
Bioinformatic Analysis of Sulfotransferases from an Unexplored Gut Microbe, Sutterella wadsworthensis 3_1_45B: Possible Roles towards Detoxification via Sulfonation by Members of the Human Gut Microbiome

Abstract
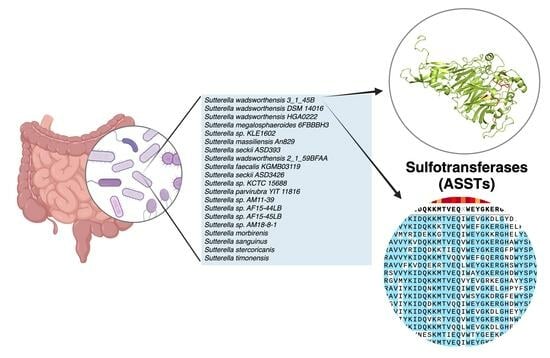
:
1. Introduction
2. Results and Discussion
2.1. Distribution of Aryl-Sulfate Sulfotransferase (asst) Genes in Human Gut Microbiota
2.2. Variability of Annotated Aryl-Sulfate Sulfotransferase (asst) Genes in the Genus Sutterella
2.3. Bioinformatic Analysis of Annotated asst Genes in S. wadsworthensis 3_1_45B
2.4. GC Content Variation in asst Genes
2.5. Analysis of Gene Neighborhoods for asst Genes in S. wadsworthensis 3_1_45B
2.6. Bioinformatic Analysis of Predicted ASST Proteins from S. wadsworthensis 3_1_45B
2.7. Analysis of Signal Peptides in SwASST Enzymes
2.8. Sequences Similarity Analysis of SwASSTs
2.9. Structural Variations in SwASST Proteins
2.10. Different Classes of Sulfotransferases from the Members of the Human Gut Microbiome and Their Comparison to Human Sulfotransferases
3. Materials and Methods
3.1. Members of the Human Gut Microbiome Harboring Annotated asst Genes
3.2. Annotated asst Genes from the Genus Sutterella
3.3. Properties of Predicted ASST Proteins from Sutterella wadsworthensis 3_1_45B
3.4. Analysis of Signal Peptides
3.5. Multiple Sequence Alignment
3.6. ASST Structure Predictions with AlphaFold2
3.7. ASST Structural Alignments with PyMOL
3.8. Search for PAPS-Dependent and PAPS-Independent Sulfotransferases in the Members of the Human Gut Microbiome
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Coughtrie, M.W.; Bamforth, K.J.; Sharp, S.; Jones, A.L.; Borthwick, E.B.; Barker, E.V.; Roberts, R.C.; Hume, R.; Burchell, A. Sulfation of endogenous compounds and xenobiotics—Interactions and function in health and disease. Chem. Biol. Interact. 1994, 92, 247–256. [Google Scholar] [CrossRef]
- Idris, M.; Mitchell, D.J.; Gordon, R.; Sidharthan, N.P.; Butcher, N.J.; Minchin, R.F. Interaction of the Brain-Selective Sulfotransferase SULT4A1 with Other Cytosolic Sulfotransferases: Effects on Protein Expression and Function. Drug Metab. Dispos. 2020, 48, 337–344. [Google Scholar] [CrossRef]
- Bairam, A.F.; Rasool, M.I.; Alherz, F.A.; Abunnaja, M.S.; El Daibani, A.A.; Gohal, S.A.; Alatwi, E.S.; Kurogi, K.; Liu, M. Impact of SULT1A3/SULT1A4 genetic polymorphisms on the sulfation of phenylephrine and salbutamol by human SULT1A3 allozymes. Pharmacogenet. Genom. 2019, 29, 99–105. [Google Scholar] [CrossRef]
- Bairam, A.F.; Rasool, M.I.; Alherz, F.A.; Abunnaja, M.S.; El Daibani, A.A.; Kurogi, K.; Liu, M. Effects of human SULT1A3/SULT1A4 genetic polymorphisms on the sulfation of acetaminophen and opioid drugs by the cytosolic sulfotransferase SULT1A3. Arch. Biochem. Biophys. 2018, 648, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Kurogi, K.; Shimohira, T.; Kouriki-Nagatomo, H.; Zhang, G.; Miller, E.R.; Sakakibara, Y.; Suiko, M.; Liu, M.-C. Human Cytosolic Sulphotransferase SULT1C3: Genomic analysis and functional characterization of splice variant SULT1C3a and SULT1C3d. J. Biochem. 2017, 162, 403–414. [Google Scholar] [CrossRef]
- Guidry, A.L.; Tibbs, Z.E.; Runge-Morris, M.; Falany, C.N. Expression, purification and characterization of human cytosolic sulfotransferase (SULT) 1C4. Horm. Mol. Biol. Clin. Investig. 2017, 29, 27–36. [Google Scholar] [CrossRef]
