
Detrimental Roles of Hypoxia-Inducible Factor-1α in Severe Hypoxic Brain Diseases

Department of Integrative Bioscience and Biotechnology, Konkuk University, Seoul 05029, Republic of Korea
Int. J. Mol. Sci. 2024, 25(8), 4465; https://doi.org/10.3390/ijms25084465
Submission received: 14 March 2024
/
Revised: 12 April 2024
/
Accepted: 17 April 2024
/
Published: 18 April 2024
(This article belongs to the Special Issue New Molecular Insights into Ischemia/Reperfusion)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Hypoxia stabilizes hypoxia-inducible factors (HIFs), facilitating adaptation to hypoxic conditions. Appropriate hypoxia is pivotal for neurovascular regeneration and immune cell mobilization. However, in central nervous system (CNS) injury, prolonged and severe hypoxia harms the brain by triggering neurovascular inflammation, oxidative stress, glial activation, vascular damage, mitochondrial dysfunction, and cell death. Diminished hypoxia in the brain improves cognitive function in individuals with CNS injuries. This review discusses the current evidence regarding the contribution of severe hypoxia to CNS injuries, with an emphasis on HIF-1α-mediated pathways. During severe hypoxia in the CNS, HIF-1α facilitates inflammasome formation, mitochondrial dysfunction, and cell death. This review presents the molecular mechanisms by which HIF-1α is involved in the pathogenesis of CNS injuries, such as stroke, traumatic brain injury, and Alzheimer’s disease. Deciphering the molecular mechanisms of HIF-1α will contribute to the development of therapeutic strategies for severe hypoxic brain diseases.
1. Introduction
Severe hypoxia affects the central nervous system (CNS) by triggering neurovascular inflammation, oxidative stress, glial activation, impaired mitochondrial function, and cell death [1]. High O2 therapy can elevate cerebral blood flow and improve cognitive behavioral performance by diminishing hypoxia involved in the pathogenesis of Alzheimer’s disease (AD) [2,3]. Hypoxia-inducible factors (HIF1–3) regulate transcriptional responses to reduce O2 availability [4]. HIFs are heterodimeric proteins that are composed of an O2-regulated HIF-α subunit