
Nocardia cyriacigeorgica Elicits Gut Disturbances in a Leaky Gut Model of Colitis, but Not the Harmful Cascade Leading to Gut-First Parkinson’s Disease
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
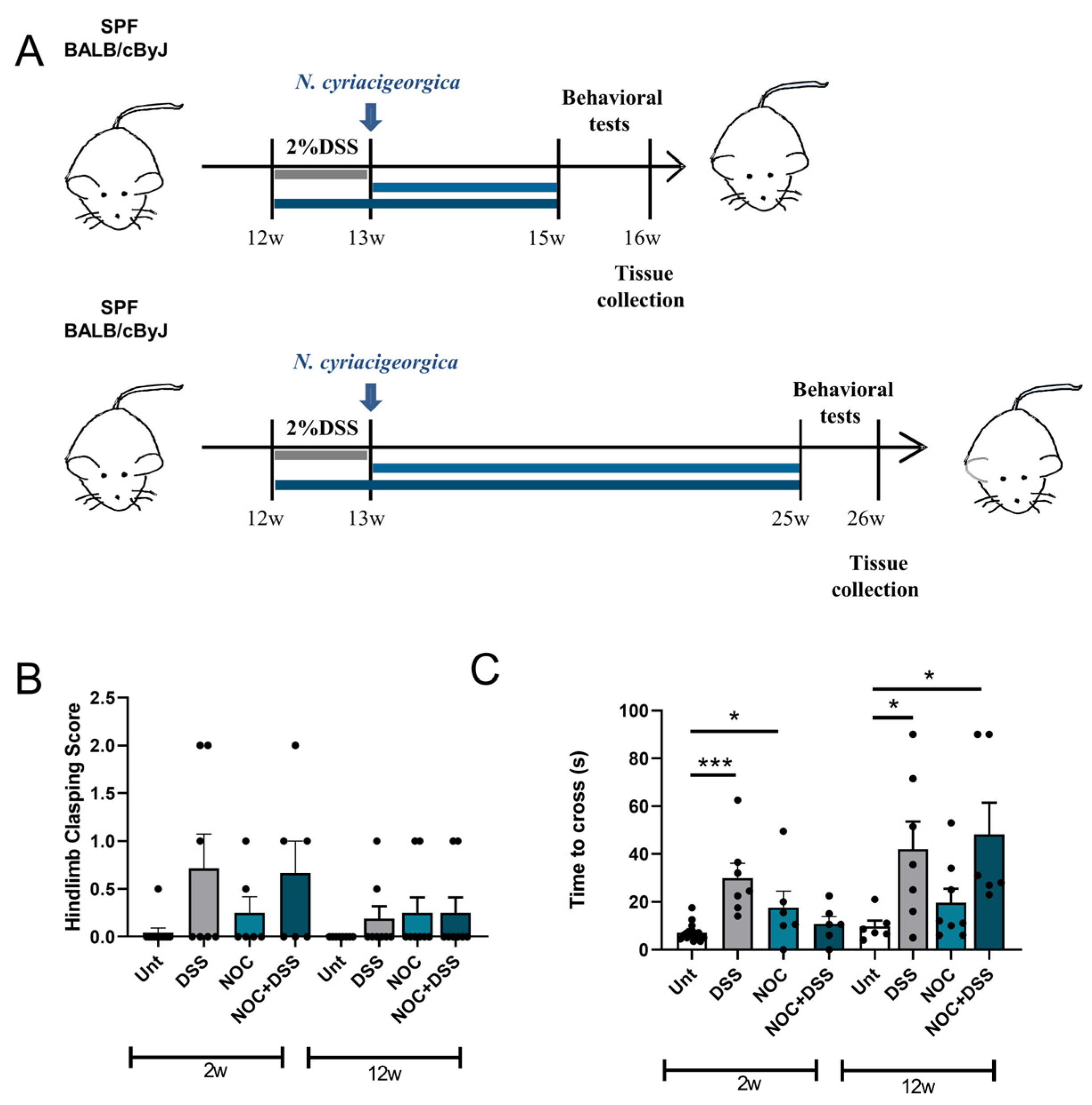
2.1. DSS Treatment Affects Early-Stage Movement, but Does Not Persist with Age
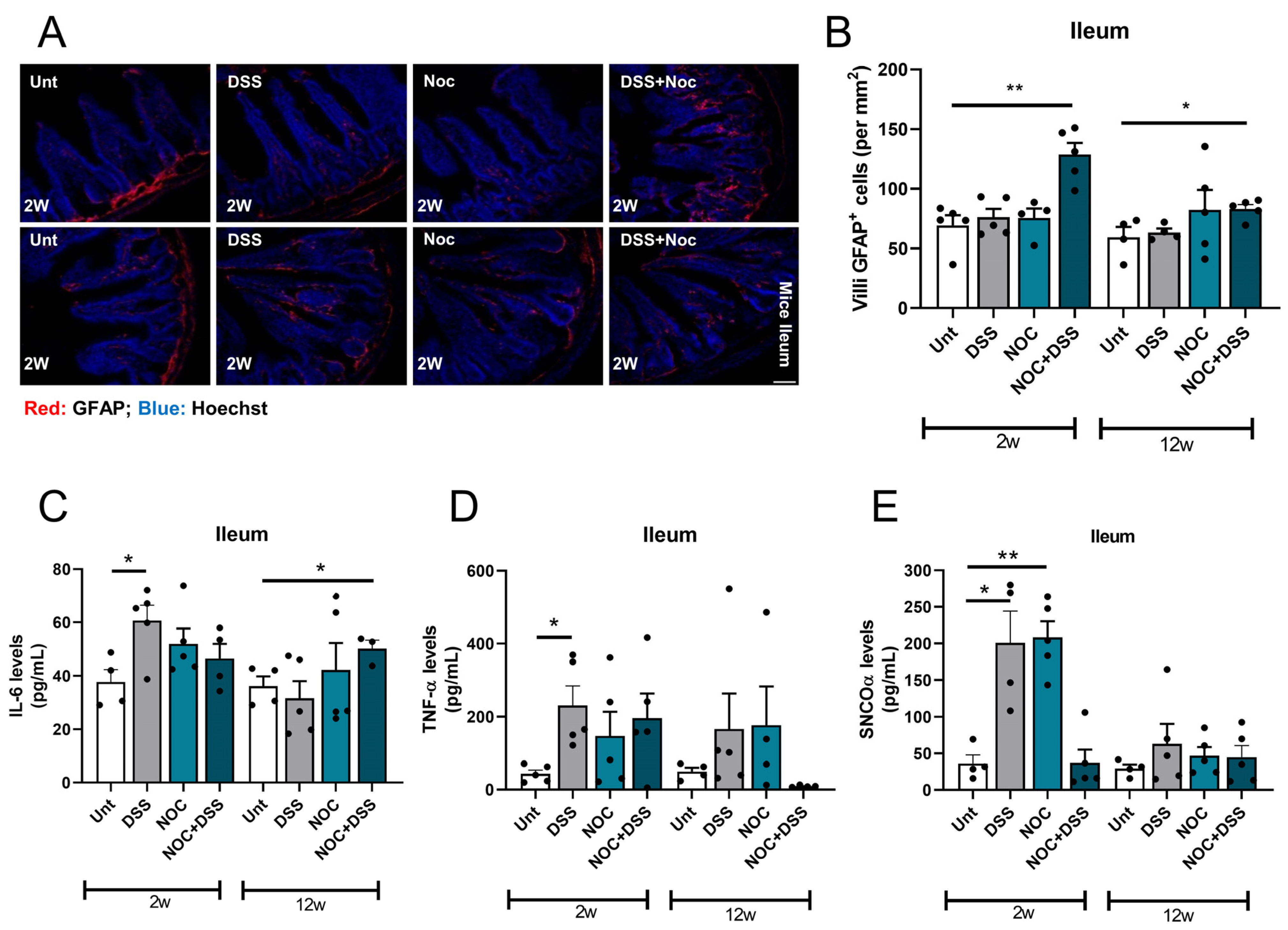
2.2. Nocardia Administration to a Colitis Model Affects Intestinal Immunity
2.3. Fitness of ENS Mitochondria Is Affected by Inflammation
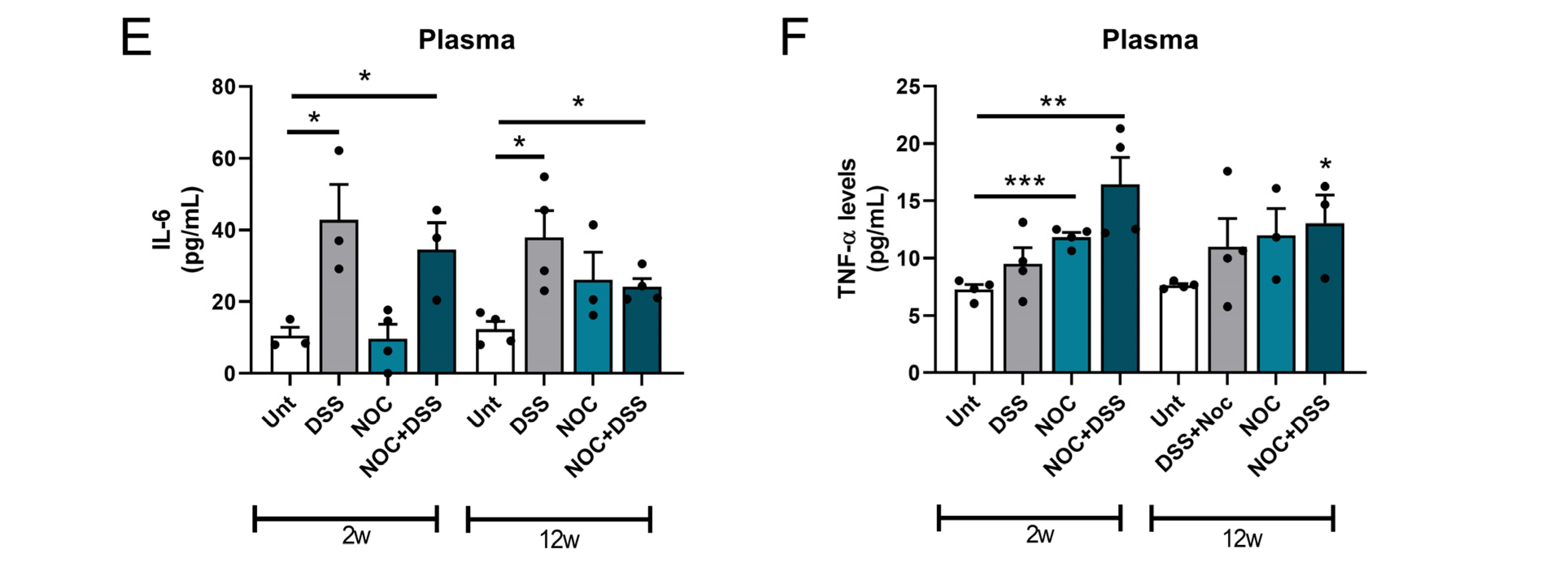
2.4. Gut Permeability and Systemic Inflammation
2.5. Midbrain Immunity Activation after Gut Inflammation
3. Discussion
4. Materials and Methods
4.1. Key Resources Table
4.2. Animal Model and Experimental Design
4.3. Bacteria and Infection
4.4. Behavior
4.5. Immunohistochemistry
4.6. ELISA Determination of Pro-Inflammatory Cytokines and aSyn and Calprotectin Fecal Levels
4.7. Microscopy
4.8. Intestinal Permeability Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Stefanis, L. α-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef] [PubMed]
- Warnecke, T.; Schäfer, K.H.; Claus, I.; Del Tredici, K.; Jost, W.H. Gastrointestinal involvement in Parkinson’s disease: Pathophysiology, diagnosis, and management. NPJ Park. Dis. 2022, 8, 31. [Google Scholar] [CrossRef]
- Stokholm, M.G.; Danielsen, E.H.; Hamilton-Dutoit, S.J.; Borghammer, P. Pathological α-synuclein in gastrointestinal tissues from prodromal Parkinson disease patients. Ann. Neurol. 2016, 79, 940–949. [Google Scholar] [CrossRef]
- Braak, H.; Rüb, U.; Gai, W.P.; Del Tredici, K. Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural Transm. 2003, 110, 517–536. [Google Scholar] [CrossRef]
- Tansey, M.G.; Wallings, R.L.; Houser, M.C.; Herrick, M.K.; Keating, C.E.; Joers, V. Inflammation and immune dysfunction in Parkinson disease. Nat. Rev. Immunol. 2022, 22, 657–673. [Google Scholar] [CrossRef]