- Lu, J.; Li, H.; Zhang, J.; Li, M.; An, X.; Liu, M.-C.; Chang, W. Crystal structures of SULT1A2 and SULT1A1 *3: Insights into the substrate inhibition and the role of Tyr149 in SULT1A2. Biochem. Biophys. Res. Commun. 2010, 396, 429–434. [Google Scholar] [CrossRef]
- Cook, I.T.; Duniec-Dmuchowski, Z.; Kocarek, T.A.; Runge-Morris, M.; Falany, C.N. 24-hydroxycholesterol sulfation by human cytosolic sulfotransferases: Formation of monosulfates and disulfates, molecular modeling, sulfatase sensitivity, and inhibition of liver x receptor activation. Drug Metab. Dispos. 2009, 37, 2069–2078. [Google Scholar] [CrossRef]
- Lu, L.-Y.; Hsieh, Y.-C.; Liu, M.-Y.; Lin, Y.-H.; Chen, C.-J.; Yang, Y.-S. Identification and characterization of two amino acids critical for the substrate inhibition of human dehydroepiandrosterone sulfotransferase (SULT2A1). Mol. Pharmacol. 2008, 73, 660–668. [Google Scholar] [CrossRef]
- Allali-Hassani, A.; Pan, P.W.; Dombrovski, L.; Najmanovich, R.; Tempel, W.; Dong, A.; Loppnau, P.; Martin, F.; Thonton, J.; Edwards, A.M.; et al. Structural and chemical profiling of the human cytosolic sulfotransferases. PLoS Biol. 2007, 5, e97. [Google Scholar]
- Gamage, N.U.; Tsvetanov, S.; Duggleby, R.G.; McManus, M.E.; Martin, J.L. The structure of human SULT1A1 crystallized with estradiol. An insight into active site plasticity and substrate inhibition with multi-ring substrates. J. Biol. Chem. 2005, 280, 41482–41486. [Google Scholar] [CrossRef]
- Pedersen, L.C.; Petrotchenko, E.; Shevtsov, S.; Negishi, M. Crystal structure of the human estrogen sulfotransferase-PAPS complex: Evidence for catalytic role of Ser137 in the sulfuryl transfer reaction. J. Biol. Chem. 2002, 277, 17928–17932. [Google Scholar] [CrossRef]
- Fuda, H.; Lee, Y.C.; Shimizu, C.; Javitt, N.B.; Strott, C.A. Mutational analysis of human hydroxysteroid sulfotransferase SULT2B1 isoforms reveals that exon 1B of the SULT2B1 gene produces cholesterol sulfotransferase, whereas exon 1A yields pregnenolone sulfotransferase. J. Biol. Chem. 2002, 277, 36161–36166. [Google Scholar] [CrossRef]
- Freimuth, R.R.; Raftogianis, R.B.; Wood, T.C.; Moon, E.; Kim, U.-J.; Xu, J.; Siciliano, M.J.; Weinshilboum, R.M. Human sulfotransferases SULT1C1 and SULT1C2: cDNA characterization, gene cloning, and chromosomal localization. Genomics 2000, 65, 157–165. [Google Scholar] [CrossRef]
- Wang, J.; Falany, J.L.; Falany, C.N. Expression and characterization of a novel thyroid hormone-sulfating form of cytosolic sulfotransferase from human liver. Mol. Pharmacol. 1998, 53, 274–282. [Google Scholar] [CrossRef]
- Sakakibara, Y.; Yanagisawa, K.; Katafuchi, J.; Ringer, D.P.; Takami, Y.; Nakayama, T.; Suiko, M.; Liu, M. Molecular cloning, expression, and characterization of novel human SULT1C sulfotransferases that catalyze the sulfonation of N-hydroxy-2-acetylaminofluorene. J. Biol. Chem. 1998, 273, 33929–33935. [Google Scholar] [CrossRef]
- Fujita, K.-I.; Nagata, K.; Ozawa, S.; Sasano, H.; Yamazoe, Y. Molecular cloning and characterization of rat ST1B1 and human ST1B2 cDNAs, encoding thyroid hormone sulfotransferases. J. Biochem. 1997, 122, 1052–1061. [Google Scholar] [CrossRef]
- Falany, C.N.; Krasnykh, V.; Falany, J.L. Bacterial expression and characterization of a cDNA for human liver estrogen sulfotransferase. J. Steroid Biochem. Mol. Biol. 1995, 52, 529–539. [Google Scholar] [CrossRef]
