P.J.; Ruland, A., Jr. NF-κB signaling in lymphoma: Mechanisms, consequences, and therapeutic implications. Blood 2006, 109, 2700–2707. [Google Scholar] [CrossRef] [PubMed]
- Vendramini, E.; Bomben, R.; Pozzo, F.; Bittolo, T.; Tissino, E.; Gattei, V.; Zucchetto, A. KRAS and RAS-MAPK Pathway Deregulation in Mature B Cell Lymphoproliferative Disorders. Cancers 2022, 14, 666. [Google Scholar] [CrossRef]
- Westin, J.R. Status of PI3K/Akt/mTOR pathway inhibitors in lymphoma. Clin. Lymphoma Myeloma Leuk. 2014, 14, 335–342. [Google Scholar] [CrossRef]
- Zhang, J.; Pan, C.; Xu, T.; Niu, Z.; Ma, C.; Xu, C. Interleukin 18 augments growth ability via NF-κB and p38/ATF2 pathways by targeting cyclin B1, cyclin B2, cyclin A2, and Bcl-2 in BRL-3A rat liver cells. Gene 2015, 563, 45–51. [Google Scholar] [CrossRef]
- Liu, T.; Zhou, Y.; Ko, K.; Yang, H. Interactions between Myc and Mediators of Inflammation in Chronic Liver Diseases. Mediat. Inflamm. 2015, 2015, 276850. [Google Scholar] [CrossRef]
- Hinz, M.; Krappmann, D.; Eichten, A.; Heder, A.; Scheidereit, C.; Strauss, M. NF-κB function in growth control: Regulation of cyclin D1 expression and G0/G1-to-S-phase transition. Mol. Cell Biol. 1999, 19, 2690–2698. [Google Scholar] [CrossRef]
- Klanova, M.; Klener, P. BCL-2 Proteins in Pathogenesis and Therapy of B-Cell Non-Hodgkin Lymphomas. Cancers 2020, 12, 938. [Google Scholar] [CrossRef]
- Gladkikh, A.; Potashnikova, D.; Korneva, E.; Khudoleeva, O.; Vorobjev, I. Cyclin D1 expression in B-cell lymphomas. Exp. Hematol. 2010, 38, 1047–1057. [Google Scholar] [CrossRef]
- Nguyen, L.; Papenhausen, P.; Shao, H. The Role of c-MYC in B-Cell Lymphomas: Diagnostic and Molecular Aspects. Genes 2017, 8, 116. [Google Scholar] [CrossRef]
- Perez-Yepez, E.A.; Ayala-Sumuano, J.T.; Lezama, R.; Meza, I. A novel β-catenin signaling pathway activated by IL-1β leads to the onset of epithelial-mesenchymal transition in breast cancer cells. Cancer Lett. 2014, 354, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Kassem, S.; Cleynen, A.; Chrétien, M.L.; Guillerey, C.; Putz, E.M.; Bald, T.; Förster, I.; Vuckovic, S.; Hill, G.R.; et al. Dysregulated IL-18 Is a Key Driver of Immunosuppression and a Possible Therapeutic Target in the Multiple Myeloma Microenvironment. Cancer Cell 2018, 33, 634–648.e635. [Google Scholar] [CrossRef]
- Tu, S.; Bhagat, G.; Cui, G.; Takaishi, S.; Kurt-Jones, E.A.; Rickman, B.; Betz, K.S.; Penz-Oesterreicher, M.; Bjorkdahl, O.; Fox, J.G.; et al. Overexpression of interleukin-1β induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell 2008, 14, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Theivanthiran, B.; Yarla, N.; Haykal, T.; Nguyen, Y.V.; Cao, L.; Ferreira, M.; Holtzhausen, A.; Al-Rohil, R.; Salama, A.K.S.; Beasley, G.M.; et al. Tumor-intrinsic NLRP3-HSP70-TLR4 axis drives premetastatic niche development and hyperprogression during anti-PD-1 immunotherapy. Sci. Transl. Med. 2022, 14, eabq7019. [Google Scholar] [CrossRef]
- Xu, C.; Xia, Y.; Zhang, B.W.; Drokow, E.K.; Li, H.Y.; Xu, S.; Wang, Z.; Wang, S.Y.; Jin, P.; Fang, T.; et al. Macrophages facilitate tumor cell PD-L1 expression via an IL-1β-centered loop to attenuate immune checkpoint blockade. MedComm (2020) 2023, 4, e242. [Google Scholar] [CrossRef] [PubMed]
- Daley, D.; Mani, V.R.; Mohan, N.; Akkad, N.; Pandian, G.; Savadkar, S.; Lee, K.B.; Torres-Hernandez, A.; Aykut, B.; Diskin, B.; et al. NLRP3 signaling drives macrophage-induced adaptive immune suppression in pancreatic carcinoma. J. Exp. Med. 2017, 214, 1711–1724. [Google Scholar] [CrossRef]
- Slager, S.L.; Parikh, S.A.; Achenbach, S.J.; Norman, A.D.; Rabe, K.G.; Boddicker, N.J.; Olson, J.E.; Kleinstern, G.; Lesnick, C.E.; Call, T.G.; et al. Progression and survival of MBL: A screening study of 10,139 individuals. Blood 2022, 140, 1702–1709. [Google Scholar] [CrossRef]