- Pope, J.L.; Bhat, A.A.; Sharma, A.; Ahmad, R.; Krishnan, M.; Washington, M.K.; Beauchamp, R.D.; Singh, A.B.; Dhawan, P. Claudin-1 regulates intestinal epithelial homeostasis through the modulation of Notch-signalling. Gut 2014, 63, 622–634. [Google Scholar] [CrossRef]
- Mankertz, J.; Tavalali, S.; Schmitz, H.; Mankertz, A.; Riecken, E.O.; Fromm, M.; Schulzke, J.D. Expression from the human occludin promoter is affected by tumor necrosis factor alpha and interferon gamma. J. Cell Sci. 2000, 113 Pt 11, 2085–2090. [Google Scholar] [CrossRef]
- Wang, F.; Graham, W.V.; Wang, Y.; Witkowski, E.D.; Schwarz, B.T.; Turner, J.R. Interferon-gamma and tumor necrosis factor-alpha synergize to induce intestinal epithelial barrier dysfunction by up-regulating myosin light chain kinase expression. Am. J. Pathol. 2005, 166, 409–419. [Google Scholar] [CrossRef]
- Kumar, M.; Leon Coria, A.; Cornick, S.; Petri, B.; Mayengbam, S.; Jijon, H.B.; Moreau, F.; Shearer, J.; Chadee, K. Increased intestinal permeability exacerbates sepsis through reduced hepatic SCD-1 activity and dysregulated iron recycling. Nat. Commun. 2020, 11, 483. [Google Scholar] [CrossRef]
- Liu, B.; Sjölander, A.; Pedersen, N.L.; Ludvigsson, J.F.; Chen, H.; Fang, F.; Wirdefeldt, K. Irritable bowel syndrome and Parkinson’s disease risk: Register-based studies. NPJ Park. Dis. 2021, 7, 5. [Google Scholar] [CrossRef] [PubMed]
- Bu, X.L.; Wang, X.; Xiang, Y.; Shen, L.L.; Wang, Q.H.; Liu, Y.H.; Jiao, S.S.; Wang, Y.R.; Cao, H.Y.; Yi, X.; et al. The association between infectious burden and Parkinson’s disease: A case-control study. Park. Relat. Disord. 2015, 21, 877–881. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Pinto, M.F.; Empadinhas, N.; Cardoso, S.M. The neuromicrobiology of Parkinson’s disease: A unifying theory. Ageing Res. Rev. 2021, 70, 101396. [Google Scholar] [CrossRef]
- Magalhães, J.D.; Candeias, E.; Melo-Marques, I.; Silva, D.F.; Esteves, A.R.; Empadinhas, N.; Cardoso, S.M. Intestinal infection triggers mitochondria-mediated α-synuclein pathology: Relevance to Parkinson’s disease. Cell. Mol. Life Sci. CMLS 2023, 80, 166. [Google Scholar] [CrossRef] [PubMed]
