
Drought Stress Induced Different Response Mechanisms in Three Dendrobium Species under Different Photosynthetic Pathways
 ,
,
Abstract
:1. Introduction
2. Results
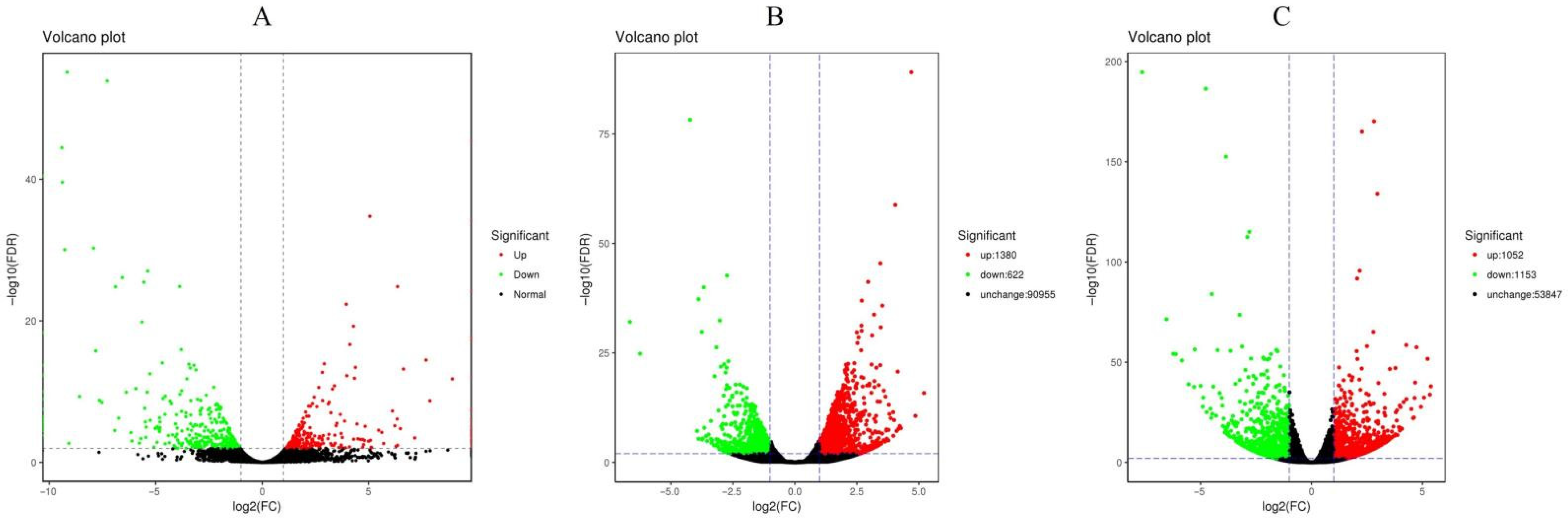
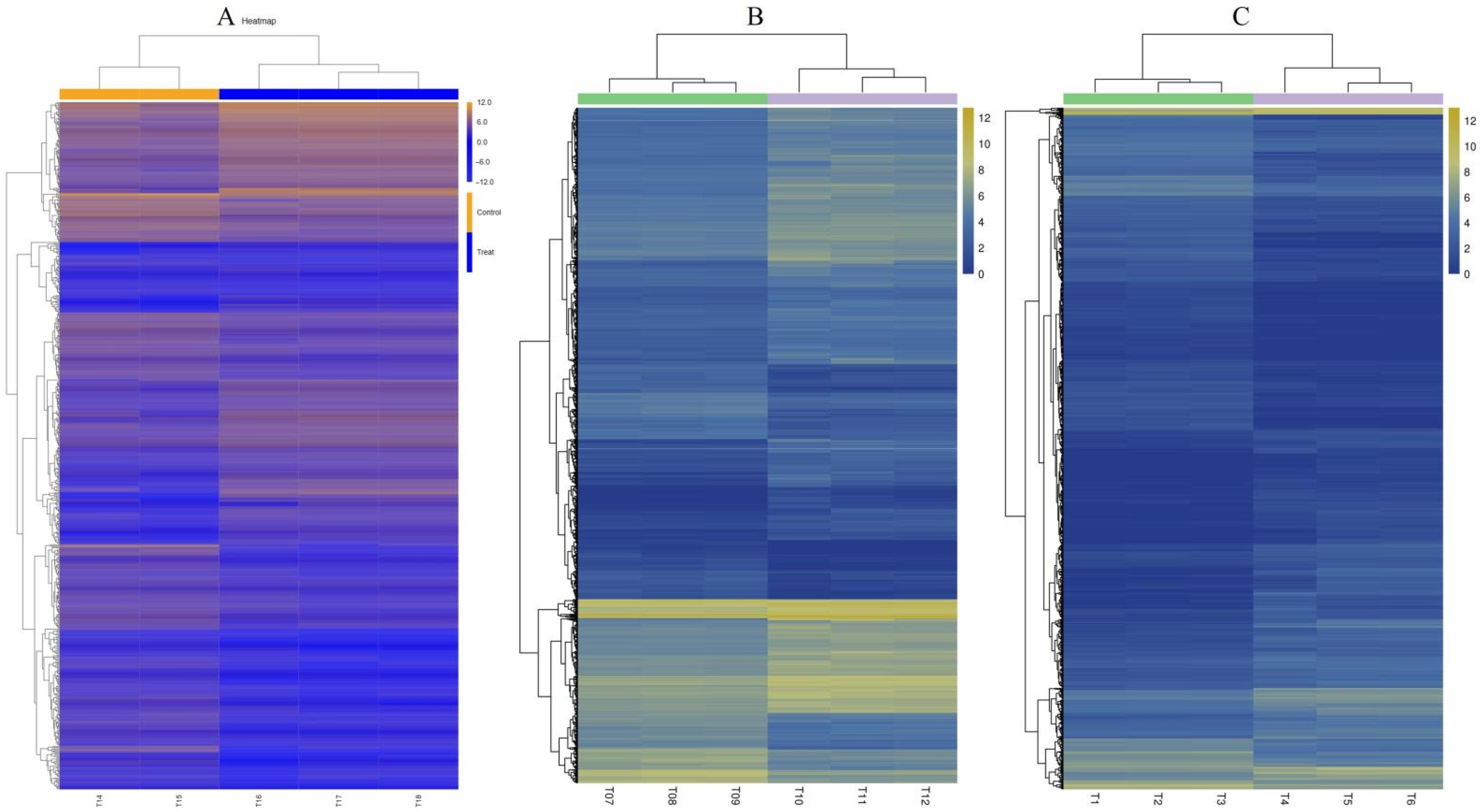
2.1. RNA-Seq Analysis under Drought Stress
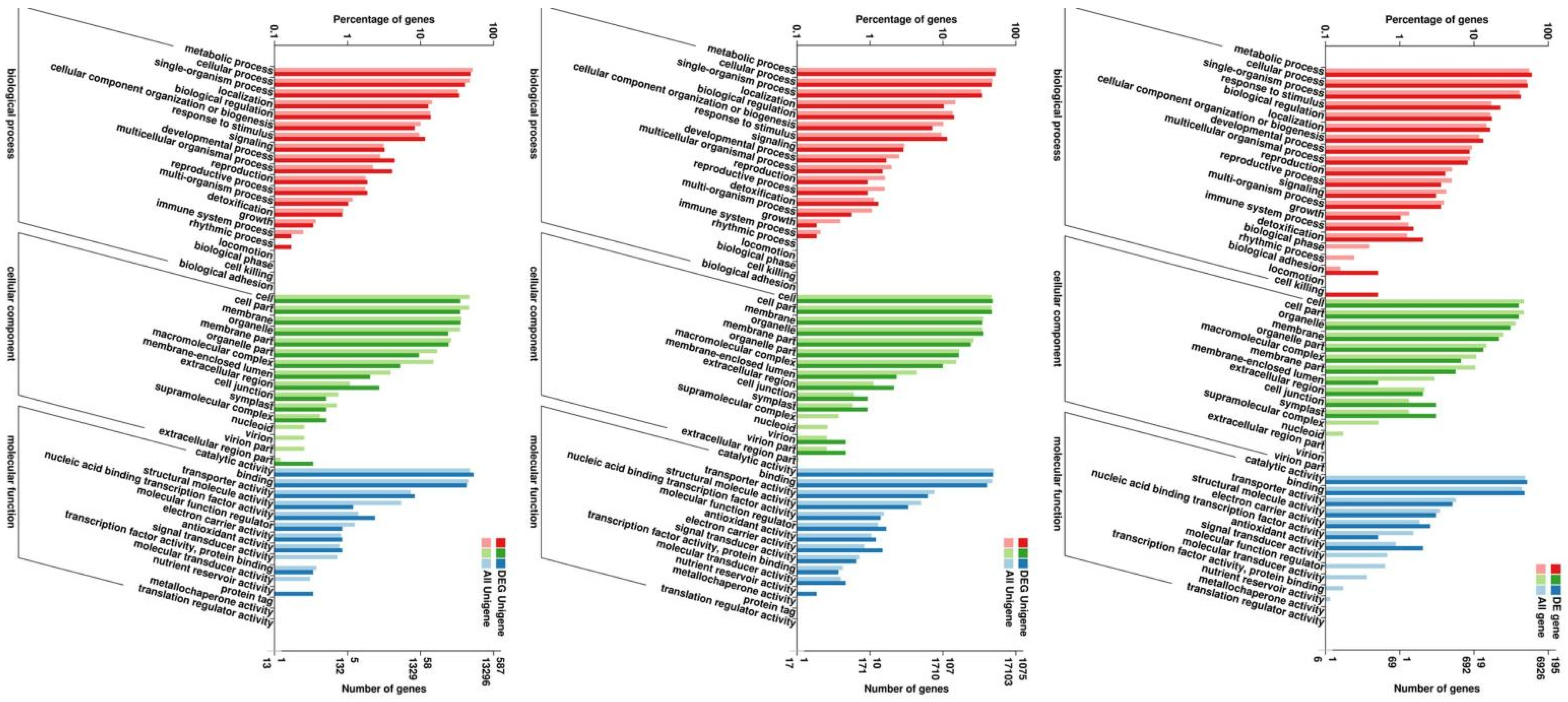
2.2. Gene Annotation and DEG Identification
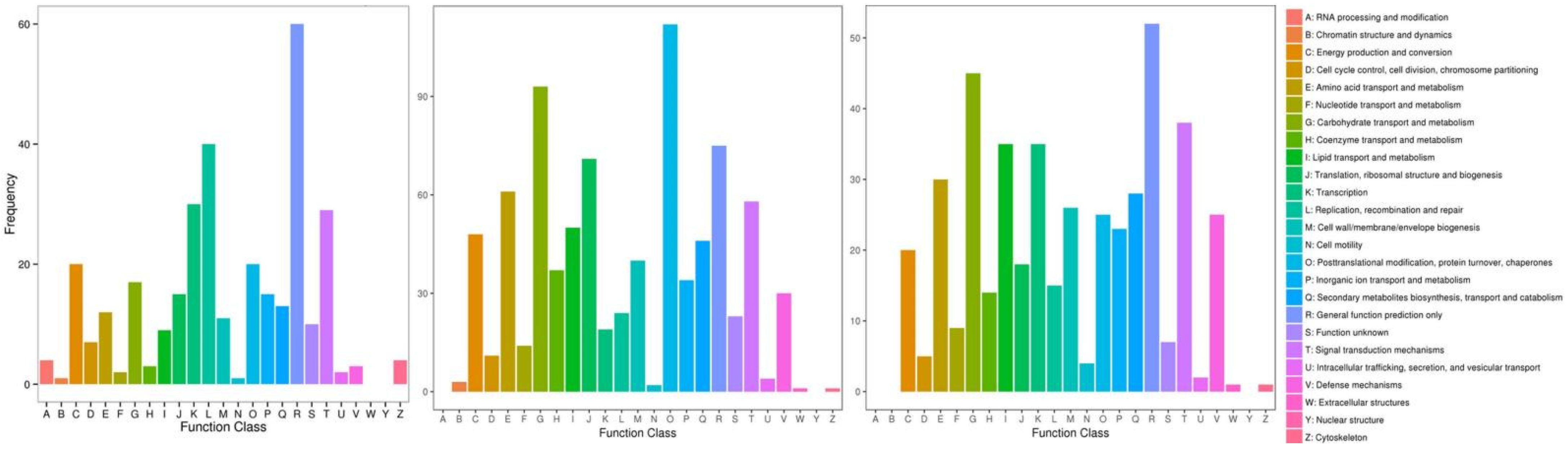
2.3. Clustering and Functional Categorization of DEGs
2.4. Multiple Sequence Alignment of the Three Dendrobium PEPCs
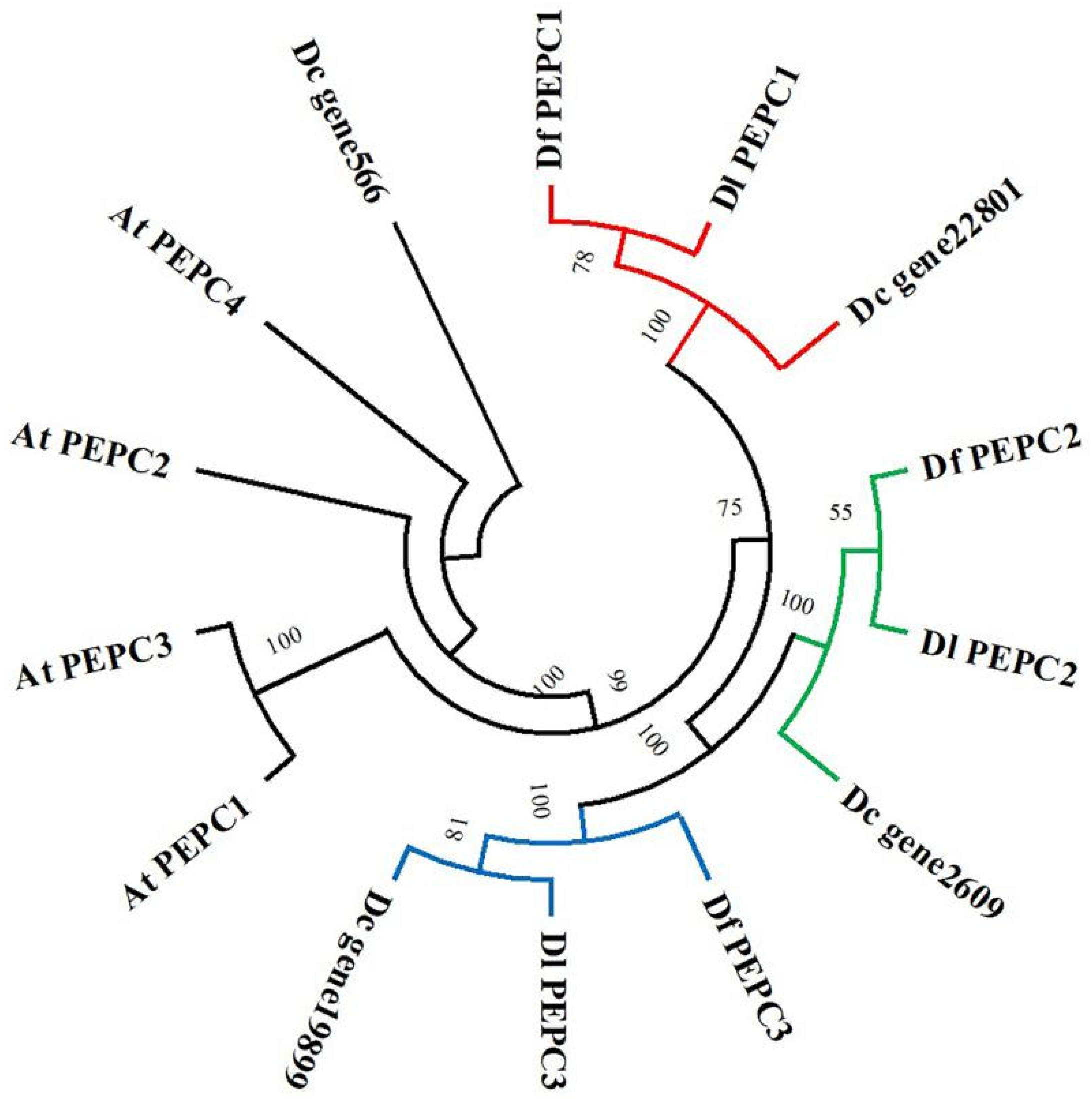
2.5. Structure and Phylogenetic Analysis of the PEPC Proteins
2.6. Analysis of miRNA Targets in the PEPC Gene of Dendrobium
2.7. Validation of the DEGs Expression
3. Discussion
4. Materials and Methods
4.1. Plant Material and Treatments
4.2. RNA Extraction, Library Construction, and Sequencing
4.3. Quality Control and Transcriptome Assembly
4.4. Quantification of Gene Expression and Differential Expression Analysis
4.5. Analyses of GO and KEGG Pathways
4.6. Phylogenetic Tree Construction, Sequence, and Structural Domain Alignment
4.7. MiRNA Target Site Analysis
4.8. Real-Time PCR Analysis of PEPC
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, Z.; Liu, C.; Zhang, Y.; Wang, B.; Ran, Q.; Zhang, J. The bHLH family member ZmPTF1 regulates drought tolerance in maize by promoting root development and abscisic acid synthesis. J. Exp. Bot. 2019, 70, 5471–5486. [Google Scholar] [CrossRef]
- Zhang, C.; Shi, S. Physiological and proteomic responses of contrasting alfalfa (Medicago sativa L.) varieties to PEG-induced osmotic stress. Front. Plant Sci. 2018, 9, 242. [Google Scholar] [CrossRef] [PubMed]
- Opitz, N.; Marcon, C.; Paschold, A.; Malik, W.A.; Lithio, A.; Brandt, R.; Piepho, H.P.; Nettleton, D.; Hochholdinger, F. Extensive tissue-specific transcriptomic plasticity in maize primary roots upon water deficit. J. Exp. Bot. 2016, 67, 1095–1107. [Google Scholar] [CrossRef]
- Tripathi, P.; Rabara, R.C.; Shen, Q.J.; Rushton, P.J. Transcriptomics analyses of soybean leaf and root samples during water-deficit. Genom. Data 2015, 5, 164–166. [Google Scholar] [CrossRef] [PubMed]
- Schuppler, U.; He, P.H.; John, P.C.L.; Munns, R. Effect of water stress on cell division and cell-division-cycle 2-like cell-cycle kinase activity in wheat leaves. Plant Physiol. 1998, 117, 667–678. [Google Scholar] [CrossRef]