- Radominska, A.; A Comer, K.; Zimniak, P.; Falany, J.; Iscan, M.; Falany, C.N. Human liver steroid sulphotransferase sulphates bile acids. Biochem. J. 1990, 272, 597–604. [Google Scholar] [CrossRef]
- Whittemore, R.M.; Pearce, L.B.; Roth, J.A. Purification and kinetic characterization of a phenol-sulfating form of phenol sulfotransferase from human brain. Arch. Biochem. Biophys. 1986, 249, 464–471. [Google Scholar] [CrossRef]
- Le, H.H.; Lee, M.-T.; Besler, K.R.; Comrie, J.M.C.; Johnson, E.L. Characterization of interactions of dietary cholesterol with the murine and human gut microbiome. Nat. Microbiol. 2022, 7, 1390–1403. [Google Scholar] [CrossRef]
- Yao, L.; D’agostino, G.D.; Park, J.; Hang, S.; Adhikari, A.A.; Zhang, Y.; Li, W.; Avila-Pacheco, J.; Bae, S.; Clish, C.B.; et al. A biosynthetic pathway for the selective sulfonation of steroidal metabolites by human gut bacteria. Nat. Microbiol. 2022, 7, 1404–1418. [Google Scholar] [CrossRef]
- Kim, D.-H.; Kobashi, K. The role of intestinal flora in metabolism of phenolic sulfate esters. Biochem. Pharmacol. 1986, 35, 3507–3510. [Google Scholar] [CrossRef]
- Kim, D.-H.; Konishi, L.; Kobashi, K. Purification, characterization and reaction mechanism of novel arylsulfotransferase obtained from an anaerobic bacterium of human intestine. Biochim. Biophys. Acta 1986, 872, 33–41. [Google Scholar] [CrossRef]
- Kim, D.-H.; Yoon, H.-K.; Koizumi, M.; Kobashi, K. Sulfation of phenolic antibiotics by sulfotransferase obtained from a human intestinal bacterium. Chem. Pharm. Bull. 1992, 40, 1056–1057. [Google Scholar] [CrossRef]
- Kobashi, K.; Fukaya, Y.; Kim, D.-H.; Akao, T.; Takebe, S. A novel type of aryl sulfotransferase obtained from an anaerobic bacterium of human intestine. Arch. Biochem. Biophys. 1986, 245, 537–539. [Google Scholar] [CrossRef]
- Suiko, M.; Kurogi, K.; Hashiguchi, T.; Sakakibara, Y.; Liu, M.-C. Updated perspectives on the cytosolic sulfotransferases (SULTs) and SULT-mediated sulfation. Biosci. Biotechnol. Biochem. 2017, 81, 63–72. [Google Scholar] [CrossRef]
- Tibbs, Z.E.; Rohn-Glowacki, K.J.; Crittenden, F.; Guidry, A.L.; Falany, C.N. Structural plasticity in the human cytosolic sulfotransferase dimer and its role in substrate selectivity and catalysis. Drug Metab. Pharmacokinet. 2015, 30, 3–20. [Google Scholar] [CrossRef]
- Malojčić, G.; Owen, R.L.; Grimshaw, J.P.A.; Brozzo, M.S.; Dreher-Teo, H.; Glockshuber, R. A structural and biochemical basis for PAPS-independent sulfuryl transfer by aryl sulfotransferase from uropathogenic Escherichia coli. Proc. Natl. Acad. Sci. USA 2008, 105, 19217–19222. [Google Scholar] [CrossRef]
- Gamage, N.; Barnett, A.; Hempel, N.; Duggleby, R.G.; Windmill, K.F.; Martin, J.L.; McManus, M.E. Human sulfotransferases and their role in chemical metabolism. Toxicol. Sci. 2006, 90, 5–22. [Google Scholar] [CrossRef]
- Zhang, H.; Varlamova, O.; Vargas, F.M.; Falany, C.N.; Leyh, T.S. Sulfuryl transfer: The catalytic mechanism of human estrogen sulfotransferase. J. Biol. Chem. 1998, 273, 10888–10892. [Google Scholar] [CrossRef]
- Konishi-Imamura, L.; Dohi, K.; Sato, M.; Kobashi, K. Improved purification of arylsulfate sulfotransferase from human intestinal bacterium by using polyclonal antibody. J. Biochem. 1994, 115, 1097–1100. [Google Scholar] [CrossRef]
- Baek, M.-C.; Kwon, A.-R.; Chung, Y.-J.; Kim, B.-K.; Choi, E.-C. Distribution of bacteria with the arylsulfate sulfotransferase activity. Arch. Pharm. Res. 1998, 21, 475–477. [Google Scholar] [CrossRef]