- Seshadri, R.; Roux, S.; Huber, K.J.; Wu, D.; Yu, S.; Udwary, D.; Call, L.; Nayfach, S.; Hahnke, R.L.; Pukall, R.; et al. Expanding the genomic encyclopedia of Actinobacteria with 824 isolate reference genomes. Cell Genom. 2022, 2, 100213. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, D.A.; LeWitt, P.A.; Camp, D.M. Nocardia asteroides-Induced movement abnormalities in mice: Relevance for Parkinson’s disease? Mov. Disord. Off. J. Mov. Disord. Soc. 2016, 31, 1134–1138. [Google Scholar] [CrossRef]
- Tam, S.; Barry, D.P.; Beaman, L.; Beaman, B.L. Neuroinvasive Nocardia asteroides GUH-2 Induces Apoptosis in the Substantia Nigra in vivo and Dopaminergic Cells in vitro. Exp. Neurol. 2002, 177, 453–460. [Google Scholar] [CrossRef]
- Zhou, Y.; Ji, G.; Yang, X.; Chen, Z.; Zhou, L. Behavioral abnormalities in C57BL/6 mice with chronic ulcerative colitis induced by DSS. BMC Gastroenterol. 2023, 23, 84. [Google Scholar] [CrossRef]
- Chassaing, B.; Aitken, J.D.; Malleshappa, M.; Vijay-Kumar, M. Dextran sulfate sodium (DSS)-induced colitis in mice. Curr. Protoc. Immunol. 2014, 104, 15.25.11–15.25.14. [Google Scholar] [CrossRef]
- Mebius, R.E.; Kraal, G. Structure and function of the spleen. Nat. Rev. Immunol. 2005, 5, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.Y.; Yang, Y.B.; Chou, D.; Xu, J.H. The Ventrolateral Periaqueductal Gray Contributes to Depressive-Like Behaviors in Recovery of Inflammatory Bowel Disease Rat Model. Front. Neurosci. 2020, 14, 254. [Google Scholar] [CrossRef] [PubMed]
- Esteves, A.R.; Munoz-Pinto, M.F.; Nunes-Costa, D.; Candeias, E.; Silva, D.F.; Magalhães, J.D.; Pereira-Santos, A.R.; Ferreira, I.L.; Alarico, S.; Tiago, I.; et al. Footprints of a microbial toxin from the gut microbiome to mesencephalic mitochondria. Gut 2023, 72, 73–89. [Google Scholar] [CrossRef] [PubMed]
