
Extracellular Vesicle Protein Expression in Doped Bioactive Glasses: Further Insights Applying Anomaly Detection
 ,
,  , ,
, ,  , and
, and
Abstract
:1. Introduction
1.1. Notes on Small Sample Inference
1.2. Observations on Distance Functions
1.3. Remark for Statistical Analysis on Primary Cell Data
1.4. Aim of the Study
- The procedure is studied for small-dimensional datasets
- The procedure should support researchers in obtaining additional information on the data under analysis, marking expression levels in actual sample space rather than considering average values
- It leverages Euclidean geometry isomorphism, taking into account the magnitude and direction of the protein level changes
- It exploits existing and already verified methods for novel and augmented interpretation of the experimental variables
- Verify the procedure on data coming from primary cells directly collected from donors
1.5. Proteomics of Extracellular Vesicles
1.6. Biomaterials Dataset Analyzed for Protein Anomaly Detection
2. Results
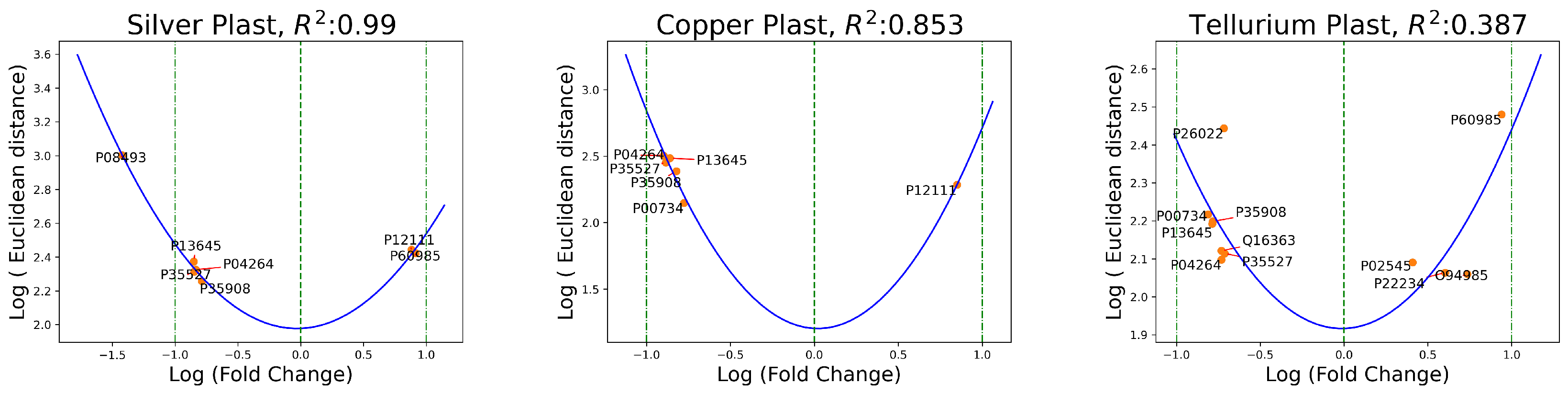
2.1. Relation between Euclidean Distances and Fold Change
2.2. Within-Group Variation
3. Discussion
3.1. Biological Interpretation of the Outcomes
3.2. Limitations of the Study
4. Materials and Methods
4.1. Bioactive Glasses Preparation
4.2. Mesenchymal Stem/Stromal Cell Isolation
4.3. Wet-Lab Experimental Conditions
- cell cultures on “SBA2”, “SBA3”, and “ST” modified bioactive glasses doped with silver, copper, and tellurium, respectively (i.e., doped).
- cell culture on “Plastic”, a baseline condition without the presence of biomaterials (i.e., plast)
4.4. Mass Spectrum Summary
4.5. Computational Resources
4.6. Dry-Lab Experimental Sequence
- The raw values from the three donors were log-transformed
- The log-transformed values were clustered, and the values of the same cluster were taken to ensure analysis of similar data representing the same biological phenomena
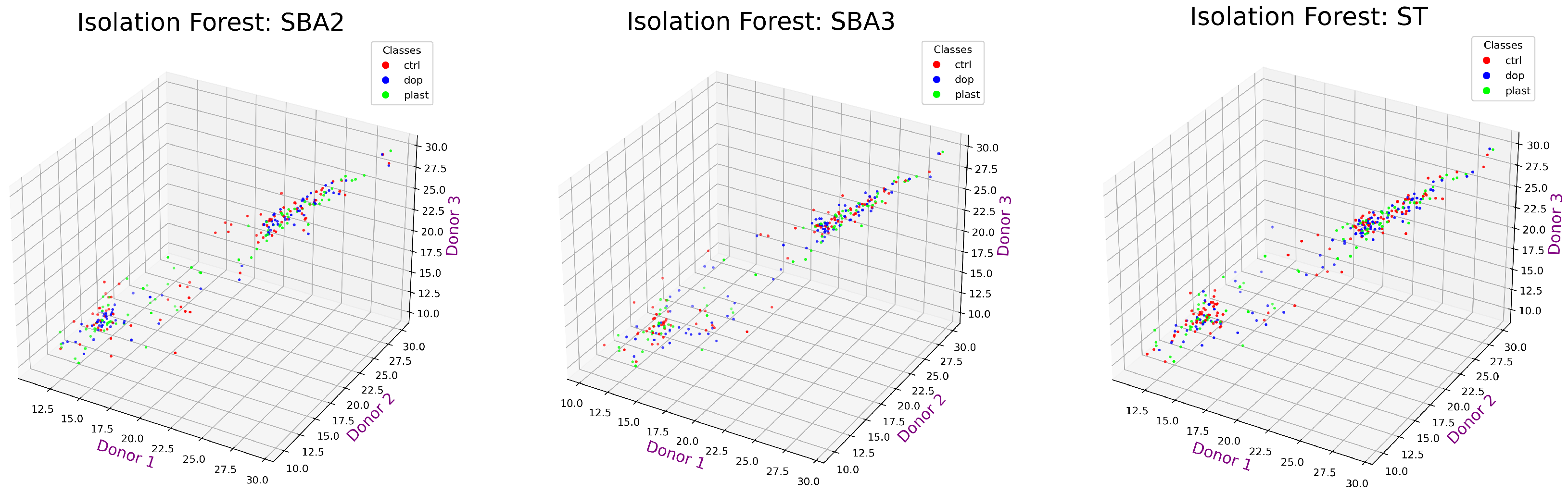
- Each value was labeled as outlier (potential “anomaly” or extreme variation) or not applying Isolation Forest
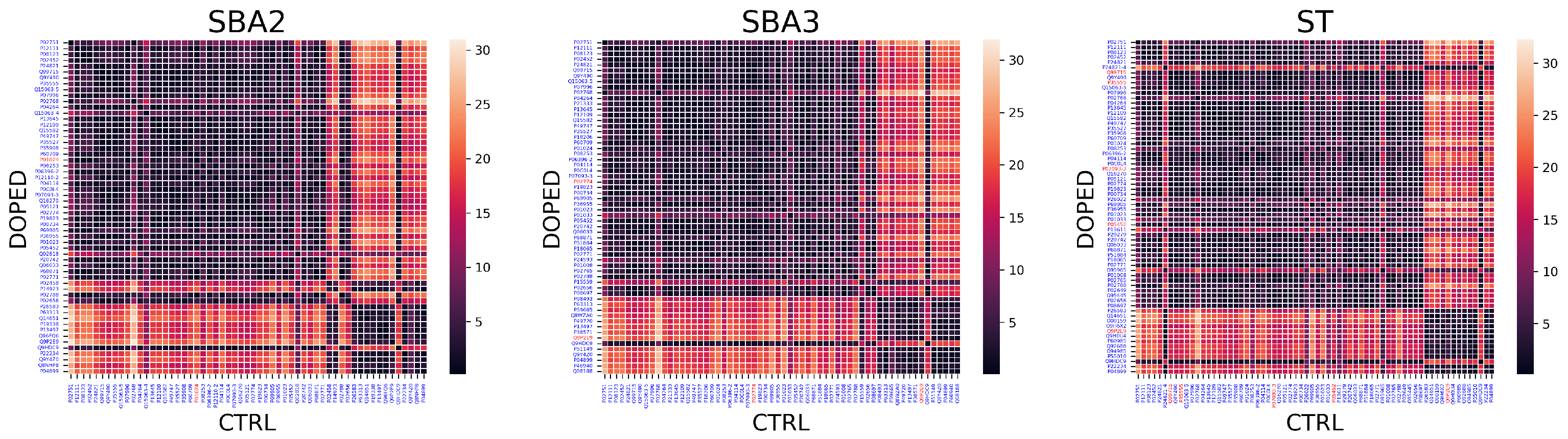
- Computed the distance between outlier proteins in the donors’ 3D space to identify abnormal variations in the EV-related protein expression