- Koppel, N.; Rekdal, V.M.; Balskus, E.P. Chemical transformation of xenobiotics by the human gut microbiota. Science 2017, 356, 6344. [Google Scholar] [CrossRef]
- Wexler, H.M.; Reeves, D.; Summanen, P.H.; Molitoris, E.; McTEAGUE, M.; Duncan, J.; Wilson, K.H.; Finegold, S.M. Sutterella wadsworthensis gen. nov., sp. nov., bile-resistant microaerophilic Campylobacter gracilis-like clinical isolates. Int. J. Syst. Bacteriol. 1996, 46, 252–258. [Google Scholar] [CrossRef]
- Engberg, J.; On, S.L.W.; Harrington, C.S.; Gerner-Smidt, P. Prevalence of Campylobacter, Arcobacter, Helicobacter, and Sutterella spp. in human fecal samples as estimated by a reevaluation of isolation methods for Campylobacters. J. Clin. Microbiol. 2000, 38, 286–291. [Google Scholar] [CrossRef]
- Sakon, H.; Nagai, F.; Morotomi, M.; Tanaka, R. Sutterella parvirubra sp. nov. and Megamonas funiformis sp. nov., isolated from human faeces. Int. J. Syst. Evol. Microbiol. 2008, 58 Pt 4, 970–975. [Google Scholar] [CrossRef]
- Ndongo, S.; Cadoret, F.; Dubourg, G.; Delerce, J.; Fournier, P.-E.; Raoult, D.; Lagier, J.-C. ‘Collinsella phocaeensis’ sp. nov., ‘Clostridium merdae’ sp. nov., ‘Sutterella massiliensis’ sp. nov., ‘Sutturella timonensis’ sp. nov., ‘Enorma phocaeensis’ sp. nov., ‘Mailhella massiliensis’ gen. nov., sp. nov., ‘Mordavella massiliensis’ gen. nov., sp. nov. and ‘Massiliprevotella massiliensis’ gen. nov., sp. nov., 9 new species isolated from fresh stool samples of healthy French patients. New Microbes New Infect. 2017, 17, 89–95. [Google Scholar]
- Dione, N.; Ngom, I.I.; Valles, C.; Cadoret, F.; Fournier, P.E.; Raoult, D.; Lagier, J.C. ‘Collinsella provencensis’ sp. nov., ‘Parabacteroides bouchesdurhonensis’ sp. nov. and ‘Sutterella seckii’ sp. nov., three new bacterial species identified from human gut microbiota. New Microbes New Infect. 2018, 23, 44–47. [Google Scholar] [CrossRef]
- Sakamoto, M.; Ikeyama, N.; Kunihiro, T.; Iino, T.; Yuki, M.; Ohkuma, M. Mesosutterella multiformis gen. nov., sp. nov., a member of the family Sutterellaceae and Sutterella megalosphaeroides sp. nov., isolated from human faeces. Int. J. Syst. Evol. Microbiol. 2018, 68, 3942–3950. [Google Scholar] [CrossRef] [PubMed]
- Oh, B.S.; Kim, J.S.; Yu, S.Y.; Ryu, S.W.; Park, S.H.; Kang, S.W.; Park, J.-E.; Choi, S.-H.; Han, K.-I.; Lee, K.C.; et al. Sutterella faecalis sp. nov., isolated from human faeces. J. Microbiol. 2020, 58, 99–104. [Google Scholar] [CrossRef]
- Kaakoush, N.O. Sutterella Species, IgA-degrading Bacteria in Ulcerative Colitis. Trends Microbiol. 2020, 28, 519–522. [Google Scholar] [CrossRef]
- Williams, B.L.; Hornig, M.; Parekh, T.; Lipkin, W.I. Application of novel PCR-based methods for detection, quantitation, and phylogenetic characterization of Sutterella species in intestinal biopsy samples from children with autism and gastrointestinal disturbances. mBio 2012, 3, e00261-11. [Google Scholar] [CrossRef]
- Wang, L.; Christophersen, C.T.; Sorich, M.J.; Gerber, J.P.; Angley, M.T.; Conlon, M.A. Increased abundance of Sutterella spp. and Ruminococcus torques in feces of children with autism spectrum disorder. Mol. Autism. 2013, 4, 42. [Google Scholar] [CrossRef]
- Kirk, K.F.; Andersen, K.L.; Tarpgaard, I.H.; Nielsen, H.L. Three cases of Sutterella wadsworthensis bacteremia secondary to abdominal infections. Anaerobe 2021, 72, 102460. [Google Scholar] [CrossRef]