- Kohbata, S.; Beaman, B.L. L-dopa-responsive movement disorder caused by Nocardia asteroides localized in the brains of mice. Infect. Immun. 1991, 59, 181–191. [Google Scholar] [CrossRef]
- Allen Reish, H.E.; Standaert, D.G. Role of α-synuclein in inducing innate and adaptive immunity in Parkinson disease. J. Park. Dis. 2015, 5, 1–19. [Google Scholar] [CrossRef]
- Baumuratov, A.S.; Antony, P.M.A.; Ostaszewski, M.; He, F.; Salamanca, L.; Antunes, L.; Weber, J.; Longhino, L.; Derkinderen, P.; Koopman, W.J.H.; et al. Enteric neurons from Parkinson’s disease patients display ex vivo aberrations in mitochondrial structure. Sci. Rep. 2016, 6, 33117. [Google Scholar] [CrossRef]
- Pan-Montojo, F.; Anichtchik, O.; Dening, Y.; Knels, L.; Pursche, S.; Jung, R.; Jackson, S.; Gille, G.; Spillantini, M.G.; Reichmann, H.; et al. Progression of Parkinson’s disease pathology is reproduced by intragastric administration of rotenone in mice. PLoS ONE 2010, 5, e8762. [Google Scholar] [CrossRef]
- Pitman, R.S.; Blumberg, R.S. First line of defense: The role of the intestinal epithelium as an active component of the mucosal immune system. J. Gastroenterol. 2000, 35, 805–814. [Google Scholar] [CrossRef]
- Kuo, W.T.; Zuo, L.; Odenwald, M.A.; Madha, S.; Singh, G.; Gurniak, C.B.; Abraham, C.; Turner, J.R. The Tight Junction Protein ZO-1 Is Dispensable for Barrier Function but Critical for Effective Mucosal Repair. Gastroenterology 2021, 161, 1924–1939. [Google Scholar] [CrossRef] [PubMed]
- Hor, J.W.; Lim, S.Y.; Khor, E.S.; Chong, K.K.; Song, S.L.; Ibrahim, N.M.; Teh, C.S.J.; Chong, C.W.; Hilmi, I.N.; Tan, A.H. Fecal Calprotectin in Parkinson’s Disease and Multiple System Atrophy. J. Mov. Disord. 2022, 15, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, S.M.; Empadinhas, N. The Microbiome-Mitochondria Dance in Prodromal Parkinson’s Disease. Front. Physiol. 2018, 9, 471. [Google Scholar] [CrossRef]
- Progatzky, F.; Pachnis, V. The role of enteric glia in intestinal immunity. Curr. Opin. Immunol. 2022, 77, 102183. [Google Scholar] [CrossRef]