and a constitutively expressed HIF-1β subunit. HIF-α subunits are subject to prolyl hydroxylation, which targets proteins for degradation under normoxic conditions [5,6]. Two HIF-α proteins, HIF-1α and HIF-2α, are stabilized under low O2 tension and dimerize with HIF-1β. Heterodimeric proteins bind to hypoxia-responsive elements in multiple target genes and regulate their transcription to facilitate adaptation to hypoxia [7].
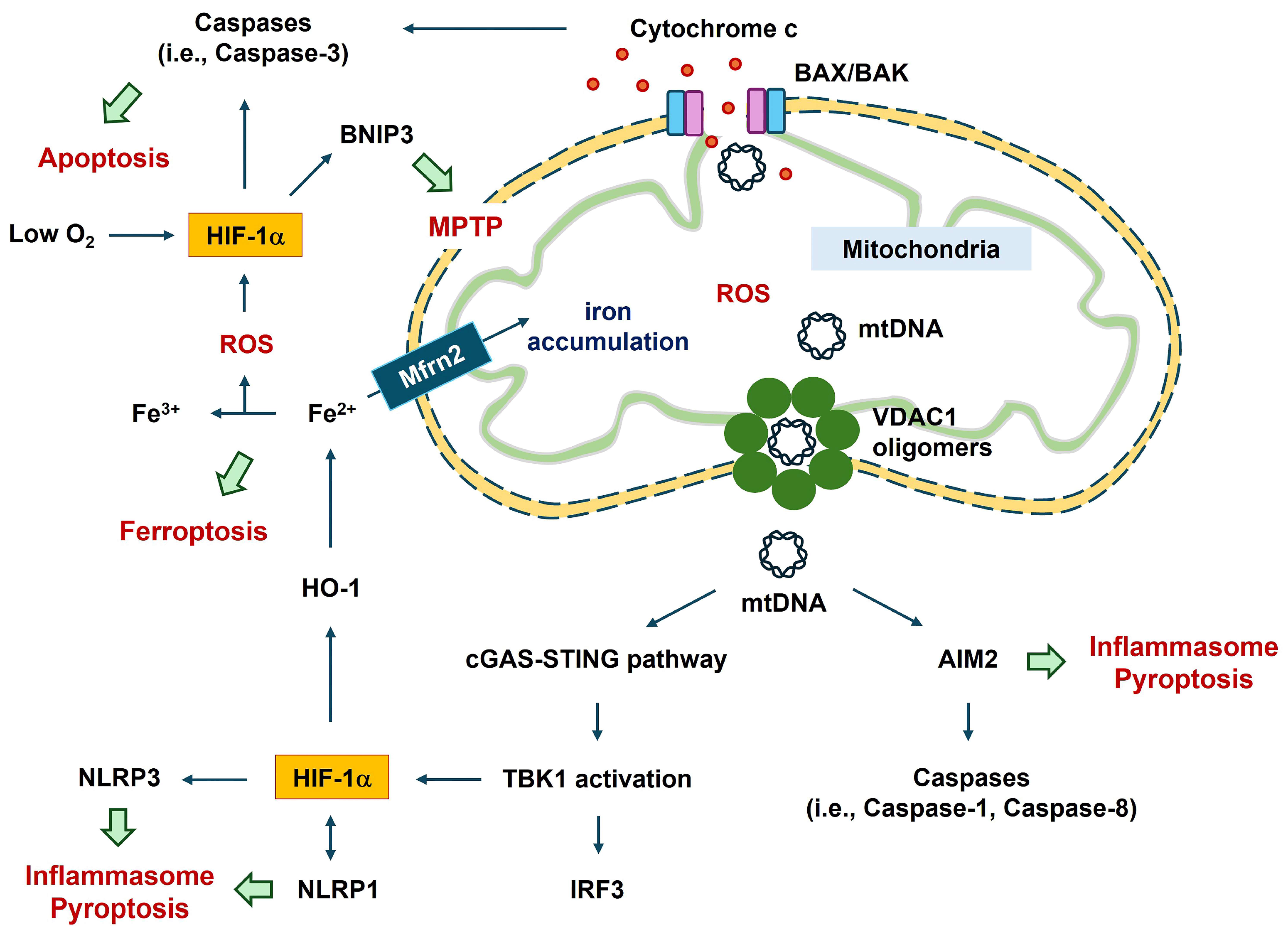
HIF-1α accumulation has dual effects, including cell death and cell survival, in neurovascular diseases such as stroke, traumatic brain injury (TBI), and AD [8,9,10,11,12,13,14]. HIF-1α has complex effects in the brain [15,16,17], which largely depend on the severity of and time-point after hypoxic damage. HIF-1α stabilization during mild hypoxia may enhance cell regeneration (i.e., angiogenesis and neurogenesis), mitochondrial biogenesis, and cell survival in the brain through HIF-1α target genes [16,17]. In severe hypoxia, HIF-1α causes various gene expression changes and post-translational modifications related to cell damage, mitochondrial dysfunction, cellular lipid peroxidation, and inflammasome formation [18,19,20,21,22] (Figure 1). Here, this review focuses on the detrimental effects of HIF-1α on cell damage under severe hypoxic conditions.
Narrowing or blockage of arteries can induce ischemic stroke, leading to reactive oxygen species (ROS)-mediated death of neurons, endothelial cells, and glia including oligodendrocytes [23,24]. TBI is an acquired brain injury caused by a mechanical impact on the head [25]. Individuals with severe ischemia or trauma are more susceptible to the development of AD [26,27]. Some cases of dementia may arise from cerebral hypoperfusion after ischemic injury due to decreased beta-amyloid (Aβ) clearance or catabolism [26,27,28]. A significant increase in microglia-specific thromboxane A synthase 1 was observed in the human AD brain [29]. Notably, thromboxane A is a potent vasoconstrictor in the cerebral circulation and is also a target for the secondary prevention of stroke [30]. Therefore, hypoxia-related diseases may share similar pathological pathways.
