
Comparative Analyses of Targeted Myeloid Cancer Next-Generation Sequencing Panel in Fresh Blood, Bone Marrow and FFPE Material


Abstract
:1. Introduction
2. Results
2.1. Assessment of Workflow Stability
2.2. Underperforming Amplicons in Fresh Bone Marrow and Blood Material
2.3. Underperforming Amplicons in FFPE Material
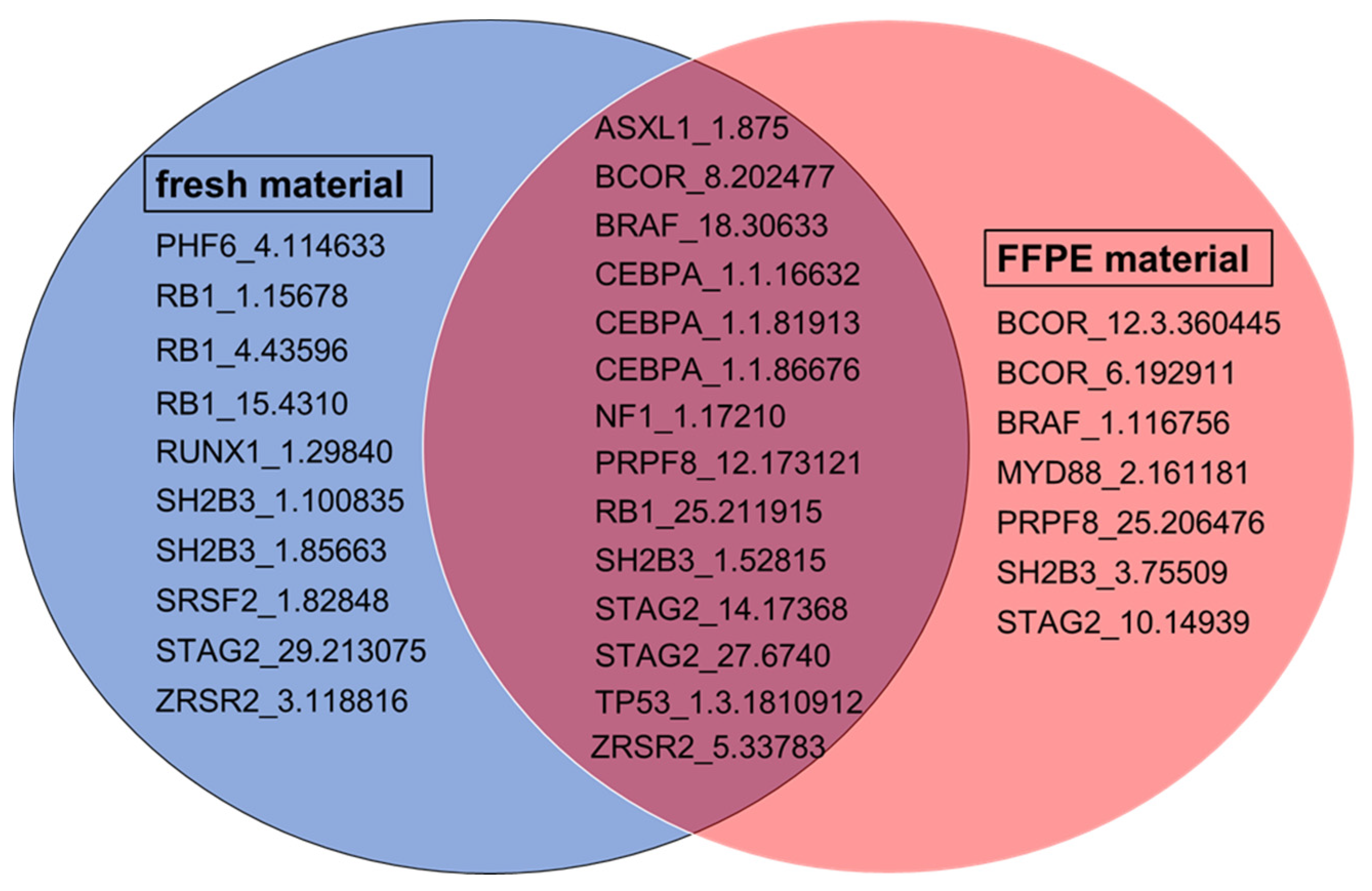
2.4. Comparison of Amplicon Performance in FFPE and Fresh Material
2.5. Clinical Relevance of Underperforming Amplicons
3. Discussion
3.1. Assessment of Workflow Stability
3.2. Comparison of Amplicon Performance in Fresh and FFPE Material
3.3. Sequencing Depth
4. Materials and Methods
4.1. Nucleic Acid Isolation and Quantification
4.2. Targeted Sequencing
4.3. Data Analysis
4.4. Assessment of Workflow Stability
4.5. Analysis of Underperforming Amplicons
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cazzola, M.; Della Porta, M.G.; Malcovati, L. The genetic basis of myelodysplasia and its clinical relevance. Blood 2013, 122, 4021–4034. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627; quiz 3699. [Google Scholar] [CrossRef]
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and management of AML in adults: 2022 Recommendations from an international expert panel on behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.-M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.N.; Bejar, R. MDS overlap disorders and diagnostic boundaries. Blood 2019, 133, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Lasho, T.L.; Finke, C.M.; Elala, Y.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Pardanani, A. Targeted deep sequencing in primary myelofibrosis. Blood Adv. 2016, 1, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Gerstung, M.; Papaemmanuil, E.; Martincorena, I.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Heuser, M.; Thol, F.; Bolli, N.; Ganly, P.; et al. Precision oncology for acute myeloid leukemia using a knowledge bank approach. Nat. Genet. 2017, 49, 332–340. [Google Scholar] [CrossRef]
- Yohe, S.; Thyagarajan, B. Review of Clinical Next-Generation Sequencing. Arch. Pathol. Lab. Med. 2017, 141, 1544–1557. [Google Scholar] [CrossRef]
- Kim, S.; Park, C.; Ji, Y.; Kim, D.G.; Bae, H.; van Vrancken, M.; Kim, D.-H.; Kim, K.-M. Deamination Effects in Formalin-Fixed, Paraffin-Embedded Tissue Samples in the Era of Precision Medicine. J. Mol. Diagn. 2017, 19, 137–146. [Google Scholar] [CrossRef]
- Do, H.; Dobrovic, A. Sequence artifacts in DNA from formalin-fixed tissues: Causes and strategies for minimization. Clin. Chem. 2015, 61, 64–71. [Google Scholar] [CrossRef]
- Xue, V.W.; Wong, C.S.C.; Cho, W.C.S. Early detection and monitoring of cancer in liquid biopsy: Advances and challenges. Expert Rev. Mol. Diagn. 2019, 19, 273–276. [Google Scholar] [CrossRef]
