
Molecular Basis of Neuronal and Microglial States in the Aging Brain and Impact on Cerebral Blood Vessels

{kind=link}
{kind=link}
Abstract
1. Introduction
2. Cellular Senescence
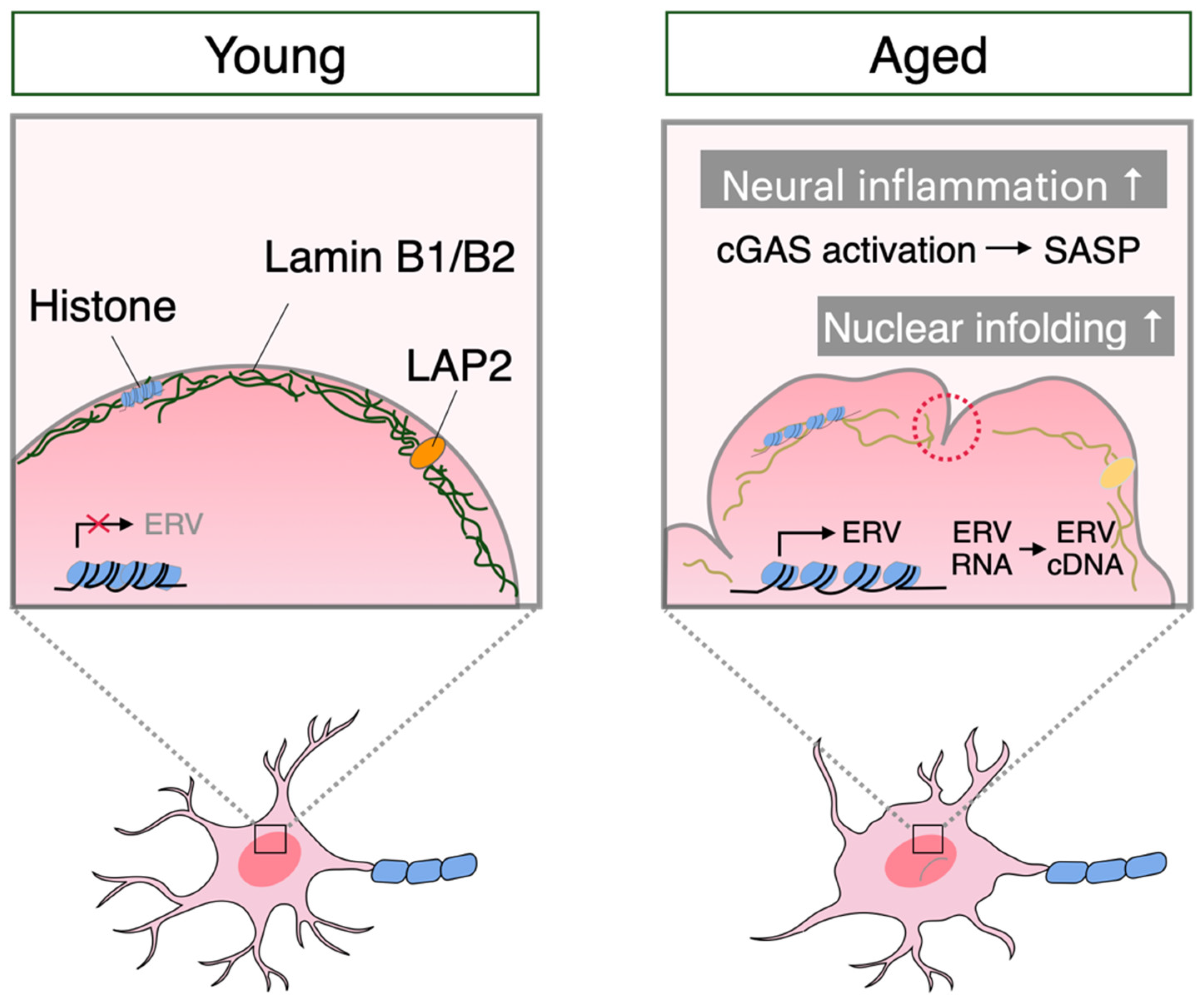
3. Neuronal Senescence
4. Microglial Senescence
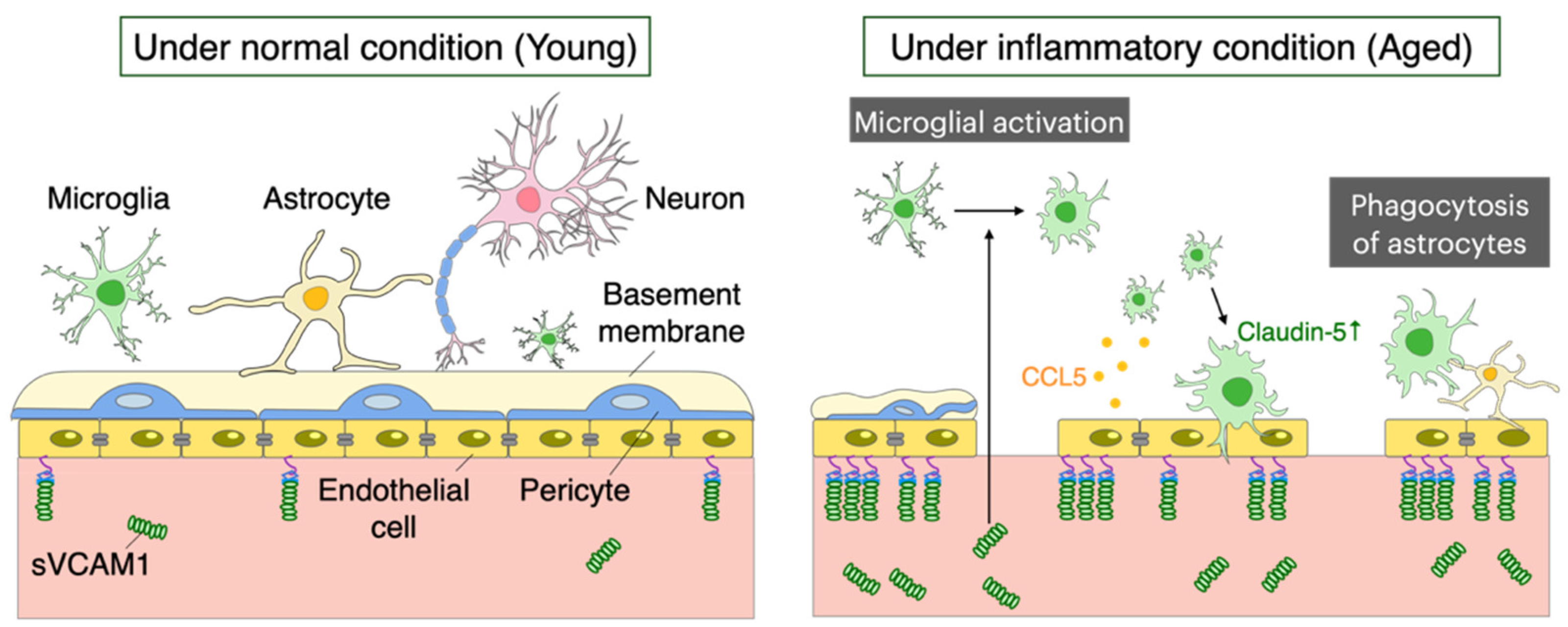
5. Aging of the Blood-Brain Barrier
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Tosato, M.; Zamboni, V.; Ferrini, A.; Cesari, M. The Aging Process and Potential Interventions to Extend Life Expectancy. Clin. Interv. Aging 2007, 2, 401–412. [Google Scholar]
- Mattson, M.P.; Arumugam, T.V. Hallmarks of Brain Aging: Adaptive and Pathological Modification by Metabolic States. Cell Metab. 2018, 27, 1176–1199. [Google Scholar] [CrossRef]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a Risk Factor for Neurodegenerative Disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef]
- Iadecola, C. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef]
- Mathiisen, T.M.; Lehre, K.P.; Danbolt, N.C.; Ottersen, O.P. The Perivascular Astroglial Sheath Provides a Complete Covering of the Brain Microvessels: An Electron Microscopic 3D Reconstruction. Glia 2010, 58, 1094–1103. [Google Scholar] [CrossRef]
- Takano, T.; Tian, G.-F.; Peng, W.; Lou, N.; Libionka, W.; Han, X.; Nedergaard, M. Astrocyte-Mediated Control of Cerebral Blood Flow. Nat. Neurosci. 2006, 9, 260–267. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The Serial Cultivation of Human Diploid Cell Strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-Damage Response in Human Biology and Disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef]
- Fumagalli, M.; Rossiello, F.; Mondello, C.; d’Adda di Fagagna, F. Stable Cellular Senescence Is Associated with Persistent DDR Activation. PLoS ONE 2014, 9, e110969. [Google Scholar] [CrossRef]
- Fumagalli, M.; Rossiello, F.; Clerici, M.; Barozzi, S.; Cittaro, D.; Kaplunov, J.M.; Bucci, G.; Dobreva, M.; Matti, V.; Beausejour, C.M.; et al. Telomeric DNA Damage Is Irreparable and Causes Persistent DNA-Damage-Response Activation. Nat. Cell Biol. 2012, 14, 355–365. [Google Scholar] [CrossRef]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An Oncogene-Induced DNA Damage Model for Cancer Development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef]
- Chapman, J.; Fielder, E.; Passos, J.F. Mitochondrial Dysfunction and Cell Senescence: Deciphering a Complex Relationship. FEBS Lett. 2019, 593, 1566–1579. [Google Scholar] [CrossRef]