4.6.1. Proposed Sequence: Log-Transformation Preprocessing
4.6.2. Proposed Sequence: Clustering
4.6.3. Proposed Sequence: Outlier Detection by Isolation Forest
4.6.4. Proposed Sequence: Distance Metric
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CPU | Central Processing Unit |
| Ctrl | Control bioactive glasses |
| CV | Coefficient of Variation |
| BMP | Bone Morphogenetic Proteins |
| DMEM | Dulbecco’s Modified Essential Medium |
| EV | Extracellular Vesicles |
| ISCT | International Society for Cell and Gene Therapy |
| ML | Machine Learning |
| MSC | Mesenchymal Stem Cells |
| MS | Mass Spectrometry |
| Plast | Experimental condition on Plastic material (baseline) |
| RAM | Random Access Memory |
| SBA2 | Experimental setting involving bioactive glass doped or not with silver |
| SBA3 | Experimental setting involving bioactive glass doped or not with copper |
| ST | Experimental setting involving bioactive glass doped or not with tellurium |
Appendix A. Theoretical Proteomic Scenario
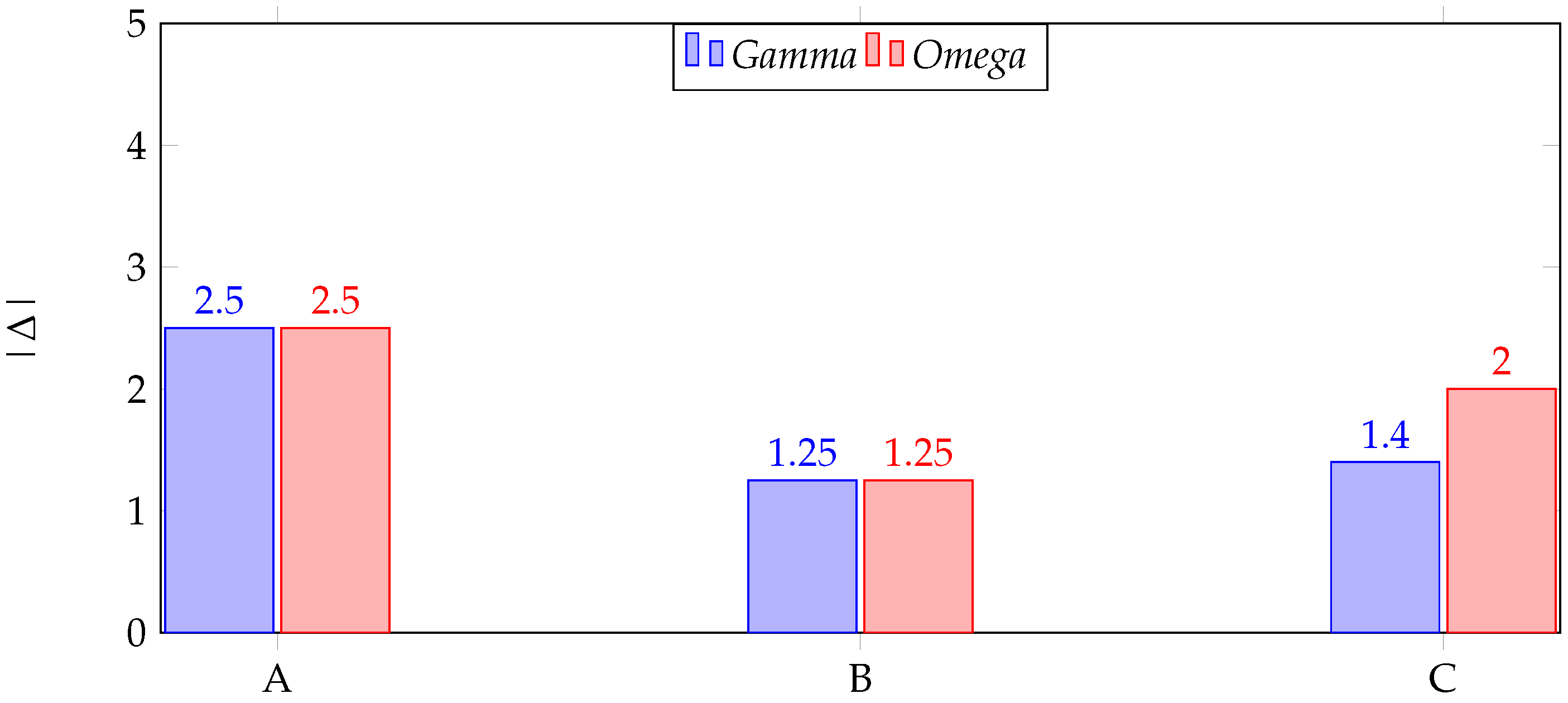
Appendix A.1. A Toy Example on Proteomics’ Small Sample Dataset

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Donor | Exp. 1 | Exp. 2 | Δ 1 | |
|---|---|---|---|---|
| A | 1 | 3.5 | 2.5 | |
| B | 2 | 3.25 | 1.25 | |
| C | 1.5 | 2.9 | 1.4 | |
| 1.5 | 3.2 | 1.71 | Mean (μ) | |
| 0.5 | 0.3 | 0.68 | St. Dev. (σ) |

| Donor | Exp. 1 | Exp. 2 | Δ 1 | |
|---|---|---|---|---|
| A | 1 | 3.5 | 2.5 | |
| B | 2 | 3.25 | 1.25 | |
| C | 2.5 | 0.5 | −2 | |
| 1.83 | 2.41 | 0.58 | Mean (μ) | |
| 0.76 | 1.66 | 2.32 | St. Dev. (σ) |


Appendix A.2. Proposed Alternative Data Evaluation Method

Appendix B. Additional Definitions and Images
Appendix B.1. Mass Spectrometry
Appendix B.2. Outliers
- outliers impacting the entire dataset and characterized by extreme values that deviate from the overall pattern of the data. They can significantly affect summary statistics and models being usually referenced as global outliers
- points considered unusual or extreme within a specific subgroup or context may not be outliers when the entire dataset is considered. Detecting these types of points requires considering subsets of the data, and are called contextual outliers.