- Seguella, L.; Gulbransen, B.D. Enteric glial biology, intercellular signalling and roles in gastrointestinal disease. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 571–587. [Google Scholar] [CrossRef]
- Chen, Q.Q.; Haikal, C.; Li, W.; Li, J.Y. Gut Inflammation in Association with Pathogenesis of Parkinson’s Disease. Front. Mol. Neurosci. 2019, 12, 218. [Google Scholar] [CrossRef]
- Grathwohl, S.A.; Steiner, J.A.; Britschgi, M.; Brundin, P. Mind the gut: Secretion of α-synuclein by enteric neurons. J. Neurochem. 2013, 125, 487–490. [Google Scholar] [CrossRef] [PubMed]
- Banoth, B.; Cassel, S.L. Mitochondria in innate immune signaling. Transl. Res. J. Lab. Clin. Med. 2018, 202, 52–68. [Google Scholar] [CrossRef]
- Van Den Berge, N.; Ferreira, N.; Gram, H.; Mikkelsen, T.W.; Alstrup, A.K.O.; Casadei, N.; Tsung-Pin, P.; Riess, O.; Nyengaard, J.R.; Tamgüney, G.; et al. Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of alpha-synuclein in rats. Acta Neuropathol. 2019, 138, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Magalhães, J.D.; Esteves, A.R.; Candeias, E.; Silva, D.F.; Empadinhas, N.; Cardoso, S.M. The Role of Bacteria-Mitochondria Communication in the Activation of Neuronal Innate Immunity: Implications to Parkinson’s Disease. Int. J. Mol. Sci. 2023, 24, 4339. [Google Scholar] [CrossRef] [PubMed]
- Chelakkot, C.; Ghim, J.; Ryu, S.H. Mechanisms regulating intestinal barrier integrity and its pathological implications. Exp. Mol. Med. 2018, 50, 1–9. [Google Scholar] [CrossRef]
- Craven, M.; Egan, C.E.; Dowd, S.E.; McDonough, S.P.; Dogan, B.; Denkers, E.Y.; Bowman, D.; Scherl, E.J.; Simpson, K.W. Inflammation drives dysbiosis and bacterial invasion in murine models of ileal Crohn’s disease. PLoS ONE 2012, 7, e41594. [Google Scholar] [CrossRef]