- Mizushima, N. A Brief History of Autophagy from Cell Biology to Physiology and Disease. Nat. Cell Biol. 2018, 20, 521–527. [Google Scholar] [CrossRef]
- Aman, Y.; Schmauck-Medina, T.; Hansen, M.; Morimoto, R.I.; Simon, A.K.; Bjedov, I.; Palikaras, K.; Simonsen, A.; Johansen, T.; Tavernarakis, N.; et al. Autophagy in Healthy Aging and Disease. Nat. Aging 2021, 1, 634–650. [Google Scholar] [CrossRef]
- Lukášová, E.; Kovařík, A.; Kozubek, S. Consequences of Lamin B1 and Lamin B Receptor Downregulation in Senescence. Cells 2018, 7, 11. [Google Scholar] [CrossRef]
- Yang, N.; Sen, P. The Senescent Cell Epigenome. Aging 2018, 10, 3590–3609. [Google Scholar] [CrossRef]
- Coppé, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the P53 Tumor Suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef]
- Morrison, J.H.; Baxter, M.G. The Ageing Cortical Synapse: Hallmarks and Implications for Cognitive Decline. Nat. Rev. Neurosci. 2012, 13, 240–250. [Google Scholar] [CrossRef]
- Kowald, A.; Kirkwood, T.B. Accumulation of Defective Mitochondria through Delayed Degradation of Damaged Organelles and Its Possible Role in the Ageing of Post-Mitotic and Dividing Cells. J. Theor. Biol. 2000, 202, 145–160. [Google Scholar] [CrossRef]
- Noda, S.; Sato, S.; Fukuda, T.; Tada, N.; Hattori, N. Aging-Related Motor Function and Dopaminergic Neuronal Loss in C57BL/6 Mice. Mol. Brain 2020, 13, 46. [Google Scholar] [CrossRef]
- Wang, C.L.; Ohkubo, R.; Mu, W.C.; Chen, W.; Fan, J.L.; Song, Z.; Maruichi, A.; Sudmant, P.H.; Pisco, A.O.; Dubal, D.B.; et al. The Mitochondrial Unfolded Protein Response Regulates Hippocampal Neural Stem Cell Aging. Cell Metab. 2023, 35, 996–1008.e7. [Google Scholar] [CrossRef]
- Lipinski, M.M.; Zheng, B.; Lu, T.; Yan, Z.; Py, B.F.; Ng, A.; Xavier, R.J.; Li, C.; Yankner, B.A.; Scherzer, C.R.; et al. Genome-Wide Analysis Reveals Mechanisms Modulating Autophagy in Normal Brain Aging and in Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2010, 107, 14164–14169. [Google Scholar] [CrossRef]
- Kovács, T.; Szinyákovics, J.; Billes, V.; Murányi, G.; Varga, V.B.; Bjelik, A.; Légrádi, Á.; Szabó, M.; Sándor, S.; Kubinyi, E.; et al. A Conserved MTMR Lipid Phosphatase Increasingly Suppresses Autophagy in Brain Neurons during Aging. Sci. Rep. 2022, 12, 21817. [Google Scholar] [CrossRef]
- Moreno-Blas, D.; Gorostieta-Salas, E.; Pommer-Alba, A.; Muciño-Hernández, G.; Gerónimo-Olvera, C.; Maciel-Barón, L.A.; Konigsberg, M.; Massieu, L.; Castro-Obregón, S. Cortical Neurons Develop a Senescence-like Phenotype Promoted by Dysfunctional Autophagy. Aging 2019, 11, 6175–6198. [Google Scholar] [CrossRef]