- Positional outliers are outliers that deviate from the typical position within a distribution. These are identified based on their location in relation to measures such as the mean, median, or quartiles.
- Outliers that cause the distribution of the data to be skewed; they can have a substantial impact on the shape of the distribution, making it asymmetrical (aka skewed outliers).
- Groups of observations that collectively deviate from the expected pattern may not be identified when examining individual data points but become apparent when considering groups of observations (aka collective outliers).
- In multivariate analysis, observations that deviate from the overall pattern in a multidimensional space may be considered outliers.
- Masked outliers are not immediately apparent in univariate analysis but become noticeable when considering interactions or relationships between variables. These outliers may affect the results of statistical models.
- Unexpected events or changes in the underlying process generating the data might produce novel occurrences that could be marked as outliers (i.e., innovational outliers).
- Points with values higher or lower than the majority of the data significantly impact the mean and standard deviation (i.e., additive outliers).
Appendix B.3. Protein Expression Points Selected by Clustering

Appendix B.4. Matrices of Euclidean Distances


Appendix B.5. Mean Variability in Each Experiment
| Biomaterial | Mean Var. Ctrl | Mean Var. Doped | Mean Abs Diff. |
|---|---|---|---|
| SBA2 | 5.22 | 1.17 | 4.54 |
| SBA3 | 1.91 | 1.3 | 1.66 |
| ST | 0.59 | 1.36 | 0.89 |
| Biomaterial | Mean Var. Plast | Mean Var. Doped | Mean Abs Diff. |
| SBA2 | 0.43 | 0.51 | 0.34 |
| SBA3 | 0.15 | 0.53 | 0.46 |
| ST | 0.65 | 0.56 | 0.68 |
| Biomaterial | Mean CV Ctrl | Mean CV Doped | Mean Abs Diff. |
|---|---|---|---|
| SBA2 | 17.563 | 8.594 | 8.969 |
| SBA3 | 8.442 | 8.394 | 0.048 |
| ST | 5.388 | 7.591 | 2.203 |
| Biomaterial | Mean CV Plast | Mean CV Doped | Mean Abs Diff. |
| SBA2 | 3.1 | 4.271 | 1.171 |
| SBA3 | 1.555 | 3.485 | 1.929 |
| ST | 4.359 | 4.262 | 0.097 |
Appendix C. Gene Ontology Enrichment Analysis
| Ontology | Accession | GO Class |
|---|---|---|
| Biological Process | - | - |
| Molecular Function | GO:0042731 | PH domain binding |
| Cellular Component | GO:0001533 | Cornified envelope |
| GO:0099080 | Supramolecular complex |
| Ontology | Accession | GO Class |
|---|---|---|
| Biological Process | GO:0071830 | triglyceride-rich lipoprotein particle clearance |
| Molecular Function | - | - |
| Cellular Component | - | - |
| Ontology | Accession | GO Class |
|---|---|---|
| Biological Process | - | - |
| Molecular Function | - | - |
| Cellular Component | GO:0099535 | synapse-associated extracellular matrix |
| GO:0005788 | endoplasmic reticulum lumen |
| Ontology | Accession | GO Class |
|---|---|---|
| Biological Process | GO:0051291 | protein hetero-oligomerization |
| GO:0018149 | peptide cross-linking | |
| GO:0045684 | positive regulation of epidermis development | |
| GO:0045103 | intermediate filament-based process | |
| GO:0097435 | supramolecular fiber organization | |
| GO:0009888 | tissue development | |
| GO:0030855 | epithelial cell differentiation | |
| GO:0048513 | organogenesis | |
| Molecular Function | GO:0005198 | structural molecule activity |
| Cellular Component | GO:0001533 | cornified envelope |
| GO:0099080 | supramolecular complex | |
| GO:0045111 | intermediate filament cytoskeleton | |
| GO:0062023 | collagen-containing extracellular matrix | |
| GO:1903561 | extracellular vesicle | |
| GO:0005615 | extracellular space |
| Ontology | Accession | GO class |
|---|---|---|
| Biological Process | GO:0051291 | protein hetero-oligomerization |
| GO:0018149 | peptide cross-linking | |
| GO:0042730 | fibrinolysis | |
| GO:0045684 | positive regulation of epidermis development | |
| GO:0045103 | cytoskeleton intermediate filament process | |
| GO:0097435 | supramolecular fiber organization | |
| GO:0008544 | epidermis development | |
| GO:0030855 | epithelial cell differentiation | |
| GO:0043588 | skin development | |
| Molecular Function | GO:0005198 | structural molecule activity |
| Cellular Component | GO:0001533 | cornified envelope |
| GO:0099080 | macromolecular complex | |
| GO:0045111 | intermediate filament cytoskeleton | |
| GO:0005788 | endoplasmic reticulum lumen | |
| GO:0062023 | collagen-containing extracellular matrix | |
| GO:0043230 | extracellular organelle | |
| GO:0031982 | extracellular vesicle | |
| GO:0005615 | extracellular space |
| Ontology | Accession | GO class |
|---|---|---|
| Biological Process | GO:0018149 | peptide cross-linking |
| GO:0045103 | cytoskeleton intermediate filament-based process | |
| GO:0008544 | epidermis development | |
| Molecular Function | GO:0005198 | structural molecule activity |
| Cellular Component | GO:0001533 | cornified envelope |
| GO:0099081 | supramolecular polymer | |
| GO:0045111 | intermediate filament cytoskeleton | |
| GO:0030312 | external encapsulating structure | |
| GO:0043230 | extracellular organelle | |
| GO:0031982 | extracellular vesicle | |
| GO:0005615 | extracellular space |
References
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Žídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef]
- Vishnoi, S.; Matre, H.; Garg, P.; Pandey, S.K. Artificial intelligence and machine learning for protein toxicity prediction using proteomics data. Chem. Biol. Drug Des. 2020, 96, 902–920. [Google Scholar] [CrossRef]