- Jumniensuk, C.; Nobori, A.; Lee, T.; Senaratne, T.N.; Rao, D.; Pullarkat, S. Concordance of Peripheral Blood and Bone Marrow Next-Generation Sequencing in Hematologic Neoplasms. Adv. Hematol. 2022, 2022, 8091746. [Google Scholar] [CrossRef]
- Lucas, F.; Michaels, P.D.; Wang, D.; Kim, A.S. Mutational analysis of hematologic neoplasms in 164 paired peripheral blood and bone marrow samples by next-generation sequencing. Blood Adv. 2020, 4, 4362–4365. [Google Scholar] [CrossRef]
- FRAENKEL-CONRAT, H.; OLCOTT, H.S. The reaction of formaldehyde with proteins; cross-linking between amino and primary amide or guanidyl groups. J. Am. Chem. Soc. 1948, 70, 2673–2684. [Google Scholar] [CrossRef]
- McGhee, J.D.; Hippel, P.H. von. Formaldehyde as a probe of DNA structure. r. Mechanism of the initial reaction of Formaldehyde with DNA. Biochemistry 1977, 16, 3276–3293. [Google Scholar] [CrossRef]
- Cappello, F.; Angerilli, V.; Munari, G.; Ceccon, C.; Sabbadin, M.; Pagni, F.; Fusco, N.; Malapelle, U.; Fassan, M. FFPE-Based NGS Approaches into Clinical Practice: The Limits of Glory from a Pathologist Viewpoint. J. Pers. Med. 2022, 12, 750. [Google Scholar] [CrossRef]
- McDonough, S.J.; Bhagwate, A.; Sun, Z.; Wang, C.; Zschunke, M.; Gorman, J.A.; Kopp, K.J.; Cunningham, J.M. Use of FFPE-derived DNA in next generation sequencing: DNA extraction methods. PLoS ONE 2019, 14, e0211400. [Google Scholar] [CrossRef] [PubMed]
- Kerick, M.; Isau, M.; Timmermann, B.; Sültmann, H.; Herwig, R.; Krobitsch, S.; Schaefer, G.; Verdorfer, I.; Bartsch, G.; Klocker, H.; et al. Targeted high throughput sequencing in clinical cancer settings: Formaldehyde fixed-paraffin embedded (FFPE) tumor tissues, input amount and tumor heterogeneity. BMC Med. Genom. 2011, 4, 68. [Google Scholar] [CrossRef] [PubMed]
- Jennings, L.J.; Arcila, M.E.; Corless, C.; Kamel-Reid, S.; Lubin, I.M.; Pfeifer, J.; Temple-Smolkin, R.L.; Voelkerding, K.V.; Nikiforova, M.N. Guidelines for Validation of Next-Generation Sequencing-Based Oncology Panels: A Joint Consensus Recommendation of the Association for Molecular Pathology and College of American Pathologists. J. Mol. Diagn. 2017, 19, 341–365. [Google Scholar] [CrossRef] [PubMed]
- Reed, S.C.; Croessmann, S.; Park, B.H. CHIP Happens: Clonal Hematopoiesis of Indeterminate Potential and Its Relationship to Solid Tumors. Clin. Cancer Res. 2023, 29, 1403–1411. [Google Scholar] [CrossRef]
- Luthra, R.; Patel, K.P.; Reddy, N.G.; Haghshenas, V.; Routbort, M.J.; Harmon, M.A.; Barkoh, B.A.; Kanagal-Shamanna, R.; Ravandi, F.; Cortes, J.E.; et al. Next-generation sequencing-based multigene mutational screening for acute myeloid leukemia using MiSeq: Applicability for diagnostics and disease monitoring. Haematologica 2014, 99, 465–473. [Google Scholar] [CrossRef]