- Zhang, H.; Li, J.; Yu, Y.; Ren, J.; Liu, Q.; Bao, Z.; Sun, S.; Liu, X.; Ma, S.; Liu, Z.; et al. Nuclear Lamina Erosion-Induced Resurrection of Endogenous Retroviruses Underlies Neuronal Aging. Cell Rep. 2023, 42, 112593. [Google Scholar] [CrossRef]
- Das, S.; Ramanan, N. Region-Specific Heterogeneity in Neuronal Nuclear Morphology in Young, Aged and in Alzheimer’s Disease Mouse Brains. Front. Cell Dev. Biol. 2023, 11, 1032504. [Google Scholar] [CrossRef]
- Frey, T.; Murakami, T.; Maki, K.; Kawaue, T.; Tani, N.; Sugai, A.; Nakazawa, N.; Ishiguro, K.; Adachi, T.; Kengaku, M.; et al. Age-Associated Reduction of Nuclear Shape Dynamics in Excitatory Neurons of the Visual Cortex. Aging Cell 2023, 22, e13925. [Google Scholar] [CrossRef]
- Chen, X.; Xie, C.; Tian, W.; Sun, L.; Wang, Z.; Hawes, S.; Chang, L.; Kung, J.; Ding, J.; Chen, S.; et al. Parkinson’s Disease-Related Leucine-Rich Repeat Kinase 2 Modulates Nuclear Morphology and Genomic Stability in Striatal Projection Neurons during Aging. Mol. Neurodegener. 2020, 15, 12. [Google Scholar] [CrossRef]
- Paonessa, F.; Evans, L.D.; Solanki, R.; Larrieu, D.; Wray, S.; Hardy, J.; Jackson, S.P.; Livesey, F.J. Microtubules Deform the Nuclear Membrane and Disrupt Nucleocytoplasmic Transport in Tau-Mediated Frontotemporal Dementia. Cell Rep. 2019, 26, 582–593.e5. [Google Scholar] [CrossRef]
- Helmut, K.; Hanisch, U.K.; Noda, M.; Verkhratsky, A. Physiology of Microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Li, Q.; Barres, B.A. Microglia and Macrophages in Brain Homeostasis and Disease. Nat. Rev. Immunol. 2018, 18, 225–242. [Google Scholar] [CrossRef]
- Barry-Carroll, L.; Greulich, P.; Marshall, A.R.; Riecken, K.; Fehse, B.; Askew, K.E.; Li, K.; Garaschuk, O.; Menassa, D.A.; Gomez-Nicola, D. Microglia Colonize the Developing Brain by Clonal Expansion of Highly Proliferative Progenitors, following Allometric Scaling. Cell Rep. 2023, 42, 112425. [Google Scholar] [CrossRef]
- Prinz, M.; Masuda, T.; Wheeler, M.A.; Quintana, F.J. Annual Review of Immunology Microglia and Central Nervous System-Associated Macrophages-From Origin to Disease Modulation. Annu. Rev. Immunol. 2021, 39, 251–277. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate Mapping Analysis Reveals That Adult Microglia Derive from Primitive Macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia Sculpt Postnatal Neural Circuits in an Activity and Complement-Dependent Manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef]