This review discusses the current evidence regarding the contribution of severe hypoxia to CNS injuries, with an emphasis on HIF-1α-mediated pathways. Understanding the role of these pathways in severe hypoxic CNS injuries, such as ischemic stroke, TBI, and AD, will provide clues for therapeutic strategies. HIF-1α-mediated pathways include neurovascular inflammation, oxidative stress, glial activation, vascular damage, mitochondrial dysfunction, and cell death (Figure 1) [1,14,22,31]. This review describes the important actions of HIF-1α in severe hypoxic CNS injuries and their potential pathogenetic mechanisms.
2. Role of HIF in Cell Damage
Several cell death pathways are associated with severe hypoxia. This section discusses the roles of HIF-1α in types of cell death in the CNS.
2.1. Apoptosis
Apoptosis refers to the process of programmed cell death, characterized by the orchestrated collapse of a cell, with membrane blebbing, cell shrinkage, chromatin condensation, and DNA fragmentation [32]. While many reports have demonstrated the protective effects of HIF-1α on metabolic adaptation during mild hypoxia, activation of the HIF-1α signaling pathway during severe hypoxia is associated with cell death [15]. Genetic neuronal HIF-1α and HIF-2α deficiencies triggered neuronal survival and sensorimotor function in an ischemic stroke model [8]. BCL-2/adenovirus E1B 19 kDa-interacting protein 3 (BNIP3), a downstream target gene of HIF-1α, is involved in apoptosis [15,33]. HIF-1 is linked to oxidative stress-induced Aβ accumulation and subsequent activation of the pro-death gene BNIP3 in primary cortical neurons [34]. In TBI, HIF-1α mediates tumor necrosis factor (TNF)-related apoptosis, inducing ligand-induced neuronal apoptosis [35]. Pericyte cell death is related to HIF-1α-mediated caspase 3 activation during TBI [9].
Apoptosis is related to mitochondrial permeability. The release of mitochondrial DNA (mtDNA) during mitochondrial outer membrane permeabilization generally involves the pro-apoptotic pore-formation proteins BCL-2-associated X, apoptosis regulator (BAX), and BCL-2 antagonist/killer 1 (BAK1) [36]. The relative availability of BAX and BAK molecules tunes apoptotic pore growth to control mtDNA-mediated inflammation [37]. BAX–BAK1 pores in the outer mitochondrial membrane enable extrusion of the inner mitochondrial membrane into the cytosol, culminating in inner mitochondrial membrane breakdown and cytosolic release of mtDNA [38]. A positive relationship between HIF-1α and pro-apoptotic proteins, such as BAX, has been reported [18,39]. Overexpression of HIF-1α upregulates active monomers of BAX in hypoxic cardiomyocyte cells [18]. HIF-1α modulates apoptosis-inducing proteins (i.e., BAX, BNIP3), leading to mitochondrial dysfunction and consequent cell death under severe hypoxic conditions [40,41].
2.2. Ferroptosis
Ferroptosis is an iron-dependent form of cell death that results from increased ROS and lipid peroxidation in the plasma and mitochondrial membranes [42]. The Fenton reaction involves the production of ROS from the reaction between H2O2 and ferrous iron (Fe2+), which can trigger lipid peroxidation. HIF-1α plays a role in CNS ferroptosis. During hypoxia, HIF-1α upregulates heme oxygenase-1 (HO-1, encoded by HMOX1) gene expression [43,44]. HO-1 resides within the endoplasmic reticulum, and its sustained expression in glial fibrillary acidic protein (GFAP)-expressing astrocytes exacerbates AD development [44]. Transgenic mice exhibiting prolonged expression of HO-1 in astrocytes (GFAP.HMOX1 transgenic) acquire abnormal iron deposition in the mitochondria of astrocytes located in the striatum as well as neuronal deficiency and reduced cognitive ability [45,46,47,48]. When GFAP.HMOX1 transgenic mice-derived astrocytes and neurons are co-cultured, active caspase-3 (an apoptotic factor) is increased in transgenic-derived neurons compared with wild-type preparations [48]. These studies suggest that overexpression of HO-1 in astrocytes may give rise to neuronal dysfunction.