- Gupta, A.; Rico-Medina, A.; Caño-Delgado, A.I. The physiology of plant responses to drought. Science 2020, 368, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.D.; Xue, J.Q.; Guo, D.W. Efficacy of planting date adjustment as a cultivation strategy to cope with drought stress and increase rainfed maize yield and water-use efficiency. Agric. Water Manag. 2017, 179, 227–235. [Google Scholar] [CrossRef]
- Wu, X.; Yuan, J.; Luo, A.; Chen, Y.; Fan, Y. Drought stress and re-watering increase secondary metabolites and enzyme activity in Dendrobium moniliforme. Ind. Crops Prod. 2016, 94, 385–393. [Google Scholar] [CrossRef]
- Mahajan, S.; Tuteja, N. Cold, salinity and drought stresses: An overview. Arch. Biochem. Biophys. 2005, 444, 139–158. [Google Scholar] [CrossRef]
- Bota, J.; Medrano, H.; Flexas, J. Is photosynthesis limited by decreased RuBisCO activity and RuBP content under progressive water stress? New Phytol. 2004, 162, 671–681. [Google Scholar] [CrossRef]
- Hoekstra, F.A.; Golovina, E.A.; Buitink, J. Mechanisms of plant desiccation tolerance. Trends Plant Sci. 2001, 6, 431–438. [Google Scholar] [CrossRef]
- Chaves, M.M.; Maroco, J.P.; Pereira, J.S. Understanding plant responses to drought—From genes to the whole plant. Funct. Plant Biol. 2003, 30, 239–264. [Google Scholar] [CrossRef]
- Zhu, J.K. Abiotic stress signaling and responses in plants. Cell 2016, 167, 313–324. [Google Scholar] [CrossRef]
- Khanna-Chopra, R.; Singh, K. Drought Resistance in Crops: Physiological and Genetic Basis of Traits for Crop Productivity Stress Responses in Plants; Tripathi, B.N., Müller, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2015; pp. 267–292. [Google Scholar]
- Qiu, S.; Xia, K.; Yang, Y.; Wu, Q.; Zhao, Z. Mechanisms underlying the C3–CAM photosynthetic shift in facultative CAM plants. Horticulturae 2023, 9, 398. [Google Scholar] [CrossRef]
- He, J.; Chua, E.L.; Qin, L. Drought does not induce crassulacean acid metabolism (CAM) but regulates photosynthesis and enhances nutritional quality of Mesembryanthemum crystallinum. PLoS ONE 2020, 15, e0229897. [Google Scholar] [CrossRef]
- Ahmad, P. Water Stress and Crop Plants: A Sustainable Approach; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2016; Volume 2. [Google Scholar] [CrossRef]