- Hill, R.A.; Damisah, E.C.; Chen, F.; Kwan, A.C.; Grutzendler, J. Targeted Two-Photon Chemical Apoptotic Ablation of Defined Cell Types In Vivo. Nat. Commun. 2017, 8, 15837. [Google Scholar] [CrossRef]
- Cunningham, C.L.; Martínez-Cerdeño, V.; Noctor, S.C. Microglia Regulate the Number of Neural Precursor Cells in the Developing Cerebral Cortex. J. Neurosci. 2013, 33, 4216–4233. [Google Scholar] [CrossRef]
- Sierra, A.; Encinas, J.M.; Deudero, J.J.P.; Chancey, J.H.; Enikolopov, G.; Overstreet-Wadiche, L.S.; Tsirka, S.E.; Maletic-Savatic, M. Microglia Shape Adult Hippocampal Neurogenesis through Apoptosis-Coupled Phagocytosis. Cell Stem Cell 2010, 7, 483–495. [Google Scholar] [CrossRef]
- Miyamoto, A.; Wake, H.; Ishikawa, A.W.; Eto, K.; Shibata, K.; Murakoshi, H.; Koizumi, S.; Moorhouse, A.J.; Yoshimura, Y.; Nabekura, J. Microglia Contact Induces Synapse Formation in Developing Somatosensory Cortex. Nat. Commun. 2016, 7, 12540. [Google Scholar] [CrossRef]
- Haruwaka, K.; Ikegami, A.; Tachibana, Y.; Ohno, N.; Konishi, H.; Hashimoto, A.; Matsumoto, M.; Kato, D.; Ono, R.; Kiyama, H.; et al. Dual Microglia Effects on Blood Brain Barrier Permeability Induced by Systemic Inflammation. Nat. Commun. 2019, 10, 5816. [Google Scholar] [CrossRef]
- Li, X.; Li, Y.; Jin, Y.; Zhang, Y.; Wu, J.; Xu, Z.; Huang, Y.; Cai, L.; Gao, S.; Liu, T.; et al. Transcriptional and Epigenetic Decoding of the Microglial Aging Process. Nat. Aging 2023, 3, 1288–1311. [Google Scholar] [CrossRef]
- Taketomi, T.; Tsuruta, F. Towards an Understanding of Microglia and Border-Associated Macrophages. Biology 2023, 12, 1091. [Google Scholar] [CrossRef]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 2019, 50, 253–271. [Google Scholar] [CrossRef]
- Safaiyan, S.; Besson-Girard, S.; Kaya, T.; Cantuti-Castelvetri, L.; Liu, L.; Ji, H.; Schifferer, M.; Gouna, G.; Usifo, F.; Kannaiyan, N.; et al. White Matter Aging Drives Microglial Diversity. Neuron 2021, 109, 1100–1117. [Google Scholar] [CrossRef]
- Marschallinger, J.; Iram, T.; Zardeneta, M.; Lee, S.E.; Lehallier, B.; Haney, M.S.; Pluvinage, J.V.; Mathur, V.; Hahn, O.; Morgens, D.W.; et al. Lipid-Droplet-Accumulating Microglia Represent a Dysfunctional and Proinflammatory State in the Aging Brain. Nat. Neurosci. 2020, 23, 194–208. [Google Scholar] [CrossRef]