Mitoferrin 2 (MFRN2) is an iron transporter found in the mitochondrial membrane. MFRN2 gene silencing inhibits mitochondrial iron overload and stabilizes the mitochondrial membrane potential in TNF-α-treated endothelial cells [49]. Abnormal iron accumulation is associated with cognitive impairment in patients with AD [50]. Induction of ferroptosis by excessive HO-1 overexpression may be associated with the reduction in the free iron-binding ability of ferritin induced by pro-oxidant conditions (i.e., heme, Aβ, H2O2, dopamine, hyperoxia, ultraviolet light, heavy metals, prostaglandins, and nitric oxide) [51,52]. HO-1 accelerates erastin-induced ferroptotic death in fibrosarcoma cells [51]. Induction of ferroptosis by the HIF-1α/HO-1 pathway has been reported in various cells, including neuronal and vascular smooth muscle cells [19,20,21]. Ferroptosis aggravated diabetic nephropathy and damaged renal tubules via the HIF-1α/HO-1 pathway in a mouse model of diabetes [53]. Compared to sodium iodate (an ROS inducer) alone, sodium iodate with hypoxia-mediated HIF-1α stabilization markedly increases retinal pigment epithelial cell death via ferroptosis [54].
BNIP3 can act as the upstream regulator of the HIF-1α-mediated glycolytic program [55]. In melanoma cells, BNIP3 deficiency results in increased intracellular iron levels caused by heightened nuclear receptor coactivator 4 (NCOA4)-mediated autophagic degradation of ferritin (ferritinophagy), which facilitates HIF-1α degradation [55]. NCOA4 is a selective cargo receptor that mediates autophagic degradation of ferritin, a cytosolic iron-storage complex [56]. Under autophagy-disrupting conditions, NCOA4 may be unable to target ferritin for lysosomal degradation, resulting in the accumulation of free iron [57]. Both HIF-1α and HIF-2α upregulate NCOA4 expression in hepatic cells treated with the iron chelator deferoxamine [58]. Taken together, HIF-1α may upregulate iron production and mitochondrial accumulation via HO-1. Additionally, HIF-α may be involved in ferritinophagy through the NCOA4-mediated pathway.
3. Role of HIF in Inflammasomes
Pyroptosis refers to an inflammatory form of cell death involving caspase-1 activation and consequent cleavage of the pore-forming protein gasdermin D [59]. Inflammation can be regulated by the nucleotide-binding domain and leucine-rich-repeat-containing receptor (NLR) family, to form large multiprotein complexes called inflammasomes [60]. Inflammasomes include NLR family pyrin domain-containing 1 (NLRP1), NLRP3, NLRP6, NLRP7, NLRP12, caspase activation and recruitment domain (CARD)-domain containing 4 (NLRC4), and absent in melanoma 2 (AIM2) [61,62].
NLRP3 interacts with the adaptor molecule apoptosis-associated speck-like protein-containing CARD (ASC) via its pyrin domain (PYD). The CARD domain of ASC recruits the CARD domain of pro-caspase-1 to form the NLRP3–ASC–pro-caspase-1 complex, which is also known as the NLRP3 inflammasome [63,64,65]. Upon activation, NLRP1 directly interacts with pro-caspase-1, without the adaptor protein ASC [66]. However, ASC can also increase NLRP1-mediated caspase-1 activation [67]. Double-stranded DNA (dsDNA) from the nucleus and mitochondria induces AIM2 activation [68]. The AIM2 inflammasome increases the conversion of gasdermin D into gasdermin D-N fragments, which leads to pyroptosis [68]. The observation that AIM2 overexpression results in greater gasdermin D activity, and vice versa, in atherosclerotic plaques of apolipoprotein E−/− (ApoE−/−) mice further supports the role of AIM2 in mediating pyroptotic cell death [69].
Caspases belong to the cysteine-dependent protease family and play key roles in various forms of cell damage [70]. Caspase-3, -6, -7, -8, and -9 are involved in apoptosis in mammals. Caspase-1, -4, -5, and -12 are associated with inflammation in humans [71]. CoCl2-induced HIF-1α induces apoptosis by upregulating caspase-3, -8, and -9 in human fibroblasts [72]. Retinal ischemia/reperfusion increases Toll-like receptor 4 (TLR4) expression, triggering caspase-8 signaling [73]. Caspase-8 promotes NLRP1 and NLRP3 inflammasome activation and interleukin (IL)-1β production in an acute glaucoma model [73]. Intravitreous injection of a caspase-8 inhibitor reduces ischemia/reperfusion-induced NLRP1 and NLRP3 inflammasomes and IL-1β production [73]. Hypoxic injury is associated with inflammasome formation in the brain. In the following sections, the relationship between HIF-1α and inflammasomes in neurovascular diseases, such as stroke, TBI, and AD, is discussed.