- Yin, Y.; Butler, C.; Zhang, Q. Challenges in the application of NGS in the clinical laboratory. Hum. Immunol. 2021, 82, 812–819. [Google Scholar] [CrossRef]
- Tawana, K.; Wang, J.; Renneville, A.; Bödör, C.; Hills, R.; Loveday, C.; Savic, A.; van Delft, F.W.; Treleaven, J.; Georgiades, P.; et al. Disease evolution and outcomes in familial AML with germline CEBPA mutations. Blood 2015, 126, 1214–1223. [Google Scholar] [CrossRef] [PubMed]
- Grunwald, L.; Grosse-Thie, C.; Sender, S.; Knuebel, G.; Krohn, S.; Roolf, C.; Junghanss, C.; Henze, L.; Murua Escobar, H. Ultradeep targeted sequencing reveals low allele frequencies of somatic JAK2 and MPL variants in patients with abdominal vein thromboses: Results of an ongoing prospective prevalence study in Mecklenburg-West Pomerania. Biomark. Res. 2020, 8, 73. [Google Scholar] [CrossRef]
- Mackenzie, R.; Kommoss, S.; Winterhoff, B.J.; Kipp, B.R.; Garcia, J.J.; Voss, J.; Halling, K.; Karnezis, A.; Senz, J.; Yang, W.; et al. Targeted deep sequencing of mucinous ovarian tumors reveals multiple overlapping RAS-pathway activating mutations in borderline and cancerous neoplasms. BMC Cancer 2015, 15, 415. [Google Scholar] [CrossRef]
- Malapelle, U.; Vigliar, E.; Sgariglia, R.; Bellevicine, C.; Colarossi, L.; Vitale, D.; Pallante, P.; Troncone, G. Ion Torrent next-generation sequencing for routine identification of clinically relevant mutations in colorectal cancer patients. J. Clin. Pathol. 2015, 68, 64–68. [Google Scholar] [CrossRef]
- Tinhofer, I.; Budach, V.; Saki, M.; Konschak, R.; Niehr, F.; Jöhrens, K.; Weichert, W.; Linge, A.; Lohaus, F.; Krause, M.; et al. Targeted next-generation sequencing of locally advanced squamous cell carcinomas of the head and neck reveals druggable targets for improving adjuvant chemoradiation. Eur. J. Cancer 2016, 57, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.T.; Lee, J.; Hong, M.; Park, K.; Park, J.O.; Ahn, T.; Park, S.H.; Park, Y.S.; Lim, H.Y.; Sun, J.-M.; et al. The NEXT-1 (Next generation pErsonalized tX with mulTi-omics and preclinical model) trial: Prospective molecular screening trial of metastatic solid cancer patients, a feasibility analysis. Oncotarget 2015, 6, 33358–33368. [Google Scholar] [CrossRef]
- Au, C.H.; Wa, A.; Ho, D.N.; Chan, T.L.; Ma, E.S.K. Clinical evaluation of panel testing by next-generation sequencing (NGS) for gene mutations in myeloid neoplasms. Diagn. Pathol. 2016, 11, 11. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.; Riccitelli, N.; Pollner, R. Abstract 1693: Comparison of commercially available myeloid next-generation sequencing assays. Cancer Res. 2019, 79, 1693. [Google Scholar] [CrossRef]