- Pluvinage, J.V.; Haney, M.S.; Smith, B.A.H.; Sun, J.; Iram, T.; Bonanno, L.; Li, L.; Lee, D.P.; Morgens, D.W.; Yang, A.C.; et al. CD22 Blockade Restores Homeostatic Microglial Phagocytosis in Ageing Brains. Nature 2019, 568, 187–192. [Google Scholar] [CrossRef]
- Linnartz-Gerlach, B.; Bodea, L.G.; Klaus, C.; Ginolhac, A.; Halder, R.; Sinkkonen, L.; Walter, J.; Colonna, M.; Neumann, H. TREM2 Triggers Microglial Density and Age-Related Neuronal Loss. Glia 2019, 67, 539–550. [Google Scholar] [CrossRef]
- Paul, B.D.; Snyder, S.H.; Bohr, V.A. Signaling by CGAS–STING in Neurodegeneration, Neuroinflammation, and Aging. Trends Neurosci. 2021, 44, 83–96. [Google Scholar] [CrossRef]
- Gulen, M.F.; Samson, N.; Keller, A.; Schwabenland, M.; Liu, C.; Glück, S.; Thacker, V.V.; Favre, L.; Mangeat, B.; Kroese, L.J.; et al. CGAS–STING Drives Ageing-Related Inflammation and Neurodegeneration. Nature 2023, 620, 374–380. [Google Scholar] [CrossRef]
- Dong, X. Current Strategies for Brain Drug Delivery. Theranostics 2018, 8, 1481–1493. [Google Scholar] [CrossRef]
- Stamatovic, S.M.; Keep, R.F.; Andjelkovic, A. V Brain Endothelial Cell-Cell Junctions: How to “Open” the Blood Brain Barrier. Curr. Neuropharmacol. 2008, 6, 179–192. [Google Scholar] [CrossRef]
- Stamatovic, S.M.; Johnson, A.M.; Keep, R.F.; Andjelkovic, A.V. Junctional Proteins of the Blood-Brain Barrier: New Insights into Function and Dysfunction. Tissue Barriers 2016, 4, e1154641. [Google Scholar] [CrossRef]
- Geranmayeh, M.H.; Rahbarghazi, R.; Farhoudi, M. Targeting Pericytes for Neurovascular Regeneration. Cell Commun. Signal. 2019, 17, 26. [Google Scholar] [CrossRef]
- Ahn, S.I.; Sei, Y.J.; Park, H.J.; Kim, J.; Ryu, Y.; Choi, J.J.; Sung, H.J.; MacDonald, T.J.; Levey, A.I.; Kim, Y.T. Microengineered Human Blood–Brain Barrier Platform for Understanding Nanoparticle Transport Mechanisms. Nat. Commun. 2020, 11, 175. [Google Scholar] [CrossRef]
- Armulik, A.; Genové, G.; Mäe, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes Regulate the Blood-Brain Barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef]
- Nielsen, S.; Nagelhus, E.A.; Amiry-Moghaddam, M.; Bourque, C.; Agre, P.; Ottersen, O.P. Specialized Membrane Domains for Water Transport in Glial Cells: High-Resolution Immunogold Cytochemistry of Aquaporin-4 in Rat Brain. J. Neurosci. 1996, 17, 171–180. [Google Scholar] [CrossRef]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-Endothelial Interactions at the Blood-Brain Barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Sengillo, J.D.; Winkler, E.A.; Walker, C.T.; Sullivan, J.S.; Johnson, M.; Zlokovic, B.V. Deficiency in Mural Vascular Cells Coincides with Blood-Brain Barrier Disruption in Alzheimer’s Disease. Brain Pathol. 2013, 23, 303–310. [Google Scholar] [CrossRef]