- Kyle, R.A.; Therneau, T.M.; Rajkumar, S.V.; Offord, J.R.; Larson, D.R.; Plevak, M.F.; Melton, L.J., 3rd. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N. Engl. J. Med. 2002, 346, 564–569. [Google Scholar] [CrossRef]
- Blanco, G.; Puiggros, A.; Sherry, B.; Nonell, L.; Calvo, X.; Puigdecanet, E.; Chiu, P.Y.; Kieso, Y.; Ferrer, G.; Palacios, F.; et al. Chronic lymphocytic leukemia-like monoclonal B-cell lymphocytosis exhibits an increased inflammatory signature that is reduced in early-stage chronic lymphocytic leukemia. Exp. Hematol. 2021, 95, 68–80. [Google Scholar] [CrossRef]
- Anagnostopoulos, A.; Evangelopoulou, A.; Sotou, D.; Gika, D.; Mitsibounas, D.; Dimopoulos, M.A. Incidence and evolution of monoclonal gammopathy of undetermined significance (MGUS) in Greece. Ann. Hematol. 2002, 81, 357–361. [Google Scholar] [CrossRef]
- Lust, J.A.; Donovan, K.A. The role of interleukin-1β in the pathogenesis of multiple myeloma. Hematol. Oncol. Clin. N. Am. 1999, 13, 1117–1125. [Google Scholar] [CrossRef]
- Bashir, M.; Bettendorf, B.; Hariman, R. A Rare but Fascinating Disorder: Case Collection of Patients with Schnitzler Syndrome. Case Rep. Rheumatol. 2018, 2018, 7041576. [Google Scholar] [CrossRef]
- Pathak, S.; Rowczenio, D.M.; Owen, R.G.; Doody, G.M.; Newton, D.J.; Taylor, C.; Taylor, J.; Cargo, C.; Hawkins, P.N.; Krause, K.; et al. Exploratory Study of MYD88 L265P, Rare NLRP3 Variants, and Clonal Hematopoiesis Prevalence in Patients with Schnitzler Syndrome. Arthritis Rheumatol. 2019, 71, 2121–2125. [Google Scholar] [CrossRef]
- Warren, J.T.; Link, D.C. Clonal hematopoiesis and risk for hematologic malignancy. Blood 2020, 136, 1599–1605. [Google Scholar] [CrossRef]
- Belizaire, R.; Wong, W.J.; Robinette, M.L.; Ebert, B.L. Clonal haematopoiesis and dysregulation of the immune system. Nat. Rev. Immunol. 2023, 23, 595–610. [Google Scholar] [CrossRef]
- Missiroli, S.; Perrone, M.; Boncompagni, C.; Borghi, C.; Campagnaro, A.; Marchetti, F.; Anania, G.; Greco, P.; Fiorica, F.; Pinton, P.; et al. Targeting the NLRP3 Inflammasome as a New Therapeutic Option for Overcoming Cancer. Cancers 2021, 13, 2297. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.D.; Samaniego, F.; Ohanian, M.; Koller, P.B.; Chandhok, N.S.; Sadiq, A.A.; Mahadevan, D.; Cherry, M.A.; Altman, J.K.; Burke, J.M.; et al. A Phase 1a/b Trial of Luxeptinib (CG-806) in Patients with Relapsed/Refractory B-Cell Malignancies or Acute Myeloid Leukemia and Evaluation of New G3 Formulation. Blood 2023, 142, 5953. [Google Scholar] [CrossRef]
- Sonowal, H.; Rice, W.G.; Howell, S.B. Luxeptinib interferes with LYN-mediated activation of SYK and modulates BCR signaling in lymphoma. PLoS ONE 2023, 18, e0277003. [Google Scholar] [CrossRef] [PubMed]
- Thieme, E.; Liu, T.; Bruss, N.; Roleder, C.; Lam, V.; Wang, X.; Nechiporuk, T.; Shouse, G.; Danilova, O.V.; Bottomly, D.; et al. Dual BTK/SYK inhibition with CG-806 (luxeptinib) disrupts B-cell receptor and Bcl-2 signaling networks in mantle cell lymphoma. Cell Death Dis. 2022, 13, 246. [Google Scholar] [CrossRef]
- Sonowal, H.; Zhang, H.; Rice, W.; Howell, S.B. Luxeptinib disables NLRP3 inflammasome-mediated IL-1β release and pathways required for secretion of inflammatory cytokines IL-6 and TNFα. Biochem. Pharmacol. 2022, 195, 114861. [Google Scholar] [CrossRef] [PubMed]
- Shui, J.W.; Boomer, J.S.; Han, J.; Xu, J.; Dement, G.A.; Zhou, G.; Tan, T.H. Hematopoietic progenitor kinase 1 negatively regulates T cell receptor signaling and T cell-mediated immune responses. Nat. Immunol. 2007, 8, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhao, Q.; Chen, T.; Liu, W.; Qiu, X.; Chen, J.; Huang, S.; Huang, R.; Dong, L. An HPK1 inhibitor enhanced the tumour response to anti-PD-1 immunotherapy in non-Hodgkin’s lymphoma. Clin. Exp. Med. 2023, 23, 3767–3780. [Google Scholar] [CrossRef]