- Giri, S.; Mangalam, A. Chapter 34—The Gut Microbiome and Metabolome in Multiple Sclerosis. In Microbiome and Metabolome in Diagnosis, Therapy, and other Strategic Applications; Faintuch, J., Faintuch, S., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 333–340. [Google Scholar]
- Cornejo-Pareja, I.; Ruiz-Limón, P.; Gómez-Pérez, A.M.; Molina-Vega, M.; Moreno-Indias, I.; Tinahones, F.J. Differential Microbial Pattern Description in Subjects with Autoimmune-Based Thyroid Diseases: A Pilot Study. J. Pers. Med. 2020, 10, 192. [Google Scholar] [CrossRef]
- Hiippala, K.; Kainulainen, V.; Kalliomäki, M.; Arkkila, P.; Satokari, R. Mucosal Prevalence and Interactions with the Epithelium Indicate Commensalism of Sutterella spp. Front. Microbiol. 2016, 7, 1706. [Google Scholar] [CrossRef]
- Raghavan, R.; Kelkar, Y.D.; Ochman, H. A selective force favoring increased G+C content in bacterial genes. Proc. Natl. Acad. Sci. USA 2012, 109, 14504–14507. [Google Scholar] [CrossRef]
- Schell, M.A. Molecular biology of the LysR family of transcriptional regulators. Annu. Rev. Microbiol. 1993, 47, 597–626. [Google Scholar] [CrossRef]
- van der Ploeg, J.R.; Eichhorn, E.; Leisinger, T. Sulfonate-sulfur metabolism and its regulation in Escherichia coli. Arch. Microbiol. 2001, 176, 1–8. [Google Scholar] [CrossRef]
- Kouzuma, A.; Endoh, T.; Omori, T.; Nojiri, H.; Yamane, H.; Habe, H. Transcription factors CysB and SfnR constitute the hierarchical regulatory system for the sulfate starvation response in Pseudomonas putida. J. Bacteriol. 2008, 190, 4521–4531. [Google Scholar] [CrossRef]
- Pokhrel, A.; Dinh, H.; Li, L.; Hassan, K.A.; Cain, A.K.; Paulsen, I.T. Identification of a novel LysR family transcriptional regulator controlling acquisition of sulfur sources in Acinetobacter baumannii. Microb. Physiol. 2023, 33, 27–35. [Google Scholar] [CrossRef]
- Wang, J.P.; Zhang, W.M.; Chao, H.J.; Zhou, N.Y. PnpM, a LysR-Type Transcriptional Regulator Activates the Hydroquinone Pathway in para-Nitrophenol Degradation in Pseudomonas sp. Strain WBC-3. Front. Microbiol. 2017, 8, 1714. [Google Scholar] [CrossRef]
- Lu, Z.; Imlay, J.A. The Fumarate Reductase of Bacteroides thetaiotaomicron, unlike That of Escherichia coli, Is Configured so that It Does Not Generate Reactive Oxygen Species. mBio 2017, 8, e01873-16. [Google Scholar] [CrossRef]
- Laue, H.; Denger, K.; Cook, A.M. Taurine reduction in anaerobic respiration of Bilophila wadsworthia RZATAU. Appl. Environ. Microbiol. 1997, 63, 2016–2021. [Google Scholar] [CrossRef]
- Häse, C.C.; A Finkelstein, R. Bacterial extracellular zinc-containing metalloproteases. Microbiol. Rev. 1993, 57, 823–837. [Google Scholar] [CrossRef]
- Garmory, H.S.; Titball, R.W. ATP-binding cassette transporters are targets for the development of antibacterial vaccines and therapies. Infect. Immun. 2004, 72, 6757–6763. [Google Scholar] [CrossRef] [PubMed]
- Kertesz, M.A. Bacterial transporters for sulfate and organosulfur compounds. Res. Microbiol. 2001, 152, 279–290. [Google Scholar] [CrossRef]
- Chen, J.; Xie, J. Role and regulation of bacterial LuxR-like regulators. J. Cell Biochem. 2011, 112, 2694–2702. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Guthrie, B.G.H.; Alexander, M.; Noecker, C.; Ramirez, L.; Glasser, N.R.; Turnbaugh, P.J.; Balskus, E.P. Genetic manipulation of the human gut bacterium Eggerthella lenta reveals a widespread family of transcriptional regulators. Nat. Commun. 2022, 13, 7624. [Google Scholar] [CrossRef]