3.1. Role of HIF in Stroke, with a Focus on Inflammasomes
Higher HIF-1α levels have been significantly correlated with the initial stroke scale score, indicating a worse outcome [13]. Necrosis is an accidental cell death that results in the uncontrolled release of inflammatory cellular contents [74]. Necroptosis mimics features of both apoptosis and necrosis. Necroptosis requires related proteins, such as receptor-interacting protein kinase-3 (RIPK3) and the effector mixed lineage kinase domain-like protein (MLKL) [75]. HIF-1α also regulates necroptosis-related proteins, such as RIPK3 and MLKL, in ischemic stroke [75]. Enhanced HIF-1α levels after ischemic stroke appear to be involved in RIPK3/MLKL activation, leading to activation of the NLRP3 inflammasome [75]. HIF-1α induces NLRP3 inflammasome-dependent pyroptotic and apoptotic cell death following ischemic stroke in adult rats [76]. Treatment with an HIF-1α inhibitor reduces macrophage and neutrophil infiltration in the ipsilateral brain [76].
The role of NLRP1 in acute ischemic brain injury has also been previously demonstrated [77]. Nuclear factor-κB (NF-κB) and mitogen-activated protein kinase (i.e., p38, JNK, and ERK) inhibitors attenuate the expression and activation of inflammasome-related proteins, such as NLRP1, NLRP3, ASC, IL-1β, and IL-18, in the brain after focal ischemic stroke [78]. In neuronal cells exposed to oxygen–glucose deprivation, increased levels of inflammasome-related proteins (i.e., NLRP1, ASC, caspase-1, IL-1β, and IL-18) were significantly reduced by microRNAs (i.e., miR-9a-5p) [79]. miR-9a-5p binds within the 3′-untranslated region of NLRP1 [79]. Thus, the downregulation of miR-9a-5p enhances NLRP1 inflammasome-mediated ischemic injury by upregulating NLRP1 expression [79].
In a mouse model of cerebral ischemia, AIM2 and NLRC4 inflammasomes, along with ASC, contributed to the development of acute brain injury [61]. Chronic cerebral hypoperfusion activates and upregulates the AIM2 and NLRP3 inflammasomes [80]. The expression of NLRP3 and AIM2 is upregulated in glial cells in the brains of patients with cerebral infarction in the chronic phase, suggesting that chronic cerebral hypoperfusion induces inflammasomes [80].
3.2. Role of HIF in TBI, with a Focus on Inflammasomes
HIF-1α aggravates TBI via NLRP3-inflammasome-mediated pyroptosis and microglial activation by 3 days after TBI [22]. NLRP3 is mainly expressed in microglia and has also been detected in endothelial cells, astrocytes, and oligodendrocytes [81,82]. Administration of an HIF-1α inhibitor to TBI model mice reduces TBI-mediated NLRP3 protein levels and blood–brain barrier (BBB) breakdown, showing improvement of behavioral functions [22]. In the TBI brain, the levels of NLRP1, NLRP3, NLRC4, and AIM2 were found to be increased in microvascular endothelial cells [82]. Administration of a caspase-1 inhibitor after TBI decreases pyroptosis, as evidenced by decreased cleaved gasdermin D and IL-1β levels, and alleviates TBI-induced BBB leakage without affecting the expression of NLRP1, NLRP3, NLRC4, and AIM2 [82]. Compared to wild-type mice, cortical samples of Nlrp1− and Asc−/− mice that had been subjected to TBI showed reduced levels of proinflammatory cytokines, such as IL-1β and IL-6 [83]. However, motor deficits did not change in Nlrp1−/− and Asc−/− mice as compared to wild-type mice after TBI [83]. Further studies revealing the relationship between inflammasome blockade and the improvement in behavioral function after TBI are required.