- Haslam, K.; Catherwood, M.A.; Dobbin, E.; Sproul, A.; Langabeer, S.E.; Mills, K.I. Inter-Laboratory Evaluation of a Next-Generation Sequencing Panel for Acute Myeloid Leukemia. Mol. Diagn. Ther. 2016, 20, 457–461. [Google Scholar] [CrossRef]
- Chin, F.Y.L.; Leung, H.C.M.; Yiu, S.M. Sequence assembly using next generation sequencing data--challenges and solutions. Sci. China Life Sci. 2014, 57, 1140–1148. [Google Scholar] [CrossRef]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Estey, E.H. Acute myeloid leukemia: 2013 update on risk-stratification and management. Am. J. Hematol. 2013, 88, 318–327. [Google Scholar] [CrossRef]
- Chen, G.; Mosier, S.; Gocke, C.D.; Lin, M.-T.; Eshleman, J.R. Cytosine deamination is a major cause of baseline noise in next-generation sequencing. Mol. Diagn. Ther. 2014, 18, 587–593. [Google Scholar] [CrossRef]
- Mathieson, W.; Thomas, G.A. Why Formalin-fixed, Paraffin-embedded Biospecimens Must Be Used in Genomic Medicine: An Evidence-based Review and Conclusion. J. Histochem. Cytochem. 2020, 68, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Bartels, S.; Schipper, E.; Kreipe, H.H.; Lehmann, U. Comprehensive Molecular Profiling of Archival Bone Marrow Trephines Using a Commercially Available Leukemia Panel and Semiconductor-Based Targeted Resequencing. PLoS ONE 2015, 10, e0133930. [Google Scholar] [CrossRef] [PubMed]
- Aird, D.; Ross, M.G.; Chen, W.-S.; Danielsson, M.; Fennell, T.; Russ, C.; Jaffe, D.B.; Nusbaum, C.; Gnirke, A. Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol. 2011, 12, R18. [Google Scholar] [CrossRef] [PubMed]
- Kozarewa, I.; Ning, Z.; Quail, M.A.; Sanders, M.J.; Berriman, M.; Turner, D.J. Amplification-free Illumina sequencing-library preparation facilitates improved mapping and assembly of (G+C)-biased genomes. Nat. Method. 2009, 6, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Puchtler, T.J.; Johnson, K.; Palmer, R.N.; Talbot, E.L.; Ibbotson, L.A.; Powalowska, P.K.; Knox, R.; Shibahara, A.; Cunha, P.M.S.; Newell, O.J.; et al. Single-molecule DNA sequencing of widely varying GC-content using nucleotide release, capture and detection in microdroplets. Nucleic Acids Res. 2020, 48, e132. [Google Scholar] [CrossRef] [PubMed]
- Minervini, C.F.; Cumbo, C.; Orsini, P.; Anelli, L.; Zagaria, A.; Specchia, G.; Albano, F. Nanopore Sequencing in Blood Diseases: A Wide Range of Opportunities. Front. Genet. 2020, 11, 76. [Google Scholar] [CrossRef] [PubMed]
- Mouradov, D.; Sloggett, C.; Jorissen, R.N.; Love, C.G.; Li, S.; Burgess, A.W.; Arango, D.; Strausberg, R.L.; Buchanan, D.; Wormald, S.; et al. Colorectal cancer cell lines are representative models of the main molecular subtypes of primary cancer. Cancer Res. 2014, 74, 3238–3247. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Sample | Material | Number of Total Variant Calls | Number of Differing Variant Calls |