- Manes, N.P.; Song, J.; Nita-Lazar, A. EnsMOD: A Software Program for Omics Sample Outlier Detection. J. Comput. Biol. 2023, 30, 726–735. [Google Scholar] [CrossRef]
- Czibula, G.; Codre, C.; Teletin, M. AnomalP: An approach for detecting anomalous protein conformations using deep autoencoders. Expert Syst. Appl. 2021, 166, 114070. [Google Scholar] [CrossRef]
- Buck, L.; Schmidt, T.; Feist, M.; Schwarzfischer, P.; Kube, D.; Oefner, P.J.; Zacharias, H.U.; Altenbuchinger, M.; Dettmer, K.; Gronwald, W.; et al. Anomaly detection in mixed high-dimensional molecular data. Bioinformatics 2023, 39, btad501. [Google Scholar] [CrossRef]
- Shetta, O.; Niranjan, M. Robust subspace methods for outlier detection in genomic data circumvents the curse of dimensionality. R. Soc. Open Sci. 2020, 7, 190714. [Google Scholar] [CrossRef]
- Hu, G.; Di Paola, L.; Pullara, F.; Liang, Z.; Nookaew, I. Network proteomics: From protein structure to protein-protein interaction. BioMed Res. Int. 2017, 2017, 8929613. [Google Scholar] [CrossRef]
- Han, S.; Hu, X.; Huang, H.; Jiang, M.; Zhao, Y. Adbench: Anomaly detection benchmark. Adv. Neural Inf. Process. Syst. 2022, 35, 32142–32159. [Google Scholar] [CrossRef]
- Wolski, W.E.; Nanni, P.; Grossmann, J.; d’Errico, M.; Schlapbach, R.; Panse, C. Prolfqua: A comprehensive R-package for Proteomics Differential Expression Analysis. J. Proteome Res. 2023, 22, 1092–1104. [Google Scholar] [CrossRef]
- Kim, S.R.; Nguyen, T.V.; Seo, N.R.; Jung, S.; An, H.J.; Mills, D.A.; Kim, J.H. Comparative proteomics: Assessment of biological variability and dataset comparability. BMC Bioinform. 2015, 16, 121. [Google Scholar] [CrossRef]
- Käll, L.; Vitek, O. Computational mass spectrometry—Based proteomics. PLoS Comput. Biol. 2011, 7, e1002277. [Google Scholar] [CrossRef]
- Jafari, M.; Ansari-Pour, N. Why, when and how to adjust your P values? Cell J. 2019, 20, 604. [Google Scholar]
- Gao, J. P-values–A chronic conundrum. BMC Med. Res. Methodol. 2020, 20, 167. [Google Scholar] [CrossRef]
- Balding, D.J. A tutorial on statistical methods for population association studies. Nat. Rev. Genet. 2006, 7, 781–791. [Google Scholar] [CrossRef]
- Sullivan, G.M.; Feinn, R. Using effect size—Or why the P value is not enough. J. Grad. Med. Educ. 2012, 4, 279–282. [Google Scholar] [CrossRef]
- Held, L.; Ott, M. How the maximal evidence of p-values against point null hypotheses depends on sample size. Am. Stat. 2016, 70, 335–341. [Google Scholar] [CrossRef]
- Huopaniemi, I.; Suvitaival, T.; Nikkilä, J.; Orešič, M.; Kaski, S. Multivariate multi-way analysis of multi-source data. Bioinformatics 2010, 26, i391–i398. [Google Scholar] [CrossRef]
- Biau, D.J.; Kernéis, S.; Porcher, R. Statistics in brief: The importance of sample size in the planning and interpretation of medical research. Clin. Orthop. Relat. Res. 2008, 466, 2282–2288. [Google Scholar] [CrossRef]
- Button, K.S.; Ioannidis, J.P.; Mokrysz, C.; Nosek, B.A.; Flint, J.; Robinson, E.S.; Munafò, M.R. Power failure: Why small sample size undermines the reliability of neuroscience. Nat. Rev. Neurosci. 2013, 14, 365–376. [Google Scholar] [CrossRef]
- Nakagawa, S.; Cuthill, I.C. Effect size, confidence interval and statistical significance: A practical guide for biologists. Biol. Rev. 2007, 82, 591–605. [Google Scholar] [CrossRef]
- Ranstam, J. Why the p-value culture is bad and confidence intervals a better alternative. Osteoarthr. Cartil. 2012, 20, 805–808. [Google Scholar] [CrossRef]
- Holmes, S.H.; Huber, W. Modern Statistics for Modern Biology; Cambridge University Press: Cambridge, UK, 2018. [Google Scholar]
- Jakobsen, J.C.; Gluud, C.; Winkel, P.; Lange, T.; Wetterslev, J. The thresholds for statistical and clinical significance–a five-step procedure for evaluation of intervention effects in randomised clinical trials. BMC Med. Res. Methodol. 2014, 14, 34. [Google Scholar] [CrossRef]
- Escalante, H.J. A comparison of outlier detection algorithms for machine learning. In Proceedings of the International Conference on Communications in Computing, New York, NY, USA, 22–26 August 2005; pp. 228–237. [Google Scholar]
- Ur Rehman, A.; Belhaouari, S.B. Unsupervised outlier detection in multidimensional data. J. Big Data 2021, 8, 80. [Google Scholar] [CrossRef]
- Boukerche, A.; Zheng, L.; Alfandi, O. Outlier detection: Methods, models, and classification. ACM Comput. Surv. 2020, 53, 1–37. [Google Scholar] [CrossRef]
- De Winter, J.C. Using the Student’s t-test with extremely small sample sizes. Pract. Assess. Res. Eval. 2019, 18, 10. [Google Scholar]
- Rusticus, S.A.; Lovato, C.Y. Impact of sample size and variability on the power and type I error rates of equivalence tests: A simulation study. Pract. Assess. Res. Eval. 2019, 19, 11. [Google Scholar]
- Serdar, C.C.; Cihan, M.; Doğan Yücel, M.A.S. Sample size, power and effect size revisited: Simplified and practical approaches in pre-clinical, clinical and laboratory studies. Biochem. Med. 2021, 31, 010502. [Google Scholar] [CrossRef]
- Ranganathan, P.; Pramesh, C.; Buyse, M. Common pitfalls in statistical analysis: Clinical versus statistical significance. Perspect. Clin. Res. 2015, 6, 169. [Google Scholar] [CrossRef]
- Amess, B.; Kluge, W.; Schwarz, E.; Haenisch, F.; Alsaif, M.; Yolken, R.H.; Leweke, F.M.; Guest, P.C.; Bahn, S. Application of meta-analysis methods for identifying proteomic expression level differences. Proteomics 2013, 13, 2072–2076. [Google Scholar] [CrossRef]
- Grimes, M.L.; Lee, W.J.; Van der Maaten, L.; Shannon, P. Wrangling phosphoproteomic data to elucidate cancer signaling pathways. PLoS ONE 2013, 8, e52884. [Google Scholar] [CrossRef] [PubMed]