- You, D.; Hillerman, S.; Locke, G.; Chaudhry, C.; Stromko, C.; Murtaza, A.; Fan, Y.; Koenitzer, J.; Chen, Y.; Briceno, S.; et al. Enhanced antitumor immunity by a novel small molecule HPK1 inhibitor. J. Immunother. Cancer 2021, 9, e001402. [Google Scholar] [CrossRef]
- Hasegawa, H.; Bissonnette, R.P.; Gillings, M.; Sasaki, D.; Taniguchi, H.; Kitanosono, H.; Tsuruda, K.; Kosai, K.; Uno, N.; Morinaga, Y.; et al. Induction of apoptosis by HBI-8000 in adult T-cell leukemia/lymphoma is associated with activation of Bim and NLRP3. Cancer Sci. 2016, 107, 1124–1133. [Google Scholar] [CrossRef]
- Majdalawieh, A.F.; Massri, M.; Nasrallah, G.K. A comprehensive review on the anti-cancer properties and mechanisms of action of sesamin, a lignan in sesame seeds (Sesamum indicum). Eur. J. Pharmacol. 2017, 815, 512–521. [Google Scholar] [CrossRef]
- Meng, Z.; Liu, H.; Zhang, J.; Zheng, Z.; Wang, Z.; Zhang, L.; Jia, Z.; Sui, Y. Sesamin promotes apoptosis and pyroptosis via autophagy to enhance antitumour effects on murine T-cell lymphoma. J. Pharmacol. Sci. 2021, 147, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Basiorka, A.A.; McGraw, K.L.; Eksioglu, E.A.; Chen, X.; Johnson, J.; Zhang, L.; Zhang, Q.; Irvine, B.A.; Cluzeau, T.; Sallman, D.A.; et al. The NLRP3 inflammasome functions as a driver of the myelodysplastic syndrome phenotype. Blood 2016, 128, 2960–2975. [Google Scholar] [CrossRef]
- Graf, J.R.; Forster, S.; Bruehl, F.K.; Banz, Y.; Hallal, M.; Brodard, J.; Bacher, V.U.; Allam, R.; Schürch, C.M.; Bonadies, N. Diagnostic and Prognostic Implications of Caspase-1 and PD-L1 Co-Expression Patterns in Myelodysplastic Syndromes. Cancers 2021, 13, 5712. [Google Scholar] [CrossRef]
- Ward, G.A.; McGraw, K.L.; Abbas-Aghababazadeh, F.; Meyer, B.S.; McLemore, A.F.; Vincelette, N.D.; Lam, N.B.; Aldrich, A.L.; Al Ali, N.H.; Padron, E.; et al. Oxidized mitochondrial DNA released after inflammasome activation is a disease biomarker for myelodysplastic syndromes. Blood Adv. 2021, 5, 2216–2228. [Google Scholar] [CrossRef]
- Zhong, C.; Wang, R.; Hua, M.; Zhang, C.; Han, F.; Xu, M.; Yang, X.; Li, G.; Hu, X.; Sun, T.; et al. NLRP3 Inflammasome Promotes the Progression of Acute Myeloid Leukemia via IL-1β Pathway. Front. Immunol. 2021, 12, 661939. [Google Scholar] [CrossRef]
- Jia, Y.; Zhang, C.; Hua, M.; Wang, M.; Chen, P.; Ma, D. Aberrant NLRP3 inflammasome associated with aryl hydrocarbon receptor potentially contributes to the imbalance of T-helper cells in patients with acute myeloid leukemia. Oncol. Lett. 2017, 14, 7031–7044. [Google Scholar] [CrossRef]
- Liu, N.; Wu, Y.; Wen, X.; Li, P.; Lu, F.; Shang, H. Chronic stress promotes acute myeloid leukemia progression through HMGB1/NLRP3/IL-1β signaling pathway. J. Mol. Med. 2021, 99, 403–414. [Google Scholar] [CrossRef]
- Zhang, A.; Yu, J.; Yan, S.; Zhao, X.; Chen, C.; Zhou, Y.; Zhao, X.; Hua, M.; Wang, R.; Zhang, C.; et al. The genetic polymorphism and expression profiles of NLRP3 inflammasome in patients with chronic myeloid leukemia. Hum. Immunol. 2018, 79, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Paugh, S.W.; Bonten, E.J.; Savic, D.; Ramsey, L.B.; Thierfelder, W.E.; Gurung, P.; Malireddi, R.K.S.; Actis, M.; Mayasundari, A.; Min, J.; et al. NALP3 inflammasome upregulation and CASP1 cleavage of the glucocorticoid receptor cause glucocorticoid resistance in leukemia cells. Nat. Genet. 2015, 47, 607–614. [Google Scholar] [CrossRef]
- Zhang, C.; Han, F.; Yu, J.; Hu, X.; Hua, M.; Zhong, C.; Wang, R.; Zhao, X.; Shi, Y.; Ji, C.; et al. Investigation of NF-κB-94ins/del ATTG and CARD8 (rs2043211) Gene Polymorphism in Acute Lymphoblastic Leukemia. Front. Endocrinol. 2019, 10, 501. [Google Scholar] [CrossRef]