- Ryan, J.; McKillen, M.; Mason, J. Sulphate/molybdate interactions: In vivo and in vitro studies on the group VI oxyanion transport system in ovine renal tubule epithelial cells. Ann. Rech. Vet. 1987, 18, 47–55. [Google Scholar]
- Self, W.T.; Grunden, A.M.; Hasona, A.; Shanmugam, K.T. Molybdate transport. Res. Microbiol. 2001, 152, 311–321. [Google Scholar] [CrossRef]
- Mason, J.; Cardin, C. The competition of molybdate and sulphate ions for a transport system in the ovine small intestine. Res. Vet. Sci. 1977, 22, 313–315. [Google Scholar] [CrossRef]
- Toth, E.A.; Yeates, T.O. The structure of adenylosuccinate lyase, an enzyme with dual activity in the de novo purine biosynthetic pathway. Structure 2000, 8, 163–174. [Google Scholar] [CrossRef]
- Pruss, K.M.; Enam, F.; Battaglioli, E.; DeFeo, M.; Diaz, O.R.; Higginbottom, S.K.; Fischer, C.R.; Hryckowian, A.J.; Van Treuren, W.; Dodd, D.; et al. Oxidative ornithine metabolism supports non-inflammatory C. difficile colonization. Nat. Metab. 2022, 4, 19–28. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.D.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 2018, 46, D624–D632. [Google Scholar] [CrossRef]
- Amedei, A.; Capasso, C.; Nannini, G.; Supuran, C.T. Microbiota, Bacterial Carbonic Anhydrases, and Modulators of Their Activity: Links to Human Diseases? Mediators Inflamm. 2021, 2021, 6926082. [Google Scholar] [CrossRef]
- Grölund, M.-M.; Lehtonen, O.-P.; Eerola, E.; Kero, P. Fecal microflora in healthy infants born by different methods of delivery: Permanent changes in intestinal flora after cesarean delivery. J. Pediatr. Gastroenterol. Nutr. 1999, 28, 19–25. [Google Scholar]
- Hall, B.M.; Breidenstein, E.B.M.; de la Fuente-Núñez, C.; Reffuveille, F.; Mawla, G.D.; Hancock, R.E.W.; Baker, T.A. Two Isoforms of Clp Peptidase in Pseudomonas aeruginosa Control Distinct Aspects of Cellular Physiology. J. Bacteriol. 2017, 199, e00568-16. [Google Scholar] [CrossRef]
- Bolam, D.N.; van den Berg, B. TonB-dependent transport by the gut microbiota: Novel aspects of an old problem. Curr. Opin. Struct. Biol. 2018, 51, 35–43. [Google Scholar] [CrossRef]
- Pollet, R.M.; Martin, L.M.; Koropatkin, N.M. TonB-dependent transporters in the Bacteroidetes: Unique domain structures and potential functions. Mol. Microbiol. 2021, 115, 490–501. [Google Scholar] [CrossRef]
- Doerfel, L.K.; Wohlgemuth, I.; Kothe, C.; Peske, F.; Urlaub, H.; Rodnina, M.V. EF-P is essential for rapid synthesis of proteins containing consecutive proline residues. Science 2013, 339, 85–88. [Google Scholar] [CrossRef]
- Kang, T.M.; Yuan, J.; Zhou, A.; Beppler, C.; Miller, J.H. Deoxycytidine deaminase-deficient Escherichia coli strains display acute sensitivity to cytidine, adenosine, and guanosine and increased sensitivity to a range of antibiotics, including vancomycin. J. Bacteriol. 2014, 196, 1950–1957. [Google Scholar] [CrossRef]
- Sugrue, E.; Fraser, N.J.; Hopkins, D.H.; Carr, P.D.; Khurana, J.L.; Oakeshott, J.G.; Scott, C.; Jackson, C.J. Evolutionary expansion of the amidohydrolase superfamily in bacteria in response to the synthetic compounds molinate and diuron. Appl. Environ. Microbiol. 2015, 81, 2612–2624. [Google Scholar] [CrossRef]
- Erb, T.J. Carboxylases in natural and synthetic microbial pathways. Appl. Environ. Microbiol. 2011, 77, 8466–8477. [Google Scholar] [CrossRef]