- Byron, A.; Griffith, B.G.; Herrero, A.; Loftus, A.E.; Koeleman, E.S.; Kogerman, L.; Dawson, J.C.; McGivern, N.; Culley, J.; Grimes, G.R.; et al. Characterisation of a nucleo-adhesome. Nat. Commun. 2022, 13, 3053. [Google Scholar] [CrossRef]
- Grimes, M.; Hall, B.; Foltz, L.; Levy, T.; Rikova, K.; Gaiser, J.; Cook, W.; Smirnova, E.; Wheeler, T.; Clark, N.R.; et al. Integration of protein phosphorylation, acetylation, and methylation data sets to outline lung cancer signaling networks. Sci. Signal. 2018, 11, eaaq1087. [Google Scholar] [CrossRef]
- Ross, K.E.; Zhang, G.; Akcora, C.; Lin, Y.; Fang, B.; Koomen, J.; Haura, E.B.; Grimes, M. Network models of protein phosphorylation, acetylation, and ubiquitination connect metabolic and cell signaling pathways in lung cancer. PLoS Comput. Biol. 2023, 19, e1010690. [Google Scholar] [CrossRef]
- Rieder, V.; Blank-Landeshammer, B.; Stuhr, M.; Schell, T.; Biß, K.; Kollipara, L.; Meyer, A.; Pfenninger, M.; Westphal, H.; Sickmann, A.; et al. DISMS2: A flexible algorithm for direct proteome-wide distance calculation of LC-MS/MS runs. BMC Bioinform. 2017, 18, 148. [Google Scholar] [CrossRef]
- Baker, M. Reproducibility: Respect your cells! Nature 2016, 537, 433–435. [Google Scholar] [CrossRef]
- Stoddart, M.; Richards, R.; Alini, M. In vitro experiments with primary mammalian cells: To pool or not to pool? Eur. Cells Mater. 2012, 24, i–ii. [Google Scholar] [CrossRef]
- Rowe, A. Recommendations to improve use and reporting of statistics in animal experiments. Lab. Anim. 2022, 57, 224–235. [Google Scholar] [CrossRef]
- Selicato, L.; Esposito, F.; Gargano, G.; Vegliante, M.C.; Opinto, G.; Zaccaria, G.M.; Ciavarella, S.; Guarini, A.; Del Buono, N. A new ensemble method for detecting anomalies in gene expression matrices. Mathematics 2021, 9, 882. [Google Scholar] [CrossRef]
- Claridge, B.; Lozano, J.; Poh, Q.H.; Greening, D.W. Development of extracellular vesicle therapeutics: Challenges, considerations, and opportunities. Front. Cell Dev. Biol. 2021, 9, 734720. [Google Scholar] [CrossRef]
- Ren, Y.; Ge, K.; Sun, D.; Hong, Z.; Jia, C.; Hu, H.; Shao, F.; Yao, B. Rapid enrichment and sensitive detection of extracellular vesicles through measuring the phospholipids and transmembrane protein in a microfluidic chip. Biosens. Bioelectron. 2022, 199, 113870. [Google Scholar] [CrossRef]
- Lischnig, A.; Bergqvist, M.; Ochiya, T.; Lässer, C. Quantitative proteomics identifies proteins enriched in large and small extracellular vesicles. Mol. Cell. Proteom. 2022, 21, 100273. [Google Scholar] [CrossRef]
- Clemmens, H.; Lambert, D.W. Extracellular vesicles: Translational challenges and opportunities. Biochem. Soc. Trans. 2018, 46, 1073–1082. [Google Scholar] [CrossRef]
- Morales-Sanfrutos, J.; Munoz, J. Unraveling the complexity of the extracellular vesicle landscape with advanced proteomics. Expert Rev. Proteom. 2022, 19, 89–101. [Google Scholar] [CrossRef]
- Van Niel, G.; d’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Abreu, H.; Canciani, E.; Raineri, D.; Cappellano, G.; Rimondini, L.; Chiocchetti, A. Extracellular vesicles in musculoskeletal regeneration: Modulating the therapy of the future. Cells 2021, 11, 43. [Google Scholar] [CrossRef]
- Qin, Y.; Wang, L.; Gao, Z.; Chen, G.; Zhang, C. Bone marrow stromal/stem cell-derived extracellular vesicles regulate osteoblast activity and differentiation in vitro and promote bone regeneration in vivo. Sci. Rep. 2016, 6, 21961. [Google Scholar] [CrossRef]
- Liu, X.; Yang, Y.; Li, Y.; Niu, X.; Zhao, B.; Wang, Y.; Bao, C.; Xie, Z.; Lin, Q.; Zhu, L. Integration of stem cell-derived exosomes with in situ hydrogel glue as a promising tissue patch for articular cartilage regeneration. Nanoscale 2017, 9, 4430–4438. [Google Scholar] [CrossRef]
- Michel, M.C.; Murphy, T.; Motulsky, H.J. New author guidelines for displaying data and reporting data analysis and statistical methods in experimental biology. J. Pharmacol. Exp. Ther. 2020, 372, 136–147. [Google Scholar] [CrossRef]
- Oveland, E.; Muth, T.; Rapp, E.; Martens, L.; Berven, F.S.; Barsnes, H. Viewing the proteome: How to visualize proteomics data? Proteomics 2015, 15, 1341–1355. [Google Scholar] [CrossRef]
- Lallukka, M.; Houaoui, A.; Miola, M.; Miettinen, S.; Massera, J.; Verné, E. In vitro cytocompatibility of antibacterial silver and copper-doped bioactive glasses. Ceram. Int. 2023, 49, 36044–36055. [Google Scholar] [CrossRef]
- Molloy, M.P.; Brzezinski, E.E.; Hang, J.; McDowell, M.T.; VanBogelen, R.A. Overcoming technical variation and biological variation in quantitative proteomics. Proteomics 2003, 3, 1912–1919. [Google Scholar] [CrossRef]
- Xu, D.; Wang, Y.; Meng, Y.; Zhang, Z. An improved data anomaly detection method based on isolation forest. In Proceedings of the 2017 10th International Symposium on Computational Intelligence and Design (ISCID), Hangzhou, China, 9–10 December 2017; IEEE: New York, NY, USA, 2017; Volume 2, pp. 287–291. [Google Scholar]
- Nassif, A.B.; Talib, M.A.; Nasir, Q.; Dakalbab, F.M. Machine learning for anomaly detection: A systematic review. IEEE Access 2021, 9, 78658–78700. [Google Scholar] [CrossRef]
- Devane, D.; Begley, C.M.; Clarke, M. How many do I need? Basic principles of sample size estimation. J. Adv. Nurs. 2004, 47, 297–302. [Google Scholar] [CrossRef]
- Columb, M.; Atkinson, M. Statistical analysis: Sample size and power estimations. BJA Educ. 2016, 16, 159–161. [Google Scholar] [CrossRef]
- Lakens, D. Sample size justification. Collabra Psychol. 2022, 8, 33267. [Google Scholar] [CrossRef]