3.3. Role of HIF in AD, Focusing on Inflammasomes
Hypoxia facilitated plaque formation in an AD transgenic mouse model, leading to memory deficits [84]. Various pathogenic mechanisms of AD have been considered, including chronic hypoxia, amyloid precursor protein (APP) expression, Aβ aggregation, and hyperphosphorylated tau protein accumulation [1,2,85]. Chronic hypoxia-mediated HIF-1α may upregulate the activity of β-site APP-cleaving enzyme 1, facilitate the β-cleavage of APP, increase Aβ deposition, and potentiate memory deficits in APP23 transgenic AD mice [1,84,86]. In addition, HIF-1α binds to the γ-secretase subunit gene promoter and subsequently induces γ-secretase-mediated Aβ production during hypoxia [87]. NLRP1 and NLRP3 are more greatly activated in monocytes obtained from patients with AD than in those from healthy controls [88]. Mitochondrial ROS drive the assembly of the NLRP3 inflammasome inside microglia [89], consequently facilitating tau pathology [90]. The transfer of miR-146a-5p from microglial exosomes into intermittently hypoxic neurons reduces the mRNA levels of HIF-1α, NLRP3, IL-1β, and IL-18 [91]. These effects are reversed by overexpression of HIF-1α [91]. Chronic intermittent hypoxia induces the formation of the neuronal NLRP3 inflammasome, which is a key regulator of neuroinflammation during cognitive impairment [91]. HIF-1α and NLRP1 protein levels were markedly increased in the endothelial cells of the brain and the retinas of triple (PS1M146V, APPswe, and tauP301L) transgenic mouse models of AD aged 16 months [14]. In this AD mouse model, HIF-1α protein expression was observed in the cytoplasm of endothelial cells in the brain and retina [14]. Cytoplasmic accumulation of HIF-1α may have been related to apoptosis and necrosis in the cerebral cortexes of 24-month-old rats exposed to intermittent hypoxia [92]. During oxygen–glucose deprivation, endothelial cells show upregulated HIF-1α and NLRP1 protein levels, while downregulation of HIF-1α reduces NLRP1 expression, and vice versa [14]. Thus, HIF-1α can affect inflammasomes in sustained hypoxic injuries.
4. Role of HIF-1α in Mitochondrial Functions
During hypoxia, ROS are generated due to mitochondrial depolarization especially through complex I and III, activation of xanthine oxidase, and NADPH oxidases at different oxygen levels in the brain [93]. Mitochondria consume O2 for ATP production. Hypoxia elicits mitochondrial ROS accumulation due to the insufficient number of electrons supplied by O2, causing imbalanced electron transfer in the electron transport chain [94]. Mitochondrial dysfunction is the main cause of energy failure in damaged tissues and is the basis for cell death. Activation of mitochondrial ATP-sensitive K+ channels promotes HIF-1α expression in ischemic injuries [95,96]. A positive feedback regulation between ROS production and the HIF pathway has also been reported [97,98]. ROS and reactive nitrogen species (RNS) contribute to oxidative stress production [99]. One RNS, peroxynitrite (ONOO−), can be formed by nitric oxide (NO) and O2− [100]. Hypoxia-induced HIF-1α can upregulate inducible nitric oxide synthase (iNOS) expression [101], leading to NO production. Uncoupling of endothelial NOS produces O2− during hypoxia [102].
The repair strategy for hypoxic neurovascular diseases involves artificial mitochondrial transfer/transplantation by transferring healthy mitochondria into damaged cells. Mitochondrial transplantation is an emerging therapeutic approach for the treatment of hypoxic neurovascular diseases [103,104,105,106]. Transfer of healthy mitochondria ameliorates cognitive deficits and neuronal damage and increases cell viability [103,104,105,106]. Mitochondrial transfer holds great potential for maintaining homeostasis during pathological processes.