- Monti, S.M.; De Simone, G.; D’Ambrosio, K. L-Histidinol Dehydrogenase as a New Target for Old Diseases. Curr. Top. Med. Chem. 2016, 16, 2369–2378. [Google Scholar] [CrossRef] [PubMed]
- Chioccioli, S.; Del Duca, S.; Vassallo, A.; Castronovo, L.M.; Fani, R. Exploring the role of the histidine biosynthetic hisF gene in cellular metabolism and in the evolution of (ancestral) genes: From LUCA to the extant (micro)organisms. Microbiol. Res. 2020, 240, 126555. [Google Scholar] [CrossRef] [PubMed]
- Lewinson, O.; Lee, A.T.; Rees, D.C. A P-type ATPase importer that discriminates between essential and toxic transition metals. Proc. Natl. Acad. Sci. USA 2009, 106, 4677–4682. [Google Scholar] [CrossRef]
- Pealing, S.L.; Black, A.C.; Manson, F.D.C.; Ward, F.B.; Chapman, S.K.; Reid, G.A. Sequence of the gene encoding flavocytochrome c from Shewanella putrefaciens: A tetraheme flavoenzyme that is a soluble fumarate reductase related to the membrane-bound enzymes from other bacteria. Biochemistry 1992, 31, 12132–12140. [Google Scholar] [CrossRef]
- He, S.H.; DerVartanian, D.V.; LeGall, J. Isolation of fumarate reductase from Desulfovibrio multispirans, a sulfate reducing bacterium. Biochem. Biophys. Res. Commun. 1986, 135, 1000–1007. [Google Scholar] [CrossRef]
- Natale, P.; Brüser, T.; Driessen, A.J. Sec- and Tat-mediated protein secretion across the bacterial cytoplasmic membrane—Distinct translocases and mechanisms. Biochim. Biophys. Acta 2008, 1778, 1735–1756. [Google Scholar] [CrossRef]
- Hinsley, A.P.; Stanley, N.R.; Palmer, T.; Berks, B.C. A naturally occurring bacterial Tat signal peptide lacking one of the ‘invariant’ arginine residues of the consensus targeting motif. FEBS Lett. 2001, 497, 45–49. [Google Scholar] [CrossRef]
- Molik, S.; Karnauchov, I.; Weidlich, C.; Herrmann, R.G.; Klosgen, R.B. The Rieske Fe/S protein of the cytochrome b6/f complex in chloroplasts: Missing link in the evolution of protein transport pathways in chloroplasts? J. Biol. Chem. 2001, 276, 42761–42766. [Google Scholar] [CrossRef]
- Frain, K.M.; Robinson, C.; van Dijl, J.M. Transport of Folded Proteins by the Tat System. Protein J. 2019, 38, 377–388. [Google Scholar] [CrossRef]
- Green, E.R.; Mecsas, J. Bacterial Secretion Systems: An Overview. Microbiol. Spectr. 2016, 4, 1. [Google Scholar] [CrossRef]
- Mariani, V.; Biasini, M.; Barbato, A.; Schwede, T. lDDT: A local superposition-free score for comparing protein structures and models using distance difference tests. Bioinformatics 2013, 29, 2722–2728. [Google Scholar] [CrossRef]
- Carugo, O.; Pongor, S. A normalized root-mean-square distance for comparing protein three-dimensional structures. Protein Sci. 2001, 10, 1470–1473. [Google Scholar] [CrossRef]
- King, C.H.; Desai, H.; Sylvetsky, A.C.; LoTempio, J.; Ayanyan, S.; Carrie, J.; Crandall, K.A.; Fochtman, B.C.; Gasparyan, L.; Gulzar, N.; et al. Baseline human gut microbiota profile in healthy people and standard reporting template. PLoS ONE 2019, 14, e0206484. [Google Scholar] [CrossRef]
- Saito, Y.; Sato, T.; Nomoto, K.; Tsuji, H. Identification of phenol- and p-cresol-producing intestinal bacteria by using media supplemented with tyrosine and its metabolites. FEMS Microbiol. Ecol. 2018, 94, fiy125. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Annotated Genes in the Neighborhood of asst Genes | Number of Occurrences Around asst Genes | Found in the Neighborhood of this asst Gene |