- Drevets, D.A.; Leenen, P.J.; Greenfield, R.A. Invasion of the central nervous system by intracellular bacteria. Clin. Microbiol. Rev. 2004, 17, 323–347. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, Y.; Zhou, J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener. 2015, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Fleming, S.M.; Salcedo, J.; Fernagut, P.O.; Rockenstein, E.; Masliah, E.; Levine, M.S.; Chesselet, M.F. Early and progressive sensorimotor anomalies in mice overexpressing wild-type human alpha-synuclein. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 9434–9440. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.E.; Stecher, B.; Labrie, V.; Brundin, L.; Brundin, P. Triggers, Facilitators, and Aggravators: Redefining Parkinson’s Disease Pathogenesis. Trends Neurosci. 2019, 42, 4–13. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reagent or Resource | Source | |
|---|---|---|
| Antibodies—IHC/IF | ||
| Mouse anti-GFAP | Santa Cruz Biotechnology (Dallas, TX, USA; cat. No. SC-71143) | |
| Rabbit anti-ZO-1 | Abcam (Cambridge, UK; cat. No. ab96587) | |
| Rabbit anti-tyrosine hydroxylase (TH) | Millipore (Burlington, MA, USA; cat. No. AB152) | |
| Rabbit anti-Iba1 | FUJIFILM Wako Chemicals (Neuss, Germany; cat. No. 019-19741) | |
| Mouse anti-β3Tubulin | Cell Signaling (Danvers, MA, USA; cat. No. 4466) | |
| Rabbit anti-Tom20 | Santa Cruz Biotechnology (Dallas, TX, USA; cat. No. SC-11415) | |
| Biotinylated anti-rabbit IgG | Vector Labs (Newark, CA, USA; cat. No. BA-1000) | |
| Goat anti-mouse Alexa Fluor 488 | Molecular Probes, Life Technologies (Waltham, MA, USA; cat. No. A11001) | |
| Goat anti-mouse Alexa Fluor 594 | Molecular Probes, Life Technologies (Waltham, MA, USA; cat. No. A11005) | |
| Goat anti-rabbit Alexa Fluor 594 | Molecular Probes, Life Technologies (Waltham, MA, USA; cat. No. A-11012) | |
| Goat anti-rabbit Alexa Fluor 488 | Molecular Probes, Life Technologies (Waltham, MA, USA; cat. No. A11008) | |
| Chemicals | ||
| Hoechst | Invitrogen (Waltham, MA, USA; cat. No. H1399) | |
| FITC-dextran sulfate sodium salt | Sigma-Aldrich (St. Louis, MO, USA; cat. No. 78331) | |
| DPX mountant | Sigma (St. Louis, MO, USA; cat. No. 06522-100 mL) | |