- Alves, F.S.; Xabregas, L.A.; Kerr, M.W.A.; Souza, G.L.; Pereira, D.S.; Magalhães-Gama, F.; Santiago, M.R.R.; Garcia, N.P.; Tarragô, A.M.; Ogusku, M.M.; et al. Genetic polymorphisms of inflammasome genes associated with pediatric acute lymphoblastic leukemia and clinical prognosis in the Brazilian Amazon. Sci. Rep. 2021, 11, 9869. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, N.; Yan, Z.; Li, H.; Chen, L.; Zhang, Z.; Fan, G.; Xu, K.; Li, Z. Dysregulation of the NLRP3 inflammasome complex and related cytokines in patients with multiple myeloma. Hematology 2016, 21, 144–151. [Google Scholar] [CrossRef]
- Hofbauer, D.; Mougiakakos, D.; Broggini, L.; Zaiss, M.; Büttner-Herold, M.; Bach, C.; Spriewald, B.; Neumann, F.; Bisht, S.; Nolting, J.; et al. β(2)-microglobulin triggers NLRP3 inflammasome activation in tumor-associated macrophages to promote multiple myeloma progression. Immunity 2021, 54, 1772–1787.e1779. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Hua, M.; Yan, S.; Yu, J.; Han, F.; Zhong, C.; Wang, R.; Zhang, C.; Hou, M.; Ma, D. The Genetic Polymorphisms of NLRP3 Inflammasome Associated with T Helper Cells in Patients with Multiple Myeloma. J. Immunol. Res. 2018, 2018, 7569809. [Google Scholar] [CrossRef]
- Nardelli, B.; Belvedere, O.; Roschke, V.; Moore, P.A.; Olsen, H.S.; Migone, T.S.; Sosnovtseva, S.; Carrell, J.A.; Feng, P.; Giri, J.G.; et al. Synthesis and release of B-lymphocyte stimulator from myeloid cells. Blood 2001, 97, 198–204. [Google Scholar] [CrossRef]
- Stergiou, I.E.; Poulaki, A.; Voulgarelis, M. Pathogenetic Mechanisms Implicated in Sjögren’s Syndrome Lymphomagenesis: A Review of the Literature. J. Clin. Med. 2020, 9, 3794. [Google Scholar] [CrossRef]
- Moossavi, M.; Parsamanesh, N.; Bahrami, A.; Atkin, S.L.; Sahebkar, A. Role of the NLRP3 inflammasome in cancer. Mol. Cancer 2018, 17, 158. [Google Scholar] [CrossRef]
- Bossaller, L.; Chiang, P.I.; Schmidt-Lauber, C.; Ganesan, S.; Kaiser, W.J.; Rathinam, V.A.; Mocarski, E.S.; Subramanian, D.; Green, D.R.; Silverman, N.; et al. Cutting edge: FAS (CD95) mediates noncanonical IL-1β and IL-18 maturation via caspase-8 in an RIP3-independent manner. J. Immunol. 2012, 189, 5508–5512. [Google Scholar] [CrossRef] [PubMed]
- Omoto, Y.; Tokime, K.; Yamanaka, K.; Habe, K.; Morioka, T.; Kurokawa, I.; Tsutsui, H.; Yamanishi, K.; Nakanishi, K.; Mizutani, H. Human mast cell chymase cleaves pro-IL-18 and generates a novel and biologically active IL-18 fragment. J. Immunol. 2006, 177, 8315–8319. [Google Scholar] [CrossRef] [PubMed]
- Akeda, T.; Yamanaka, K.; Tsuda, K.; Omoto, Y.; Gabazza, E.C.; Mizutani, H. CD8+ T cell granzyme B activates keratinocyte endogenous IL-18. Arch. Dermatol. Res. 2014, 306, 125–130. [Google Scholar] [CrossRef]
- Grondona, P.; Bucher, P.; Schulze-Osthoff, K.; Hailfinger, S.; Schmitt, A. NF-κB Activation in Lymphoid Malignancies: Genetics, Signaling, and Targeted Therapy. Biomedicines 2018, 6, 38. [Google Scholar] [CrossRef]
- Poulaki, A.; Giannouli, S. Metabolic Swifts Govern Normal and Malignant B Cell Lymphopoiesis. Int. J. Mol. Sci. 2021, 22, 8269. [Google Scholar] [CrossRef] [PubMed]
- Bauernfried, S.; Scherr, M.J.; Pichlmair, A.; Duderstadt, K.E.; Hornung, V. Human NLRP1 is a sensor for double-stranded RNA. Science 2021, 371, eabd0811. [Google Scholar] [CrossRef]
- Robinson, K.S.; Toh, G.A.; Rozario, P.; Chua, R.; Bauernfried, S.; Sun, Z.; Firdaus, M.J.; Bayat, S.; Nadkarni, R.; Poh, Z.S.; et al. ZAKα-driven ribotoxic stress response activates the human NLRP1 inflammasome. Science 2022, 377, 328–335. [Google Scholar] [CrossRef]
- Broz, P.; von Moltke, J.; Jones, J.W.; Vance, R.E.; Monack, D.M. Differential requirement for Caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe 2010, 8, 471–483. [Google Scholar] [CrossRef]
- Lee, S.; Karki, R.; Wang, Y.; Nguyen, L.N.; Kalathur, R.C.; Kanneganti, T.D. AIM2 forms a complex with pyrin and ZBP1 to drive PANoptosis and host defence. Nature 2021, 597, 415–419. [Google Scholar] [CrossRef]