|---|---|---|
| LysR transcriptional regulator protein | 7 | 1, 2, 6, 9, 10, 16, 17 |
| Fumarate reductase flavoprotein subunit | 3 | 1, 5, 13 |
| Putative peptide zinc metalloprotease | 1 | 1 |
| Putative ATP-binding cassette transporter | 1 | 2 |
| LuxR transcriptional regulator protein | 1 | 3 |
| modC molybdate ABC transporter | 1 | 3 |
| Adenylosuccinate lyase | 1 | 3 |
| Ornithine carbamoyltransferase | 1 | 4 |
| Oligopeptidase metallo peptidase MEROPS family | 1 | 4 |
| Carbonic anhydrase | 1 | 6 |
| ATP-dependent Clp protease proteolytic subunit | 1 | 8 |
| Outer membrane transport energization protein TonB | 1 | 9 |
| Translation elongation factor P | 1 | 10 |
| Deoxycytidylate deaminase | 1 | 10 |
| Amidohydrolase | 1 | 10 |
| Pyruvate carboxylase | 1 | 11 |
| ATP phosphoribosyltransferase | 1 | 14 |
| Predicted Protein | Type of SP | Signal Peptide Sequence |
|---|---|---|
| SwASST1 | Tat type | MKFQRRQLVAAMAGLMLGLSAGAASA |
| SwASST2 | Sec type | MRLFAKSLIAASIAAASMGALA |
| SwASST3 | Tat type | MSKMPTSPISRRTLCGLAGSALFLSQFPRAAFA |
| SwASST4 | Sec type | MSKQKAHQKLNQLTAVLMMAAAAALAAPAVQA |
| SwASST5 | Tat type | MKNITIGRRSFFLGAVAAAVSSALPATAAHA |
| SwASST6 | Tat type | MIQRRTLVKAVIATLAPLSLTSAFA |
| SwASST7 | Sec type | MRISKTAIAVSVLSLFAATMVPATSMA |
| SwASST8 | Sec type | MKMKWIALAVAAAIPAAAMA |
| SwASST9 | Sec type | MKLNFHPTRAAVLVGAALLSATFAFPAMA |
| SwASST10 | Sec type | MQFQKTAFALCAAGLLSLPGLVAA |
| SwASST12 | Sec type | MNIKAKITPVALAIALAAGFGLPAAVQA |
| SwASST13 | Sec type | MKSMKLSKIAAFAAFLGFSAAVA |
| SwASST14 | Tat type | MQLTKRAFLLSGAALAVSAASGAFA |
| SwASST15 | Sec type | MKKIVPICAAVAAAAMMLSISTPAMA |
| SwASST16 | Tat type | MLLTKRQFLTSVLALSVSAAARA |
| SwASST17 | Sec type | MKFKSTVIAASVLAGIMSLSAGAYA |
| Predicted Protein | Signal Peptide Type | Sec Guided (Unfolded State) Periplasmic/Extracellular Proteins | Tat Guided (Folded State) Periplasmic/Extracellular Proteins |
|---|---|---|---|
| SwASST1 | Tat | ||
| SwASST2 | Sec | + | |
| SwASST3 | Tat | + | |
| SwASST4 | Sec | ||
| SwASST5 | Tat | + | |
| SwASST6 | Tat | + | |
| SwASST7 | Sec | ||
| SwASST8 | Sec | ||
| SwASST9 | Sec | ||
| SwASST10 | Sec | + | |
| SwASST11 | - | ||
| SwASST12 | Sec | ||
| SwASST13 | Sec | + | |
| SwASST14 | Tat | ||
| SwASST15 | Sec | ||
| SwASST16 | Tat | + | |
| SwASST17 | Sec |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Langford, L.; Shah, D.D. Bioinformatic Analysis of Sulfotransferases from an Unexplored Gut Microbe, Sutterella wadsworthensis 3_1_45B: Possible Roles towards Detoxification via Sulfonation by Members of the Human Gut Microbiome. Int. J. Mol. Sci. 2024, 25, 2983. https://doi.org/10.3390/ijms25052983
Langford L, Shah DD. Bioinformatic Analysis of Sulfotransferases from an Unexplored Gut Microbe, Sutterella wadsworthensis 3_1_45B: Possible Roles towards Detoxification via Sulfonation by Members of the Human Gut Microbiome. International Journal of Molecular Sciences. 2024; 25(5):2983. https://doi.org/10.3390/ijms25052983
Chicago/Turabian StyleLangford, Lauryn, and Dhara D. Shah. 2024. "Bioinformatic Analysis of Sulfotransferases from an Unexplored Gut Microbe, Sutterella wadsworthensis 3_1_45B: Possible Roles towards Detoxification via Sulfonation by Members of the Human Gut Microbiome" International Journal of Molecular Sciences 25, no. 5: 2983. https://doi.org/10.3390/ijms25052983