| Vectastain Elite ABC Perox standard kit | Vector Labs (Newark, CA, USA; cat. No. VCPK-6100) | |
| Normal goat serum | Abbkine (Georgia, USA; cat. No. BMS0050) | |
| MOM blocking reagent | Vector Labs (Newark, CA, USA; cat. No. MKB-2213-1) | |
| OCT mounting medium | Carl Roth (Karlsruhe, Germany; cat. No. KMA-0100-51A) | |
| Kits | Sensitivity | |
| Mouse IL-6 ELISA kit | R&D Systems (Minneapolis, MN, USA; cat. No. M6000B) | 1.3–1.8 pg/mL |
| α-Synuclein oligomer (SNCO α) ELISA kit | MyBioSource (San Diego, CA, USA; cat. No. MBS724099) | 1.0 pg/mL |
| Calprotectin kit | Invitrogen (Waltham, MA, USA; cat. EM67RB) | 0.65 ng/mL |
| Mouse TNF-α Quantikine ELISA | R&D Systems (Minneapolis, MN, USA; cat. No. MTA00B) | 0.36–7.21 pg/mL |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magalhães, J.D.; Candeias, E.; Melo-Marques, I.; Abreu, A.E.; Pereira-Santos, A.R.; Esteves, A.R.; Cardoso, S.M.; Empadinhas, N. Nocardia cyriacigeorgica Elicits Gut Disturbances in a Leaky Gut Model of Colitis, but Not the Harmful Cascade Leading to Gut-First Parkinson’s Disease. Int. J. Mol. Sci. 2024, 25, 3423. https://doi.org/10.3390/ijms25063423
Magalhães JD, Candeias E, Melo-Marques I, Abreu AE, Pereira-Santos AR, Esteves AR, Cardoso SM, Empadinhas N. Nocardia cyriacigeorgica Elicits Gut Disturbances in a Leaky Gut Model of Colitis, but Not the Harmful Cascade Leading to Gut-First Parkinson’s Disease. International Journal of Molecular Sciences. 2024; 25(6):3423. https://doi.org/10.3390/ijms25063423
Chicago/Turabian StyleMagalhães, João Duarte, Emanuel Candeias, Inês Melo-Marques, António E. Abreu, Ana Raquel Pereira-Santos, Ana Raquel Esteves, Sandra Morais Cardoso, and Nuno Empadinhas. 2024. "Nocardia cyriacigeorgica Elicits Gut Disturbances in a Leaky Gut Model of Colitis, but Not the Harmful Cascade Leading to Gut-First Parkinson’s Disease" International Journal of Molecular Sciences 25, no. 6: 3423. https://doi.org/10.3390/ijms25063423