- Lantz, B. The large sample size fallacy. Scand. J. Caring Sci. 2013, 27, 487–492. [Google Scholar] [CrossRef]
- Ioannidis, J.P.; Hozo, I.; Djulbegovic, B. Optimal type I and type II error pairs when the available sample size is fixed. J. Clin. Epidemiol. 2013, 66, 903–910. [Google Scholar] [CrossRef]
- Turner, B.O.; Paul, E.J.; Miller, M.B.; Barbey, A.K. Small sample sizes reduce the replicability of task-based fMRI studies. Commun. Biol. 2018, 1, 62. [Google Scholar] [CrossRef]
- Voelkl, B.; Vogt, L.; Sena, E.S.; Würbel, H. Reproducibility of preclinical animal research improves with heterogeneity of study samples. PLoS Biol. 2018, 16, e2003693. [Google Scholar] [CrossRef]
- Stockwell, D.R.; Peterson, A.T. Effects of sample size on accuracy of species distribution models. Ecol. Model. 2002, 148, 1–13. [Google Scholar] [CrossRef]
- Altman, D.G.; Bland, J.M. Statistics Notes: Comparing several groups using analysis of variance. BMJ 1996, 312, 1472–1473. [Google Scholar] [CrossRef]
- Nagaraj, N.; Mann, M. Quantitative analysis of the intra- and inter-individual variability of the normal urinary proteome. J. Proteome Res. 2011, 10, 637–645. [Google Scholar] [CrossRef]
- Thongboonkerd, V. The variability in tissue proteomics. Proteom.—Clin. Appl. 2012, 6, 340–342. [Google Scholar] [CrossRef]
- Pakharukova, N.A.; Pastushkova, L.K.; Moshkovskii, S.A.; Larina, I.M. Variability of the healthy human proteome. Biochem. Suppl. Ser. B Biomed. Chem. 2011, 5, 203–212. [Google Scholar] [CrossRef]
- Bischoff, R.; Permentier, H.; Guryev, V.; Horvatovich, P. Genomic variability and protein species—Improving sequence coverage for Proteogenomics. J. Proteom. 2016, 134, 25–36. [Google Scholar] [CrossRef]
- Dudzik, D.; Macioszek, S.; Struck-Lewicka, W.; Kordalewska, M.; Buszewska-Forajta, M.; Waszczuk-Jankowska, M.; Wawrzyniak, R.; Artymowicz, M.; Raczak-Gutknecht, J.; Siluk, D.; et al. Perspectives and challenges in extracellular vesicles untargeted metabolomics analysis. TrAC Trends Anal. Chem. 2021, 143, 116382. [Google Scholar] [CrossRef]
- Trentin, G.; Bitencourt, T.A.; Guedes, A.; Pessoni, A.M.; Brauer, V.S.; Pereira, A.K.; Costa, J.H.; Fill, T.P.; Almeida, F. Mass Spectrometry Analysis Reveals Lipids Induced by Oxidative Stress in Candida albicans Extracellular Vesicles. Microorganisms 2023, 11, 1669. [Google Scholar] [CrossRef]
- Jia, Y.; Yu, L.; Ma, T.; Xu, W.; Qian, H.; Sun, Y.; Shi, H. Small extracellular vesicles isolation and separation: Current techniques, pending questions and clinical applications. Theranostics 2022, 12, 6548. [Google Scholar] [CrossRef]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Théry, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef]
- Rosa-Fernandes, L.; Rocha, V.B.; Carregari, V.C.; Urbani, A.; Palmisano, G. A perspective on extracellular vesicles proteomics. Front. Chem. 2017, 5, 102. [Google Scholar] [CrossRef]
- Qadan, M.A.; Piuzzi, N.S.; Boehm, C.; Bova, W.; Moos, M., Jr.; Midura, R.J.; Hascall, V.C.; Malcuit, C.; Muschler, G.F. Variation in primary and culture-expanded cells derived from connective tissue progenitors in human bone marrow space, bone trabecular surface and adipose tissue. Cytotherapy 2018, 20, 343–360. [Google Scholar] [CrossRef]
- Foster, L.J.; Zeemann, P.A.; Li, C.; Mann, M.; Jensen, O.N.; Kassem, M. Differential expression profiling of membrane proteins by quantitative proteomics in a human mesenchymal stem cell line undergoing osteoblast differentiation. Stem Cells 2005, 23, 1367–1377. [Google Scholar] [CrossRef]
- Thompson, H.G.R.; Mih, J.D.; Krasieva, T.B.; Tromberg, B.J.; George, S.C. Epithelial-derived TGF-β2 modulates basal and wound-healing subepithelial matrix homeostasis. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2006, 291, L1277–L1285. [Google Scholar] [CrossRef]
- Jensen, S.A.; Handford, P.A. New insights into the structure, assembly and biological roles of 10–12 nm connective tissue microfibrils from fibrillin-1 studies. Biochem. J. 2016, 473, 827–838. [Google Scholar] [CrossRef]
- Tiedemann, K.; Boraschi-Diaz, I.; Rajakumar, I.; Kaur, J.; Roughley, P.; Reinhardt, D.P.; Komarova, S.V. Fibrillin-1 directly regulates osteoclast formation and function by a dual mechanism. J. Cell Sci. 2013, 126, 4187–4194. [Google Scholar] [CrossRef]
- Li, S.X.; Tong, Y.P.; Xie, X.C.; Wang, Q.H.; Zhou, H.N.; Han, Y.; Zhang, Z.Y.; Gao, W.; Li, S.G.; Zhang, X.C.; et al. Octameric structure of the human bifunctional enzyme PAICS in purine biosynthesis. J. Mol. Biol. 2007, 366, 1603–1614. [Google Scholar] [CrossRef]
- Zimmermann, D.R.; Ruoslahti, E. Multiple domains of the large fibroblast proteoglycan, versican. EMBO J. 1989, 8, 2975–2981. [Google Scholar] [CrossRef]
- Wight, T.N.; Kang, I.; Evanko, S.P.; Harten, I.A.; Chang, M.Y.; Pearce, O.M.; Allen, C.E.; Frevert, C.W. Versican—A critical extracellular matrix regulator of immunity and inflammation. Front. Immunol. 2020, 11, 512. [Google Scholar] [CrossRef]
- Starkova, T.; Polyanichko, A.; Tomilin, A.N.; Chikhirzhina, E. Structure and Functions of HMGB2 Protein. Int. J. Mol. Sci. 2023, 24, 8334. [Google Scholar] [CrossRef]
- Tiller, G.E.; Polumbo, P.A.; Weis, M.A.; Bogaert, R.; Lachman, R.S.; Cohn, D.H.; Rimoin, D.L.; Eyre, D.R. Dominant mutations in the type II collagen gene, COL2A1, produce spondyloepimetaphyseal dysplasia, Strudwick type. Nat. Genet. 1995, 11, 87–89. [Google Scholar] [CrossRef]