- Huang, H.; Wang, H.; Tong, Y.; Wang, Y. Insights into the superoxide dismutase gene family and its roles in Dendrobium catenatum under abiotic stresses. Plants 2020, 9, 1452. [Google Scholar] [CrossRef] [PubMed]
- Lim, G.H.; Kim, S.W.; Ryu, J.; Kang, S.Y.; Kim, J.B.; Kim, S.H. Upregulation of the MYB2 transcription factor Is associated with increased accumulation of anthocyanin in the leaves of Dendrobium bigibbum. Int. J. Mol. Sci. 2020, 21, 5653. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Jiang, S.; Tian, H.; Xiang, D.; Zhang, J. Polyphenols in the fermentation liquid of Dendrobium candidum relieve intestinal inflammation in zebrafish through the intestinal microbiome-mediated immune response. Front. Immunol. 2020, 11, 1542. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.Z.; Shan, Y.Y.; Chen, X.M.; Zhang, C.F.; Li, Z.J. Current development situation of dendrobium industry in China. Mod. Chin. Med. 2022, 24, 1395–1402. [Google Scholar]
- Wang, S.S.; Liu, J.M.; Sun, J.; Sun, Y.F.; Liu, J.N.; Jia, N.; Fan, B.; Dai, X.F. Diversity of culture-independent bacteria and antimicrobial activity of culturable endophytic bacteria isolated from different Dendrobium stems. Sci. Rep. 2019, 9, 10389. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Y.Y.; Chen, X.M.; Guo, S.X.; Lee, Y.I. Effect of different mycobionts on symbiotic germination and seedling growth of Dendrobium officinale, an important medicinal orchid. Bot. Stud. 2020, 61, 2. [Google Scholar] [CrossRef]
- Yeow, L.C.; Chew, B.L.; Sreeramanan, S. Elevation of secondary metabolites production through light-emitting diodes (LEDs) illumination in protocorm-like bodies (PLBs) of Dendrobium hybrid orchid rich in phytochemicals with therapeutic effects. Biotechnol. Rep. 2020, 27, e00497. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.K.Y.; Hew, C.S. Orchid pseudobulbs—‘False’ bulbs with a genuine importance in orchid growth and survival! Sci. Hortic. 2000, 83, 165–172. [Google Scholar] [CrossRef]
- Zimmerman, J.K. Role of pseudobulbs in growth and flowering of Catasetum viridiflavum (Orchidaceae). Am. J. Bot. 1990, 77, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.B.; Dai, Y.; Hao, G.Y.; Li, J.W.; Fu, X.W.; Zhang, J.L. Differentiation of water-related traits in terrestrial and epiphytic Cymbidium species. Front. Plant Sci. 2015, 6, 260. [Google Scholar] [CrossRef]