- Bostan, E.; Gokoz, O.; Atakan, N. The role of NLRP1 and NLRP3 inflammasomes in the etiopathogeneses of pityriasis lichenoides chronica and mycosis fungoides: An immunohistochemical study. Arch. Dermatol. Res. 2023, 315, 231–239. [Google Scholar] [CrossRef]
- Lin, S.-C.; Lo, Y.-C.; Wu, H. Helical assembly in the MyD88-IRAK4-RAK2 complex in TLR/IL-1R signalling. Nature 2010, 465, 885–890. [Google Scholar] [CrossRef]
- Parrondo, R.D.; Iqbal, M.; Von Roemeling, R.; Von Roemeling, C.; Tun, H.W. IRAK-4 inhibition: Emavusertib for the treatment of lymphoid and myeloid malignancies. Front. Immunol. 2023, 14, 1239082. [Google Scholar] [CrossRef]
- Yu, L.; Li, L.; Medeiros, L.J.; Young, K.H. NF-κB signaling pathway and its potential as a target for therapy in lymphoid neoplasms. Blood Rev. 2017, 31, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Helal, K.F.; Badr, M.S.; Rafeek, M.E.; Elnagar, W.M.; Lashin, M.E. Can glyburide be advocated over subcutaneous insulin for perinatal outcomes of women with gestational diabetes? A systematic review and meta-analysis. Arch. Gynecol. Obs. 2020, 301, 19–32. [Google Scholar] [CrossRef]
- Adinolfi, E.; Raffaghello, L.; Giuliani, A.L.; Cavazzini, L.; Capece, M.; Chiozzi, P.; Bianchi, G.; Kroemer, G.; Pistoia, V.; Di Virgilio, F. Expression of P2X7 receptor increases in vivo tumor growth. Cancer Res. 2012, 72, 2957–2969. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Guo, Q.; Zhu, Q.; Tan, R.; Bai, D.; Bu, X.; Lin, B.; Zhao, K.; Pan, C.; Chen, H.; et al. Flavonoid VI-16 protects against DSS-induced colitis by inhibiting Txnip-dependent NLRP3 inflammasome activation in macrophages via reducing oxidative stress. Mucosal Immunol. 2019, 12, 1150–1163. [Google Scholar] [CrossRef] [PubMed]
- Guarda, G.; Braun, M.; Staehli, F.; Tardivel, A.; Mattmann, C.; Förster, I.; Farlik, M.; Decker, T.; Du Pasquier, R.A.; Romero, P.; et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 2011, 34, 213–223. [Google Scholar] [CrossRef]
- Zhang, L.; Tai, Y.T.; Ho, M.Z.G.; Qiu, L.; Anderson, K.C. Interferon-alpha-based immunotherapies in the treatment of B cell-derived hematologic neoplasms in today’s treat-to-target era. Exp. Hematol. Oncol. 2017, 6, 20. [Google Scholar] [CrossRef]
- Olsen, E.A. Interferon in the treatment of cutaneous T-cell lymphoma. Dermatol. Ther. 2003, 16, 311–321. [Google Scholar] [CrossRef]
- Liu, X.; Pichulik, T.; Wolz, O.O.; Dang, T.M.; Stutz, A.; Dillen, C.; Delmiro Garcia, M.; Kraus, H.; Dickhöfer, S.; Daiber, E.; et al. Human NACHT, LRR, and PYD domain-containing protein 3 (NLRP3) inflammasome activity is regulated by and potentially targetable through Bruton tyrosine kinase. J. Allergy Clin. Immunol. 2017, 140, 1054–1067.e1010. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Guo, H.; Yang, J.; Liu, Y.; Liu, X.; Zhang, Q.; Zhou, K. Bruton tyrosine kinase inhibitors in B-cell lymphoma: Beyond the antitumour effect. Exp. Hematol. Oncol. 2022, 11, 60. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.-L.; Yin, H.-R.; He, Q.-Y.; Wang, Y. Targeting the NLRP3 inflammasome as new therapeutic avenue for inflammatory bowel disease. Biomed. Pharmacother. 2021, 138, 111442. [Google Scholar] [CrossRef] [PubMed]
- Shim, D.W.; Shin, W.Y.; Yu, S.H.; Kim, B.H.; Ye, S.K.; Koppula, S.; Won, H.S.; Kang, T.B.; Lee, K.H. BOT-4-one attenuates NLRP3 inflammasome activation: NLRP3 alkylation leading to the regulation of its ATPase activity and ubiquitination. Sci. Rep. 2017, 7, 15020. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.H.; Min, Y.S.; Choi, J.S.; Baeg, G.H.; Kim, Y.S.; Shin, J.W.; Kim, T.Y.; Ye, S.K. Benzoxathiol derivative BOT-4-one suppresses L540 lymphoma cell survival and proliferation via inhibition of JAK3/STAT3 signaling. Exp. Mol. Med. 2011, 43, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Lust, J.A.; Lacy, M.Q.; Zeldenrust, S.R.; Witzig, T.E.; Moon-Tasson, L.L.; Dinarello, C.A.; Donovan, K.A. Reduction in C-reactive protein indicates successful targeting of the IL-1/IL-6 axis resulting in improved survival in early stage multiple myeloma. Am. J. Hematol. 2016, 91, 571–574. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lymphoma Subtype | Type of Sample Studied | Effect of NLRP3 Inflammasome or Related Molecules | Studied NLRP3 Inflammasome Components or Related Molecules | Proposed Mechanism | Findings | Possible Clinical Relevance | Ref. |