- Lee, C.C.; Bowman, B.H.; Yang, F.M. Human alpha 2-HS-glycoprotein: The A and B chains with a connecting sequence are encoded by a single mRNA transcript. Proc. Natl. Acad. Sci. USA 1987, 84, 4403–4407. [Google Scholar] [CrossRef]
- Moll, R.; Divo, M.; Langbein, L. The human keratins: Biology and pathology. Histochem. Cell Biol. 2008, 129, 705–733. [Google Scholar] [CrossRef]
- Dos Santos, J.F.; Borçari, N.R.; da Silva Araújo, M.; Nunes, V.A. Mesenchymal stem cells differentiate into keratinocytes and express epidermal kallikreins: Towards an in vitro model of human epidermis. J. Cell. Biochem. 2019, 120, 13141–13155. [Google Scholar] [CrossRef]
- Rashtbar, M.; Hadjati, J.; Ai, J.; Shirian, S.; Jahanzad, I.; Azami, M.; Asadpuor, S.; Sadroddiny, E. Critical-sized full-thickness skin defect regeneration using ovine small intestinal submucosa with or without mesenchymal stem cells in rat model. J. Biomed. Mater. Res. Part B Appl. Biomater. 2018, 106, 2177–2190. [Google Scholar] [CrossRef]
- Komori, T.; Pham, H.; Jani, P.; Perry, S.; Wang, Y.; Kilts, T.M.; Li, L.; Young, M.F. The Role of Type VI Collagen in Alveolar Bone. Int. J. Mol. Sci. 2022, 23, 14347. [Google Scholar] [CrossRef]
- Cescon, M.; Gattazzo, F.; Chen, P.; Bonaldo, P. Collagen VI at a glance. J. Cell Sci. 2015, 128, 3525–3531. [Google Scholar] [CrossRef]
- Alcorta-Sevillano, N.; Macías, I.; Rodríguez, C.I.; Infante, A. Crucial role of Lamin A/C in the migration and differentiation of MSCs in bone. Cells 2020, 9, 1330. [Google Scholar] [CrossRef]
- Ponomareva, O.Y.; Holmen, I.C.; Sperry, A.J.; Eliceiri, K.W.; Halloran, M.C. Calsyntenin-1 regulates axon branching and endosomal trafficking during sensory neuron development in vivo. J. Neurosci. 2014, 34, 9235–9248. [Google Scholar] [CrossRef]
- Vlachos, M.; Kollios, G.; Gunopulos, D. Discovering similar multidimensional trajectories. In Proceedings of the 18th International Conference on Data Engineering, San Jose, CA, USA, 26 February–1 March 2002; IEEE: New York, NY, USA, 2002; pp. 673–684. [Google Scholar]
- Miola, M.; Verné, E. Bioactive and antibacterial glass powders doped with copper by ion-exchange in aqueous solutions. Materials 2016, 9, 405. [Google Scholar] [CrossRef]
- Miola, M.; Massera, J.; Cochis, A.; Kumar, A.; Rimondini, L.; Vernè, E. Tellurium: A new active element for innovative multifunctional bioactive glasses. Mater. Sci. Eng. C 2021, 123, 111957. [Google Scholar] [CrossRef]
- Cochis, A.; Barberi, J.; Ferraris, S.; Miola, M.; Rimondini, L.; Vernè, E.; Yamaguchi, S.; Spriano, S. Competitive surface colonization of antibacterial and bioactive materials doped with strontium and/or silver ions. Nanomaterials 2020, 10, 120. [Google Scholar] [CrossRef] [PubMed]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Taye, M.B. Biomedical applications of ion-doped bioactive glass: A review. Appl. Nanosci. 2022, 12, 3797–3812. [Google Scholar] [CrossRef]
- Ankerst, M.; Breunig, M.M.; Kriegel, H.P.; Sander, J. OPTICS: Ordering points to identify the clustering structure. ACM Sigmod Rec. 1999, 28, 49–60. [Google Scholar] [CrossRef]
- Khan, K.; Rehman, S.U.; Aziz, K.; Fong, S.; Sarasvady, S. DBSCAN: Past, present and future. In Proceedings of the Fifth International Conference on the Applications of Digital Information and Web Technologies (ICADIWT 2014), Chennai, India, 17–19 February 2014; IEEE: New York, NY, USA, 2014; pp. 232–238. [Google Scholar]
- Kustatscher, G.; Hödl, M.; Rullmann, E.; Grabowski, P.; Fiagbedzi, E.; Groth, A.; Rappsilber, J. Higher-order modular regulation of the human proteome. Mol. Syst. Biol. 2023, 19, e9503. [Google Scholar] [CrossRef]
- Liu, F.T.; Ting, K.M.; Zhou, Z.H. Isolation forest. In Proceedings of the 2008 Eighth IEEE International Conference on Data Mining, Washington, DC, USA, 15–19 December 2008; IEEE: New York, NY, USA, 2008; pp. 413–422. [Google Scholar]
- Yadav, S.K.; Singh, S.; Gupta, R. Biomedical Statistics; Springer: Berlin/Heidelberg, Germany, 2019. [Google Scholar]
- Xia, S.; Xiong, Z.; Luo, Y.; Xu, W.; Zhang, G. Effectiveness of the Euclidean distance in high dimensional spaces. Optik 2015, 126, 5614–5619. [Google Scholar] [CrossRef]
- Baker, M. Statisticians issue warning on p values. Nature 2016, 531, 151. [Google Scholar] [CrossRef]
- Aebersold, R.; Mann, M. Mass spectrometry-based proteomics. Nature 2003, 422, 198–207. [Google Scholar] [CrossRef]
- Han, X.; Aslanian, A.; Yates, J.R., III. Mass spectrometry for proteomics. Curr. Opin. Chem. Biol. 2008, 12, 483–490. [Google Scholar] [CrossRef]
- Dubitzky, W.; Granzow, M.; Berrar, D.P. Fundamentals of Data Mining in Genomics and Proteomics; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2007. [Google Scholar]
- Wilkins, M. Proteomics data mining. Expert Rev. Proteom. 2009, 6, 599–603. [Google Scholar] [CrossRef]
- Zimek, A.; Filzmoser, P. There and back again: Outlier detection between statistical reasoning and data mining algorithms. Wiley Interdiscip. Rev. Data Min. Knowl. Discov. 2018, 8, e1280. [Google Scholar] [CrossRef]
- Schessner, J.P.; Voytik, E.; Bludau, I. A practical guide to interpreting and generating bottom-up Proteomics Data Visualizations. Proteomics 2022, 22, 103. [Google Scholar] [CrossRef]









| Accession Number | Gene Name | Protein Name |
|---|---|---|
| P02458 | COL2A1 | Collagen alpha-1(II) chain |
| P19338 | NCL | Nucleolin |
| P14923 | JUP | Junction plakoglobin |
| Q9Y4Z0 | LSM4 | U6 snRNA-associated Sm-like protein LSm4 |
| Accession Number | Gene Name | Protein Name |
|---|---|---|
| P02656 | APOC3 | Apolipoprotein C-III |
| P38571 | LIPA | Lysosomal acid lipase/cholesteryl ester hydrolase |
| Q9P2E9 ** | RRBP1 | Ribosome-binding protein 1 |