4.1. Mitochondrial DNA
The released mtDNA, cytosolic double-stranded DNA, can act as a ligand for various detrimental signal sensors, activating an innate immune response in a caspase-independent manner [107]. These signal sensors include the NLRP3 inflammasome, AIM2 inflammasome, and the cytosolic cyclic GMP–AMP synthase (cGAS)-stimulator of interferon genes (STING) pathway [68,70,107]. cGAS-STING signaling cascades facilitate the mtDNA-mediated secretion of inflammatory cytokines and activate immune cells [70,107] (Figure 2). mtDNA can be oxidized by ROS to generate fragments [107,108]. The binding of cytosolic oxidized mtDNA to the NF-κB-mediated NLRP3 inflammasome suggests a link between apoptosis and the inflammasome [109]. Autophagy proteins (i.e., microtubule-associated protein 1 light chain 3B [LC3B] and beclin1) regulate the innate immune response by inhibiting NLRP3-inflammasome-mediated mtDNA release, leading to the preservation of mitochondrial integrity [110]. Autophagy is an evolutionarily conserved, lysosome-dependent mechanism through which eukaryotic cells eliminate potentially cytotoxic or superfluous materials from the cytoplasm, thereby maintaining homeostasis [70]. Depletion of autophagic proteins promotes the accumulation of dysfunctional mtDNA in the cytosol in response to lipopolysaccharides and ATP in macrophages [110]. CoCl2-induced hypoxia upregulates the expression of autophagy-related genes (ATGs), such as BNIP3, BECN1, LC3, ATG5, and ATG7 [111]. HIF-1α inhibitors reduce hypoxic preconditioning-mediated enhancement of BNIP3 and beclin1 protein levels [112].
4.2. HIF-1α–BNIP3 Axis in Mitochondrial Functions
HIF-1α regulates BNIP3 in various cells during hypoxia; however, the HIF-1α–BNIP3 axis can be beneficial or detrimental in a cell-type-dependent manner. Recent reports have shown that HIF-1α and BNIP3 can be translocated to the mitochondria [113,114]. Mitochondrial HIF-1α may play protective roles by reducing ROS generation in response to hypoxia [115]. Under hypoxia/reoxygenation (H/R) conditions, HIF-1α–BNIP3-mediated mitophagy protects tubular cells from renal injury [116]. HIF1A knockout attenuates H/R-induced mitophagy and aggravates H/R-induced apoptosis; these effects are reversed by BNIP3 overexpression in acute kidney damage [116]. Mitophagy is an autophagic response that preferentially degrades permeabilized or otherwise dysfunctional mitochondria [70]. In various hypoxic cells, the signal transducer and activator of transcription 3 (STAT3) and HIF-1α cooperate in the nucleus to transcribe HIF-1α target genes, such as vascular endothelial growth factor (VEGF) and hexokinase1 (HK1) [117,118]. STAT3 transcriptionally activates target genes, including HIF1A, possibly increasing BNIP3 expression [119]. Mitochondrial translocation of STAT3 suppresses autophagy induced by oxidative stress [113]. STAT3 may protect the mitochondria from degradation by mitophagy [113].
Mitochondrial damage may enhance the HIF-1α–BNIP3 axis, thereby promoting mitophagy during hypoxia [120]. Hippocampal neurons show hypoxia- and aging-associated disruption of mitochondrial cristae (reviewed in [121]), possibly resulting in mitophagy. Mitophagy can be evaluated by measuring the mtDNA copy number and changes in mitophagy-related proteins, including translocase of the outer mitochondrial membrane complex subunit 20, cytochrome c oxidase IV, LC3B, and the mitochondrial adaptor nucleoporin p62 in a human cell line [116]. Hypoxia regulates mitophagy through the HIF-1α–BNIP3 pathway in nucleus pulposus cells [122]. Mitochondria in nucleus pulposus cells undergo HIF-1α-dependent fragmentation by modulating dynamin-related protein 1 (DRP1) and mitochondrial dynamin-like GTPase [122]. HIF-1α positively regulates mitochondrial fission through DRP1 [123]. Similar to HIF-1α, mitophagy via BNIP3 signaling involves DRP1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes [124]. Translocation of BNIP3 to the mitochondria causes mitochondrial depolarization by inducing the mitochondrial permeability transition pore (MPTP) [125]. This leads to cytosolic accumulation of mitochondrial molecules. The cytosolic accumulation of mtDNA or mtRNA triggers the activation of TANK-binding kinase 1 (TBK1) by phosphorylation, leading to the stimulation of interferon (IFN)-regulatory factor 3 (IRF3)-mediated immune activation via IFN1β, IL-6, and TNF [70]. Hyperglycemia may aggravate the complexity of coronary atherosclerosis by activation of TBK1–HIF-1α-mediated IL-17/IL-10 signaling in macrophages [126]. The TBK1 inhibitor reverses hyperglycemia-induced HIF-1α expression [126]. Thus, mitochondrial damage in macrophages may enhance HIF-1α expression, thereby promoting inflammation (Figure 2).