|---|---|---|---|---|---|---|---|
| DLBCL | Pfeiffer cell line | Tumor-promoting | IL-18 | NLRP3 inflammasome activation induces:
| NLRP3 inflammasome activation inhibited dexamethasone-induced apoptosis via shifting the balance of BCL-2/BAX expression | Resistance to dexamethasone | [83] |
| DLBCL | 73 patients: serum samples | Tumor-promoting | IL-18 | IL-18-mediated pro-tumorigenic effect | Higher mean IL-18 levels correlated with lower CR (p = 0.465) and 3-year DFS rates (p = 0.127) after R-CHOP Lower mean IL-18 levels correlated with more favorable OS rates (p = 0.008) | Prognostic value for survival and treatment response | [85] |
| DLBCL | 35 patients and 35 healthy controls: lymphoid tissue samples | Tumor-promoting | IL-18, PD-L1 | IL-18-mediated pro-tumorigenic effect IL-18-mediated immune exhaustion | Higher IL-18 expression in DLBCL tissues compared to normal lymphoid tissues Higher IL-18 expression in DLBCLs with non-GCB phenotype compared to those with GCB phenotype Higher PD-L1 expression in DLBCL tissues compared to normal lymphoid tissues | Association with more aggressive disease Association with immune exhaustion—possible application for ICB therapy | [86] |
| DLBCL | Cell lines, murine model | Tumor-promoting | NLRP3, PD-L1, pSTAT3 | NLRP3 induced IL-18 secretion drives PD-L1 upregulation via pSTAT3 | NLRP3 inflammasome activation led to PD-L1 upregulation and T-cell decrease NLRP3 inflammasome blockade suppressed lymphoma growth and ameliorated antitumor immunity by downregulating PD-L1 in the TME and decreasing the proportion of immunosuppressive immune cells pSTAT3 protein levels decreased after IL-18 and NLPR3 inflammasome inhibition | Association with immune exhaustion—possible application for ICB therapy | [86] |
| DLBCL | GEO and TCGA gene expression datasets for DLBCL and normal B cells | Tumor-promoting | PRGs | Upregulated: PYCARD, IRF1, GZMA, GSDMD, GSDMC, GPX4, CHMP2A, CASP5, CASP1, IL-18 Amplified CNV status: BAK1 Three distinct clusters identified and associated with prognosis | Risk stratification | [102] | |
| DLBCL | TCGA dataset: dataset including 48 tissue samples of DLBCL patient | Tumor-promoting | Multiple inflammasome components | NLRP3 inflammasome upregulation in macrophages drives a proinflammatory TME that favors lymphomagenesis | Significant upregulation in AIM2, ALK, IRF3, 4 and 8, NFKB1 and 2, NOD2, NLRP1 and 3, CASP1 and 5, CARD8 and 9 in M0 and M1 macrophages | [100] | |
| Aggressive B-cell NHL | 46 patients and 20 healthy controls: serum samples | Inconclusive | IL-18 | No significant difference in IL-18 serum levels between patients and controls (p = 0.261) Νο difference in IL-18 serum levels before and after chemotherapy Higher IL-18 serum levels were higher in patients with higher LDH compared to those with normal LDH (p = 0.045) | Possible association with high-risk lymphoma features | [88] | |
| B-cell NHL | 281 patients with B-cell NHL (BM samples) and 385 healthy controls (PB samples) | Tumor-promoting | IL-1β, IL-18, NF-κΒ, CARD8 | NF-κΒ mediated NLRP3 inflammasome activation IL-18-mediated pro-tumorigenic effect | IL-18 (rs1946518) and NF-κB-94 ins/del (rs28362491) gene polymorphisms contributed to increased risk for B-cell NHL (p < 0.0001 and p = 0.0029, respectively) The AA genotype of the CARD8 rs2043211 polymorphism correlated with poorer survival (p = 0.0381) Τhe TT genotype of CARD8 (rs2043211) was observed in patients with higher LDH levels, clinical stages III-IV, and IPI scores 3–5 | Biomarkers for lymphoma susceptibility Possible association with high-risk lymphoma features | [101] |