|---|---|---|---|
| 1 | Fresh | 65 | 14 |
| 2 | Fresh | 69 | 20 |
| 3 | Fresh | 57 | 11 |
| 4 | FFPE | 61 | 11 |
| 5 | FFPE | 73 | 23 |
| 6 | FFPE | 62 | 14 |
| Sample Material | Amplicons with Differing Variant Calls | ||
|---|---|---|---|
| Fresh material | ASXL1_13.3.346675, | ASXL1_13.3.496068, | ASXL1_6.4574, |
| ASXL1_7.4911, | CBL_2.191578, | FLT3_14.216122, | |
| FLT3_14.74296, | IKZF1_3.61448, | MYD88_1.157213, | |
| NF1_13.8604, | NF1_14.128695, | NF1_26.236704, | |
| NF1_38.255547, | NF1_7.195821, | NPM1_1.84536, | |
| PRPF8_38.455070, | RB1_15.4310, | RB1_22.8039, | |
| RB1_25.211915, | RUNX1_3.80512, | RUNX1_9.14780, | |
| SF3B1_2.54904, | SF3B1_3.232163, | SF3B1_4.60700, | |
| SH2B3_1.100835, | SH2B3_3.75509, | STAG2_13.37196, | |
| STAG2_15.14340, | TET2_9.1.839229, | TP53_10.202027, | |
| TP53_10.97990 | |||
| FFPE material | ASXL1_13.1.451988, | BCOR_1.134332, | CBL_2.191578, |
| CEBPA_1.1.16632, | EZH2_5.133393, | EZH2_15.42505, | |
| EZH2_17.30704, | HRAS_2.41391, | KIT_17.49555, | |
| MYD88_1.157213, | NF1_26.236704, | NF1_38.255547, | |
| NPM1_1.84536, | PRPF8_25.206476, | PRPF8_38.455070, | |
| RB1_3.18575, | RB1_4.43596, | RB1_22.8039, | |
| RB1_15.4310, | RB1_17.165046, | RUNX1_239.65068, | |
| RUNX1_1.136369, | RUNX1_1.29840, | SF3B1_4.60700, | |
| RUNX1_7.76102, | SF3B1_3.232163, | SH2B3_7.132396, | |
| SH2B3_1.100835, | SH2B3_3.75509, | STAG2_18.267240, | |
| STAG2_10.14939, | STAG2_10.43800, | TP53_9.99685, | |
| TP53_1.3.1810912, | TP53_8.616, | ZRSR2_5.33783 | |
| TP53_10.202027, | WT1_2.145305, | ||
| Category | ||||
|---|---|---|---|---|
| 0 | I | II | III | IV |
| Amplicons underperforming in percentage of all samples (n = 55) | ||||
| 1–9% | 10–25% | 26–50% | 51–75% | 76–100% |
| Amplicons | ||||
| BCOR_6.192911 | PHF6_4.114633 | RB1_4.43596 | CEBPA_1.1.16632 | ASXL1_1.875 |
| BCOR_10.16915 | RB1_1.15678 | RB1_25.211915 | CEBPA_1.1.81913 | BCOR_8.202477 |
| JAK2_4.68202 | RB1_15.4310 | RUNX1_1.29840 | CEBPA_1.1.86676 | BRAF_18.30633 |
| NF1_55.95970 | SH2B3_1.100835 | TP53_1.3.1810912 | NF1_1.17210 | PRPF8_12.173121 |
| NPM1_1.84536 | SRSF2_1.82848 | SH2B3_1.85663 | SH2B3_1.52815 | |
| PHF6_2.86405 | STAG2_14.17368 | |||
| PHF6_6.45032 | STAG2_27.6740 | |||
| PRPF8_23.351225 | STAG2_29.213075 | |||
| PRPF8_25.206476 | ZRSR2_3.118816 | |||
| PRPF8_42.75713 | ZRSR2_5.33783 | |||
| RB1_6.27164 | ||||
| RB1_9.72315 | ||||
| SF3B1_5.8195 | ||||
| SH2B3_1.49539 | ||||
| STAG2_6.223906 | ||||
| STAG2_9.168558 | ||||
| STAG2_10.14939 | ||||
| STAG2_10.43800 | ||||
| STAG2_15.14340 | ||||
| STAG2_26.25139 | ||||
| STAG2_33.62661 | ||||
| TET2_3.23136 | ||||
| TP53_1.3.1590469 | ||||
| ZRSR2_7.7640 | ||||
| ZRSR2_11.253014 | ||||
| Category | |||
|---|---|---|---|