- Cai, J.; Liu, X.; Vanneste, K.; Proost, S.; Tsai, W.C.; Liu, K.W.; Chen, L.J.; He, Y.; Xu, Q.; Bian, C.; et al. The genome sequence of the orchid Phalaenopsis equestris. Nat. Genet. 2015, 47, 65–72. [Google Scholar] [CrossRef]
- Zhang, G.Q.; Xu, Q.; Bian, C.; Tsai, W.C.; Yeh, C.M.; Liu, K.W.; Yoshida, K.; Zhang, L.S.; Chang, S.B.; Chen, F.; et al. The Dendrobium catenatum Lindl. Genome sequence provides insights into polysaccharide synthase, floral development and adaptive evolution. Sci. Rep. 2016, 6, 19029. [Google Scholar] [CrossRef]
- Zhang, Z.; He, D.; Niu, G.H.; Gao, R.F. Concomitant CAM and C3 photosynthetic pathways in Dendrobium officinale Plants. J. Am. Soc. Hortic. Sci. 2014, 139, 290–298. [Google Scholar] [CrossRef]
- Qiu, S.; Sultana, S.; Liu, Z.D.; Yin, L.Y.; Wang, C.Y. Identification of obligate C3 photosynthesis in Dendrobium. Photosynthetica 2015, 53, 168–176. [Google Scholar] [CrossRef]
- Davis, S.C.; Simpson, J.; Gil-Vega, K.D.C.; Niechayev, N.A.; Tongerlo, E.V.; Castano, N.H.; Dever, L.V.; Búrquez, A. Undervalued potential of crassulacean acid metabolism for current and future agricultural production. J. Exp. Bot. 2019, 70, 6521–6537. [Google Scholar] [CrossRef] [PubMed]
- Bayramov, S. Changes in protein quantities of phosphoenolpyruvate carboxylase and RuBisCO Activase in various wheat genotypes. Saudi J. Biol. Sci. 2017, 24, 1529–1533. [Google Scholar] [CrossRef]
- Ping, C.Y.; Chen, F.C.; Cheng, T.C.; Lin, H.L.; Lin, T.S.; Yang, W.J.; Lee, Y.I. Expression profiles of phosphoenolpyruvate carboxylase and phosphoenolpyruvate carboxylase kinase genes in Phalaenopsis, implications for regulating the performance of crassulacean acid metabolism. Front. Plant Sci. 2018, 9, 1587. [Google Scholar] [CrossRef] [PubMed]
- Punchkhon, C.; Plaimas, K.; Buaboocha, T.; Siangliw, J.L.; Toojinda, T.; Comai, L.; De Diego, N.; Spíchal, L.; Chadchawan, S.; Gene, D.-T. Drought-tolerance gene identification using genome comparison and co-expression network analysis of chromosome substitution lines in rice. Genes 2020, 11, 1197. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Wang, X.; Liu, H.; Tian, Y.L.; Lian, J.M.; Yang, R.J.; Hao, S.M.; Wang, X.; Yang, S.; Li, Q.; et al. The genome of Dendrobium officinale illuminates the biology of the important traditional Chinese orchid herb. Mol. Plant 2015, 8, 922–934. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.Q.; Liu, K.W.; Li, Z.; Lohaus, R.; Hsiao, Y.Y.; Niu, S.C.; Wang, J.Y.; Lin, Y.C.; Xu, Q.; Chen, L.J.; et al. The Apostasia genome and the evolution of orchids. Nature 2017, 549, 379–383. [Google Scholar] [CrossRef]
- Zhang, J.X.; He, C.M.; Wu, K.L.; Teixeira da Silva, J.A.; Zeng, S.J.; Zhang, X.H.; Yu, Z.M.; Xia, H.Q.; Duan, J. Transcriptome analysis of Dendrobium officinale and its application to the identification of genes associated with polysaccharide synthesis. Front. Plant Sci. 2016, 7, 5. [Google Scholar] [CrossRef] [PubMed]
- Winter, K.; Wallace, B.J.; Stocker, G.C.; Roksandic, Z. Crassulacean acid metabolism in Australian vascular epiphytes and somerelated species. Oecologia 1983, 57, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Silvera, K.; Santiago, L.S.; Winter, K. Distribution ofcrassulacean acid metabolism in orchids of Panama:evidence of selection of weak and strong modes. Funct. Plant Biol. 2005, 32, 397–407. [Google Scholar] [CrossRef]
- Yuan, G.; Hassan, M.M.; Liu, D.; Lim, S.D.; Yim, W.C.; Cushman, J.C.; Markel, K.; Shih, P.M.; Lu, H.; Weston, D.J.; et al. Biosystems design to accelerate C3-to-CAM progression. BioDesign Res. 2020, 2020, 3686791. [Google Scholar] [CrossRef]
- Li, Q.; Liu, C.; Huang, C.; Wang, M.; Long, T.; Liu, J.; Shi, J.; Shi, J.; Li, L.; He, Y.; et al. Transcriptome and metabonomics analysis revealed the molecular mechanism of differential metabolite production of Dendrobium nobile under different epiphytic patterns. Front. Plant Sci. 2022, 13, 868472. [Google Scholar] [CrossRef]
- Yuan, Y.; Zuo, J.; Zhang, H.; Li, R.; Yu, M.; Liu, S. Integration of transcriptome and metabolome provides new insights to flavonoids biosynthesis in Dendrobium huoshanense. Front. Plant Sci. 2022, 13, 850090. [Google Scholar] [CrossRef]
- Wan, X.; Zou, L.H.; Zheng, B.Q.; Tian, Y.Q.; Wang, Y. Transcriptomic profiling for prolonged drought in Dendrobium catenatum. Sci. Data 2018, 5, 180233. [Google Scholar] [CrossRef]
- Rabas, A.R.; Martin, C.E. Movement of water from old to young leaves in three species of succulents. Ann. Bot. 2003, 92, 529–536. [Google Scholar] [CrossRef]