| P02765 | AHSG | Alpha-2-HS-glycoprotein |
| P49720 | PSMB3 | Proteasome subunit beta type-3 |
| P46940 | IQGAP1 | Ras GTPase-activating-like protein IQGAP1 |
| P59665 | DEFA1 | Neutrophil defensin 1 |
| Accession Number | Gene Name | Protein Name |
|---|---|---|
| P24821-4 | TNC | Isoform 4 of Tenascin |
| P26583 | HMGB2 | High mobility group protein B2 |
| P22234 * | PAICS | Multifunctional protein ADE2 |
| P35555 ** | FBN1 | Fibrillin-1 |
| P24821 * | TNC | Tenascin |
| Q9P2E9 ** | RRBP1 | Ribosome-binding protein 1 |
| P13611 | VCAN | Versican core protein |
| Accession Number | Gene Name | Protein Name |
|---|---|---|
| P35908 ** | KRT2 | Keratin, Type II cytoskeletal 2 epidermal |
| P35527 ** | KRT9 | Keratin, Type I cytoskeletal 9 |
| P04264 ** | KRT1 | Keratin, Type II cytoskeletal 1 |
| P13645 ** | KRT10 | Keratin, Type I cytoskeletal 10 |
| P60985 * | KRTDAP | Keratinocyte differentiation-associated protein |
| P12111 ** | COL6A3 | Collagen alpha-3(VI) chain |
| P08493 | MGP | Matrix Gla protein |
| Accession Number | Gene Name | Protein Name |
|---|---|---|
| P00734 ** | F2 | Prothrombin |
| P12111 * | COL6A3 | Collagen alpha-3(VI) chain |
| P35908 ** | KRT2 | Keratin, Type II cytoskeletal 2 epidermal |
| P35527 ** | KRT9 | Keratin, Type I cytoskeletal 9 |
| P13645 ** | KRT10 | Keratin, Type I cytoskeletal 10 |
| P04264 ** | KRT1 | Keratin, Type II cytoskeletal 1 |
| Accession Number | Gene Name | Protein Name |
|---|---|---|
| O94985 ** | CLSTN1 | Calsyntenin-1 |
| P22234 ** | PAICS | Multifunctional protein ADE2 |
| P02545 * | LMNA | Prelamin-A/C |
| P04264 ** | KRT1 | Keratin, Type II cytoskeletal 1 |
| P35527 ** | KRT9 | Keratin, Type I cytoskeletal 9 |
| Q16363 | LAMA4 | Laminin subunit alpha-4 |
| P13645 ** | KRT10 | Keratin, Type I cytoskeletal 10 |
| P35908 ** | KRT2 | Keratin, Type II cytoskeletal 2 epidermal |
| P00734 ** | F2 | Prothrombin |
| P26022 * | PTX3 | Pentraxin-related protein PTX3 |
| P60985 ** | KRTDAP | Keratinocyte differentiation-associated protein |
| Experiment | Protein | p-Value | Ctrl Var. | Doped Var. | Abs Var. * |
|---|---|---|---|---|---|
| SBA2 | P02458 | 0.77209 | 1.27 | 2.26 | 0.99 |
| SBA2 | P19338 | 0.7934 | 2.76 | 0.16 | 2.6 |
| SBA2 | P14923 | 0.90066 | 3.85 | 1.55 | 2.3 |
| SBA2 | Q9Y4Z0 | 0.37355 | 13.0 | 0.72 | 12.28 |
| SBA3 | P02656 | 0.19829 | 0.73 | 0.18 | 0.55 |
| SBA3 | P38571 | 0.85016 | 0.21 | 2.61 | 2.4 |
| SBA3 | Q9P2E9 | 0.03166 | 0.22 | 0.08 | 0.14 |
| SBA3 | P02765 | 0.51954 | 1.8 | 0.2 | 1.6 |
| SBA3 | P49720 | 0.80732 | 1.44 | 1.55 | 0.11 |
| SBA3 | P46940 | 0.67908 | 0.51 | 1.66 | 1.15 |
| SBA3 | P59665 | 0.40182 | 8.47 | 2.79 | 5.68 |
| ST | P24821-4 | 0.4643 | 1.02 | 1.73 | 0.71 |
| ST | P26583 | 0.20189 | 0.1 | 0.71 | 0.61 |
| ST | P22234 | 0.08797 | 0.5 | 0.17 | 0.33 |
| ST | P35555 | 0.04938 | 0.22 | 0.34 | 0.12 |
| ST | P24821 | 0.05846 | 0.13 | 0.96 | 0.83 |
| ST | Q9P2E9 | 0.03078 | 0.32 | 0.23 | 0.09 |
| ST | P13611 | 0.16264 | 1.83 | 5.37 | 3.54 |
| Experiment | Protein | p-Value | Ctrl Var. | Doped Var. | Abs Var. * |
|---|---|---|---|---|---|
| SBA2 | P35908 | 0.00245 | 0.07 | 0.28 | 0.21 |
| SBA2 | P35527 | 0.00029 | 0.02 | 0.14 | 0.12 |
| SBA2 | P04264 | 0.00015 | 0.01 | 0.25 | 0.24 |
| SBA2 | P13645 | 0.00041 | 0.02 | 0.35 | 0.33 |
| SBA2 | P60985 | 0.08047 | 0.27 | 0.84 | 0.57 |
| SBA2 | P12111 | 0.00145 | 0.54 | 0.04 | 0.5 |
| SBA2 | P08493 | 0.10463 | 2.05 | 1.65 | 0.4 |
| SBA3 | P00734 | 0.0291 | 0.25 | 0.01 | 0.24 |
| SBA3 | P12111 | 0.05029 | 0.54 | 0.63 | 0.09 |
| SBA3 | P35908 | 0.00252 | 0.07 | 0.57 | 0.5 |
| SBA3 | P35527 | 0.00038 | 0.02 | 0.53 | 0.51 |
| SBA3 | P13645 | 0.00059 | 0.02 | 0.79 | 0.77 |
| SBA3 | P04264 | 0.00018 | 0.01 | 0.66 | 0.65 |
| ST | O94985 | 0.02078 | 0.01 | 0.26 | 0.25 |
| ST | P22234 | 0.03636 | 1.17 | 0.17 | 1.0 |
| ST | P02545 | 0.08999 | 2.02 | 0.17 | 1.85 |
| ST | P04264 | 0.00015 | 0.01 | 0.07 | 0.06 |
| ST | P35527 | 0.00079 | 0.02 | 0.29 | 0.27 |
| ST | Q16363 | 0.25842 | 2.89 | 1.69 | 1.2 |
| ST | P13645 | 0.00037 | 0.02 | 0.07 | 0.05 |
| ST | P35908 | 0.00244 | 0.07 | 0.16 | 0.09 |
| ST | P00734 | 0.02785 | 0.25 | 0.03 | 0.22 |
| ST | P26022 | 0.08427 | 0.41 | 2.51 | 2.1 |
| ST | P60985 | 0.04855 | 0.27 | 0.69 | 0.42 |
| Components | SBA2 % mol | SBA3 % mol |
|---|---|---|
| SiO2 | 48 | 48 |
| Na2CO3 | 18 | 26 |
| CaCO3 | 30 | 22 |
| Ca3(PO4)2 | 3 | 3 |
| H3BO3 | 0.43 | 0.43 |
| Al2O3 | 0.57 | 0.57 |
| Components | ST 0% mol | ST 5% mol |
|---|---|---|
| SiO2 | 48.6 | 43.6 |
| Na2CO3 | 16.7 | 16.7 |
| CaCO3 | 34.2 | 34.2 |
| Ca3(PO4)2 | 0.5 | 0.5 |
| TeO2 | 0.0 | 5.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nascimben, M.; Abreu, H.; Manfredi, M.; Cappellano, G.; Chiocchetti, A.; Rimondini, L. Extracellular Vesicle Protein Expression in Doped Bioactive Glasses: Further Insights Applying Anomaly Detection. Int. J. Mol. Sci. 2024, 25, 3560. https://doi.org/10.3390/ijms25063560
Nascimben M, Abreu H, Manfredi M, Cappellano G, Chiocchetti A, Rimondini L. Extracellular Vesicle Protein Expression in Doped Bioactive Glasses: Further Insights Applying Anomaly Detection. International Journal of Molecular Sciences. 2024; 25(6):3560. https://doi.org/10.3390/ijms25063560
Chicago/Turabian StyleNascimben, Mauro, Hugo Abreu, Marcello Manfredi, Giuseppe Cappellano, Annalisa Chiocchetti, and Lia Rimondini. 2024. "Extracellular Vesicle Protein Expression in Doped Bioactive Glasses: Further Insights Applying Anomaly Detection" International Journal of Molecular Sciences 25, no. 6: 3560. https://doi.org/10.3390/ijms25063560