| B-cell NHL | Primary B cells and lymphoma cell lines | Not investigated Possibly tumor-promoting | NLRP3, caspase-1, IL-1β, cIAP1-TRAF2, Src | BAFF/BAFF-R axis induces cIAP1-TRAF2-mediated NLRP3 priming signals and Src activity-dependent ROS mediated NLRP3 inflammasome activation signals | Treatment with BAFF led to an increase in NLRP3 mRNA expression but also in increased NLRP3 inflammasome activation (as proven by the increase in active caspase-1 and IL-1β levels) | [113] | |
| NHL | 27 patients: serum samples | Tumor-promoting | IL-18 | IL-18-mediated pro-tumorigenic effect | Patients with IL-18 serum levels >2000 pg/mL compared to those with IL-18 serum levels <1000 pg/mL showed lower CR rates (33.3% versus 85.7%) and lower median OS (3.5 months versus 45.5 months) after CHOP chemotherapy | Prognostic value for survival and treatment response | [84] |
| MALT lymphoma | 45 SS patients and 25 sicca controls: salivary gland tissue specimens | Tumor-promoting | P2X7R, NLRP3, caspase-1, IL-18, IL-1β | P2X7R-mediated NLRP3 inflammasome activation IL-18-mediated pro-tumorigenic effect | Higher mRNA expression for P2X7R, NLRP3, caspase-1, IL-18, and IL-1β in patients with SS who developed MALT-NHL over the follow-up More pronounced P2X7R protein expression in SS patients developing MALT-NHL Three-fold higher glandular expression of IL-18 in MALT-NHL than in controls or the other patients with SS. | Biomarker for lymphoma development in SS | [89] |
| MALT lymphoma | 76 SS patients, 11 non-SS disease controls, and 30 healthy controls: salivary gland tissue specimens and serum samples | Tumor-promoting | NLRP3, caspase-1, IL-18, IL-1β, ASC | Cell-free and extranuclear DNA-mediated NLRP3 inflammasome activation IL-18-mediated pro-tumorigenic effect | Systemic activation of the NLRP3 inflammasome and significantly increased serum IL-18 and ASC levels in SS patients at high risk for lymphoma development and those with established lymphoma | Biomarker for lymphoma development in SS | [118] |
| MCL | 7 lymphoid tissue samples from patients | Tumor-promoting | IL-18 | IL-18-mediated pro-tumorigenic effect | IL-18 gene upregulation | Further MCL subtyping | [90] |
| PEL | BJAB cells, BCBL-1 PEL cells | Not investigated Possibly tumor-promoting | Caspase-1, IL-1β, IL-18 | Activation of caspase-1 and cleavage of pro-IL-1β and pro-IL-18 detected in lymphoma cells induced by KSHV | [91] | ||
| CLL/SLL | 23 patients: B cells from PB samples | Tumor-suppressive | P2X7R, ASC, NLRP3 | P2X7R-mediated NLRP3 inflammasome activation | NLRP3 overexpression correlated with inhibition of cell proliferation and induction of apoptosis NLRP3 downregulation contributed to lymphomagenesis | Biomarker for disease aggressiveness or progression | [92] |
| Burkitt lymphoma | HH514-16 and CLIX-FZ cell lines | Tumor-suppressive | NLRP3, HMGB1 | HMGB1 protein sustains ZEBRA expression via the NLRP3 inflammasome | EBV lytic phase Burkitt lymphoma cells express high levels of HMGB1 HMGB1 regulates NLRP3 inflammasome-mediated ZEBRA expression | [93] | |
| CTCL | 95 patients: plasma samples 20 patients: skin tissues | Tumor-promoting | IL-18, caspase-1 | Increased IL-18 level can favor skewing of CD4+ T cells to the characteristic Th2 phenotype | Increased IL-18 and caspase-1 plasma levels in patients compared to healthy controls Increased IL-18 and caspase-1 mRNA levels in skin lesions from patients compared to healthy skin | Biomarker | [120] |