| I | II | III | IV |
| Amplicons underperforming in percentage of all samples (n = 59) | |||
| 10–25% | 26–50% | 51–75% | 76–100% |
| Amplicons | |||
| BCOR_6.192911 | BCOR_12.3.360445 | PRPF8_25.206476 | ASXL1_1.875 |
| BRAF_1.116756 | CEBPA_1.1.86676 | BCOR_8.202477 | |
| CEBPA_1.1.16632 | BRAF_18.30633 | ||
| MYD88_2.161181 | CEBPA_1.1.81913 | ||
| NF1_1.17210 | PRPF8_12.173121 | ||
| SH2B3_1.52815 | RB1_25.211915 | ||
| SH2B3_3.75509 | STAG2_27.6740 | ||
| STAG2_10.14939 | ZRSR2_5.33783 | ||
| STAG2_14.17368 | |||
| TP53_1.3.1810912 | |||
| Clinical Relevance. | Clinically Relevant in Cancer Diseases with Hematopoietic/Lymphoid Origin and Solid Cancer Types [n = 23] | Clinically Relevant Solely in Solid Cancer Types [n = 5] | No Clinical Relevance Known to Date [n = 3] |
|---|---|---|---|
| Amplicons | BCOR_6.192911 | BRAF_1.116756 | ASXL1_1.875 |
| BCOR_8.202477 | PRPF8_12.173121 | BRAF_18.30633 | |
| BCOR_12.3.360445 | PRPF8_25.206476 | NF1_1.17210 | |
| CEBPA_1.1.16632 | RB1_4.43596 | ||
| CEBPA_1.1.81913 | TP53_1.3.1810912 | ||
| CEBPA_1.1.86676 | |||
| MYD88_2.161181 | |||
| PHF6_4.114633 | |||
| RB1_1.15678 | |||
| RB1_15.4310 | |||
| RB1_25.211915 | |||
| RUNX1_1.29840 | |||
| SH2B3_1.100835 | |||
| SH2B3_1.52815 | |||
| SH2B3_1.85663 | |||
| SH2B3_3.75509 | |||
| SRSF2_1.82848 | |||
| STAG2_10.14939 | |||
| STAG2_14.17368 | |||
| STAG2_27.6740 | |||
| STAG2_29.213075 | |||
| ZRSR2_3.118816 | |||
| ZRSR2_5.33783 |
| Performance Categories | Amplicons Underperforming in Percentage of All Samples |
|---|---|
| Category 0 | 1 to 9% |
| Category I | 10 to 25% |
| Category II | 26 to 50% |
| Category III | 51 to 75% |
| Category IV | 76 to 100% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hobeck, A.D.; Wendt, S.; Krohn, S.; Knuebel, G.; Bartels, S.; Schipper, E.; Junghanss, C.; Murua Escobar, H. Comparative Analyses of Targeted Myeloid Cancer Next-Generation Sequencing Panel in Fresh Blood, Bone Marrow and FFPE Material. Int. J. Mol. Sci. 2024, 25, 3534. https://doi.org/10.3390/ijms25063534
Hobeck AD, Wendt S, Krohn S, Knuebel G, Bartels S, Schipper E, Junghanss C, Murua Escobar H. Comparative Analyses of Targeted Myeloid Cancer Next-Generation Sequencing Panel in Fresh Blood, Bone Marrow and FFPE Material. International Journal of Molecular Sciences. 2024; 25(6):3534. https://doi.org/10.3390/ijms25063534
Chicago/Turabian StyleHobeck, Andrea Daniela, Sophia Wendt, Saskia Krohn, Gudrun Knuebel, Stephan Bartels, Elisa Schipper, Christian Junghanss, and Hugo Murua Escobar. 2024. "Comparative Analyses of Targeted Myeloid Cancer Next-Generation Sequencing Panel in Fresh Blood, Bone Marrow and FFPE Material" International Journal of Molecular Sciences 25, no. 6: 3534. https://doi.org/10.3390/ijms25063534