- Borland, A.M.; Taybi, T. Synchronization of metabolic processes in plants with crassulacean acid metabolism. J. Exp. Bot. 2004, 55, 1255–1265. [Google Scholar] [CrossRef] [PubMed]
- Arias-Moreno, D.M.; Jiménez-Bremont, J.F.; Maruri-López, I.; Delgado-Sánchez, P. Effects of catalase on chloroplast arrangement in Opuntia streptacantha chlorenchyma cells under salt stress. Sci. Rep. 2017, 7, 8656. [Google Scholar] [CrossRef] [PubMed]
- Chi, Z.; Wang, Z.P.; Wang, G.Y.; Khan, I.; Chi, Z.M. Microbial biosynthesis and secretion of l-malic acid and its applications. Crit. Rev. Biotechnol. 2016, 36, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Müller, G.L.; Lara, M.V.; Oitaven, P.; Andreo, C.S.; Maurino, V.G.; Drincovich, M.F. Improved water use efficiency and shorter life cycle of Nicotiana tabacum due to modification of guard and vascular companion cells. Sci. Rep. 2018, 8, 4380. [Google Scholar] [CrossRef]
- Stevens, G.G.; Pérez-Fernández, M.A.; Morcillo, R.J.L.; Kleinert, A.; Hills, P.; Brand, D.J.; Steenkamp, E.T.; Valentine, A.J. Roots and nodules response differently to P starvation in the Mediterranean-type legume Virgilia divaricata. Front. Plant Sci. 2019, 10, 73. [Google Scholar] [CrossRef] [PubMed]
- Izui, K.; Matsumura, H.; Furumoto, T.; Kai, Y. Phosphoenolpyruvate carboxylase: A new era of structural biology. Annu. Rev. Plant Biol. 2004, 55, 69–84. [Google Scholar] [CrossRef]
- Doubnerová Hýsková, V.; Miedzińska, L.; Dobrá, J.; Vankova, R.; Ryšlavá, H. Phosphoenolpyruvate carboxylase, NADP-malic enzyme, and pyruvate, phosphate dikinase are involved in the acclimation of Nicotiana tabacum L. to drought stress. J. Plant Physiol. 2014, 171, 19–25. [Google Scholar] [CrossRef]
- Cheng, G.; Wang, L.; Lan, H. Cloning of PEPC-1 from a C4 halophyte Suaeda aralocaspica without Kranz anatomy and its recombinant enzymatic activity in responses to abiotic stresses. Enzyme Microb. Technol. 2016, 83, 57–67. [Google Scholar] [CrossRef]
- Helwak, A.; Kudla, G.; Dudnakova, T.; Tollervey, D. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell 2013, 153, 654–665. [Google Scholar] [CrossRef]
- Li, Y.; Wang, X.; Guo, Q.; Zhang, X.; Zhou, L.; Zhang, Y.; Zhang, C. Conservation and diversity of miR166 family members from highbush blueberry (Vaccinium corymbosum) and their potential functions in abiotic stress. Front. Genet. 2022, 13, 919856. [Google Scholar] [CrossRef]