| CTCL | 53 patients, 12 healthy controls, 10 patients with psoriasis, 12 patients with atopic dermatitis: skin tissues | Tumor-promoting | NLRP3, IL-4 | NLRP3 translocation to the nucleus of the malignant CD4+ T cells, where it binds to the human IL-4 promoter in duces IL-4 production, which promotes the characteristic Th2 phenotype | IL-4 production mediated by NLRP3 increased with lesion severity and associated with disease progression | Association with disease severity and progression | [124] |
| Sézary syndrome | 28 patients, 19 erythroderma patients, and 40 healthy donors: skin tissues, PB samples, serum samples, lymphoid tissue samples | Tumor-promoting (dependent on the affected tissue) | NLRP3, IL-1β, IL-18, AIM2, NLRP1 | IL-18-mediated pro-tumorigenic effect—though its production might not be exclusively mediated by NLRP3 inflammasome | Increased IL-1β and low IL-18 levels in the epidermal skin layer of patients Increased IL-18 expression and no difference in IL-1β in the dermal skin layers of patients compared to controls Equal NLRP3 and AIM2 expression in the skin among the different groups Increased NLRP1 expression in the skin of patients Increased IL-18 serum levels in patients Upregulation in IL-18 and downregulation in IL-1β in the LNs of patients with advanced-stage disease | Diagnostic biomarker Association with disease severity | [127] |
| NK/T-cell lymphoma | 114 patients: serum samples | Tumor-promoting | IL-18 | IL-18-mediated pro-tumorigenic effect | High IL-18 serum levels associated with stage III/IV disease, presence of hemophagocytosis, and poor treatment outcomes. OS and PFS were significantly lower for the high IL-18 group compared to the low IL-18 groups (p < 0.001) High IL-18 serum levels were independently prognostic for survival in multivariate analysis | Biomarker of hemophagocytosis Prognostic biomarker | [128] |
| NK/T-cell lymphoma | 3 NK/T-cell lymphomas, 7 infectious mononucleosis cases, 6 chronic active EBV infection cases: lymphoid tissue miRNA expression datasets | Not investigated | hsa-miR-372-5p | Hsa-miR-372-5p may target NIMA-related kinase 7 to regulate NLRP3 inflammasome activation | Hsa-miR-372-5p regulates the NIMA-related kinase 7 and IL-1β release | Biomarker for EBV-associated disease | [129] |
| Various lymphoma subtypes (B-cell NHL, T-cell lymphoma, HL) | Lymphoma tissues: 68 patients (46 newly diagnosed, 22 treated) and 40 controls Plasma samples: 35 lymphoma patients and 15 controls | Tumor-promoting | IL-18 | IL-18-mediated pro-tumorigenic effect | Higher IL-18 mRNA (p = 0.0288) and protein levels (p < 0.0001) in tissues of newly diagnosed lymphoma patients compared to controls Decrease in IL-18 mRNA (p = 0.0366) and protein levels (p = 0.0098) in tissues of patients with remission after chemotherapy Elevated plasma IL-18 protein levels in newly diagnosed lymphoma patients compared to controls Decreased plasma IL-18 protein levels after chemotherapy remission (p = 0.0098) | Diagnostic and prognostic biomarker | [83] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stergiou, I.E.; Tsironis, C.; Papadakos, S.P.; Tsitsilonis, O.E.; Dimopoulos, M.A.; Theocharis, S. Unraveling the Role of the NLRP3 Inflammasome in Lymphoma: Implications in Pathogenesis and Therapeutic Strategies. Int. J. Mol. Sci. 2024, 25, 2369. https://doi.org/10.3390/ijms25042369
Stergiou IE, Tsironis C, Papadakos SP, Tsitsilonis OE, Dimopoulos MA, Theocharis S. Unraveling the Role of the NLRP3 Inflammasome in Lymphoma: Implications in Pathogenesis and Therapeutic Strategies. International Journal of Molecular Sciences. 2024; 25(4):2369. https://doi.org/10.3390/ijms25042369
Chicago/Turabian StyleStergiou, Ioanna E., Christos Tsironis, Stavros P. Papadakos, Ourania E. Tsitsilonis, Meletios Athanasios Dimopoulos, and Stamatios Theocharis. 2024. "Unraveling the Role of the NLRP3 Inflammasome in Lymphoma: Implications in Pathogenesis and Therapeutic Strategies" International Journal of Molecular Sciences 25, no. 4: 2369. https://doi.org/10.3390/ijms25042369