- Yang, A.C.; Stevens, M.Y.; Chen, M.B.; Lee, D.P.; Stähli, D.; Gate, D.; Contrepois, K.; Chen, W.; Iram, T.; Zhang, L.; et al. Physiological Blood–Brain Transport Is Impaired with Age by a Shift in Transcytosis. Nature 2020, 583, 425–430. [Google Scholar] [CrossRef]
- Salloway, S.; Gur, T.; Berzin, T.; Zipser, B.; Correia, S.; Hovanesian, V.; Fallon, J.; Kuo-Leblanc, V.; Glass, D.; Hulette, C.; et al. Effect of APOE Genotype on Microvascular Basement Membrane in Alzheimer’s Disease. J. Neurol. Sci. 2002, 203–204, 183–187. [Google Scholar] [CrossRef]
- Mendiola, A.S.; Yan, Z.; Dixit, K.; Johnson, J.R.; Bouhaddou, M.; Meyer-Franke, A.; Shin, M.G.; Yong, Y.; Agrawal, A.; MacDonald, E.; et al. Defining Blood-Induced Microglia Functions in Neurodegeneration through Multiomic Profiling. Nat. Immunol. 2023, 24, 1173–1187. [Google Scholar] [CrossRef]
- Yousef, H.; Czupalla, C.J.; Lee, D.; Chen, M.B.; Burke, A.N.; Zera, K.A.; Zandstra, J.; Berber, E.; Lehallier, B.; Mathur, V.; et al. Aged Blood Impairs Hippocampal Neural Precursor Activity and Activates Microglia via Brain Endothelial Cell VCAM1. Nat. Med. 2019, 25, 988–1000. [Google Scholar] [CrossRef]
- Pan, J.; Ma, N.; Zhong, J.; Yu, B.; Wan, J.; Zhang, W. Age-Associated Changes in Microglia and Astrocytes Ameliorate Blood-Brain Barrier Dysfunction. Mol. Ther. Nucleic Acids 2021, 26, 970–986. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maeda, C.; Tsuruta, F. Molecular Basis of Neuronal and Microglial States in the Aging Brain and Impact on Cerebral Blood Vessels. Int. J. Mol. Sci. 2024, 25, 4443. https://doi.org/10.3390/ijms25084443
Maeda C, Tsuruta F. Molecular Basis of Neuronal and Microglial States in the Aging Brain and Impact on Cerebral Blood Vessels. International Journal of Molecular Sciences. 2024; 25(8):4443. https://doi.org/10.3390/ijms25084443
Chicago/Turabian StyleMaeda, Chihiro, and Fuminori Tsuruta. 2024. "Molecular Basis of Neuronal and Microglial States in the Aging Brain and Impact on Cerebral Blood Vessels" International Journal of Molecular Sciences 25, no. 8: 4443. https://doi.org/10.3390/ijms25084443
APA StyleMaeda, C., & Tsuruta, F. (2024). Molecular Basis of Neuronal and Microglial States in the Aging Brain and Impact on Cerebral Blood Vessels. International Journal of Molecular Sciences, 25(8), 4443. https://doi.org/10.3390/ijms25084443