- Vaasen, A.; Begerow, D.; Hampp, R. Phosphoenolpyruvate carboxylase genes in C3, crassulacean acid metabolism(CAM) and C3/CAM intermediate species of the genus Clusia: Rapid reversible C3/CAM switches are based on the C3 housekeeping gene. Plant Cell Environ. 2006, 29, 2113–2123. [Google Scholar] [CrossRef]
- Gehrig, H.; Faist, K.; Kluge, M. Identification of phosphoenolpyruvate carboxylase isoforms in leaf, stemand roots of the obligate CAM plant Vanilla planifolia Salib.(Orchidaceae): A physiological and molecular approach. Plant Mol. Biol. 1998, 38, 1215–1223. [Google Scholar] [CrossRef]
- Hoagland, D.R.; Snyder, W.C. Nutrition of strawberry plant under controlled conditions. (a) Effects of deficiencies of boron and certain other elements, (b) Susceptibility to injury from sodium salts. Proc. Am. Soc. Hortic. Sci. 1933, 30, 288–294. [Google Scholar]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.D.; et al. Full-length transcriptome assembly from RNA Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Bailey, T.L.; Elkan, C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994, 2, 28–36. [Google Scholar] [PubMed]
- Zhang, Y. miRU: An automated plant miRNA target prediction server. Nucleic Acids Res. 2005, 33, 701–704. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Species | Treatment |
|---|---|---|
| T1 | Dendrobium fimbriatum Hook | WW |
| T2 | Dendrobium fimbriatum Hook | WW |
| T3 | Dendrobium fimbriatum Hook | WW |
| T4 | Dendrobium fimbriatum Hook | DS |
| T5 | Dendrobium fimbriatum Hook | DS |
| T6 | Dendrobium fimbriatum Hook | DS |
| T07 | Dendrobium loddigesii Rolfe | WW |
| T08 | Dendrobium loddigesii Rolfe | WW |
| T09 | Dendrobium loddigesii Rolfe | WW |
| T10 | Dendrobium loddigesii Rolfe | DS |
| T11 | Dendrobium loddigesii Rolfe | DS |
| T12 | Dendrobium loddigesii Rolfe | DS |
| T13 | Dendrobium catenatum Lindley | WW |
| T14 | Dendrobium catenatum Lindley | WW |
| T15 | Dendrobium catenatum Lindley | WW |
| T16 | Dendrobium catenatum Lindley | DS |
| T17 | Dendrobium catenatum Lindley | DS |
| T18 | Dendrobium catenatum Lindley | DS |
| Sample | T13 | T14 | T15 | T16 | T17 | T18 |
|---|---|---|---|---|---|---|
| Clean Reads | 25,736,675 | 24,698,887 | 23,658,162 | 25,059,306 | 23,723,106 | 22,974,697 |
| Clean Bases | 7,704,250,842 | 7,387,541,456 | 7,080,339,178 | 7,503,039,742 | 7,100,687,882 | 6,874,766,972 |
| GC Content | 45.57% | 45.28% | 45.82% | 45.64% | 46.07% | 46.04% |
| ≥Q30 | 92.66% | 92.54% | 92.75% | 92.58% | 92.34% | 92.60% |
| Mapped Reads | 84.81% | 83.72% | 84.90% | 85.16% | 85.31% | 85.26% |
| Uniq Mapped Reads | 82.13% | 81.65% | 82.21% | 82.92% | 82.69% | 82.55% |
| Multiple Map Reads | 2.68% | 2.07% | 2.69% | 2.24% | 2.62% | 2.71% |
| Reads Map to “+” | 42.09% | 41.70% | 42.15% | 42.45% | 42.45% | 42.32% |
| Reads Map to “−” | 42.18% | 41.74% | 42.25% | 42.45% | 42.51% | 42.44% |
| Samples | Read Number | Base Number | GC Content | ≥Q30 |
|---|---|---|---|---|
| T1 | 25,222,133 | 7,549,261,904 | 45.97% | 92.48% |
| T2 | 24,720,125 | 7,393,655,992 | 45.87% | 92.03% |
| T3 | 24,779,891 | 7,403,415,432 | 45.63% | 92.58% |
| T4 | 24,867,538 | 7,444,820,532 | 45.97% | 92.49% |
| T5 | 26,247,245 | 7,850,055,066 | 45.85% | 92.34% |
| T6 | 27,132,721 | 8,116,884,478 | 45.65% | 92.27% |
| T07 | 25,019,702 | 7,491,393,560 | 45.56% | 92.44% |
| T08 | 26,684,679 | 7,987,805,304 | 45.53% | 92.48% |
| T09 | 24,734,656 | 7,404,300,212 | 45.25% | 92.69% |
| T10 | 24,058,684 | 7,200,859,834 | 45.81% | 92.61% |
| T11 | 23,864,004 | 7,139,039,344 | 45.14% | 92.50% |
| T12 | 26,046,625 | 7,792,409,468 | 45.36% | 92.40% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xia, K.; Wu, Q.; Yang, Y.; Liu, Q.; Wang, Z.; Zhao, Z.; Li, J.; He, J.; Chai, S.; Qiu, S. Drought Stress Induced Different Response Mechanisms in Three Dendrobium Species under Different Photosynthetic Pathways. Int. J. Mol. Sci. 2024, 25, 2731. https://doi.org/10.3390/ijms25052731
Xia K, Wu Q, Yang Y, Liu Q, Wang Z, Zhao Z, Li J, He J, Chai S, Qiu S. Drought Stress Induced Different Response Mechanisms in Three Dendrobium Species under Different Photosynthetic Pathways. International Journal of Molecular Sciences. 2024; 25(5):2731. https://doi.org/10.3390/ijms25052731
Chicago/Turabian StyleXia, Ke, Qiaofen Wu, Yanni Yang, Qiao Liu, Zaihua Wang, Zhiguo Zhao, Jie Li, Jinxiang He, Shengfeng Chai, and Shuo Qiu. 2024. "Drought Stress Induced Different Response Mechanisms in Three Dendrobium Species under Different Photosynthetic Pathways" International Journal of Molecular Sciences 25, no. 5: 2731. https://doi.org/10.3390/ijms25052731