4.3. Role of HIF-1α in VDAC1-Mediated Mitochondrial Functions
Three voltage-dependent anion channel (VDAC) family members (VDAC1, VDAC2, and VDAC3) have been identified in mammalian mitochondria [127,128]. Among these three, a relationship between VDAC1 and HIF-1α has been reported [129]. HIF-1α and nuclear respiratory factor 1 can act as transcriptional activators of the VDAC1 promoter following serum starvation and hypoxia [129]. Hypoxia induces ROS generation from the respiratory complex in the inner mitochondrial membrane [127]. ROS production and the consequent mtDNA release through VDAC1 oligomerization can facilitate apoptosis and inflammation. VDAC1 oligomerization into dimers, trimers, tetramers, and higher-order oligomers induces apoptosis by increasing mitochondrial outer membrane permeability, allowing the release of mtDNA into the cytoplasmic matrix [130,131]. In HeLa cells, inhibition of VDAC1 oligomerization reduced selenite-mediated cell apoptosis and mitochondrial dysfunction (i.e., cytochrome c release from the mitochondria to the cytosol, ROS levels, and decreased mitochondrial membrane potential) [132]. VDAC1 forms a macromolecule-sized pore in the outer membranes of the mitochondria, and its oligomerization mediates the transport of proteins and mtDNA [133] (Figure 2). A link between VDAC1 oligomerization and inflammation in inflammatory diseases has also been reported [127].
Mitochondrial HIF-1α plays a somewhat beneficial role in cell survival. Oxidative stress induces mitochondrial translocation of endogenous HIF-1α in HeLa cells [115]. Mitochondria-localized HIF-1α reduces oxidative stress and increases cell survival [115]. HIF-1α associated with the outer mitochondrial membrane protects the integrity of mitochondrial membrane potential and prevents apoptosis by directly regulating VDAC1 and hexokinase 2, leading to the production of a C-terminally truncated active form of VDAC1 [114].
HIF-1α modulates cell metabolism by hypoxia, regulating glucose transporter-1 and hexokinase 2 expression in various cell types [134,135,136]. Hexokinase 2 catalyzes the first stage of glycolysis and suppresses apoptosis by binding to VDAC on the mitochondrial membrane [137]. HIF-1α may be associated with the regulation of mitochondrial functions via direct interactions with hexokinase 2 [137]. A recent report showed that hexokinase 2 dissociation from VDAC triggers the activation of inositol triphosphate receptors, leading to the release of Ca2+ from the endoplasmic reticulum, which is taken up by mitochondria [138]. This influx of Ca2+ into the mitochondria leads to the oligomerization of VDAC, which facilitates NLRP3 inflammasome assembly and activation [138]. The relationship between Aβ-mediated toxicity and VDAC1 has been reported previously. While the VDAC1-N-terminal peptide shows protective effects against Aβ-mediated human neuroblastoma cell apoptosis, VDAC1 facilitates Aβ-mediated cell toxicity, demonstrating mitochondrial dysfunction and apoptosis induction [139]. VDAC1 inhibition enhances mitochondrial function and synaptic activity [140]. Hence, while mitochondrial HIF-1α may protect against oxidative stress, transcriptional activity of HIF-1α may enhance VDAC1 expression leading to VDAC1 oligomerization and consequent inflammasome formation.
5. Role of HIF-1α in Cellular Activation
HIF-1α is closely related to glial activation and consequent release of proinflammatory factors (Figure 3). Inflammatory factors induce BBB leakage by changing the structures of tight junction proteins [141,142]. Endothelial damage, pericyte apoptosis, reactive glial activation (gliosis), and inflammatory cytokines exacerbate CNS neurodegeneration by uncoupling normal cell–cell communication [9]. The infiltration of immune cells through leaky vessels further stimulates various brain cells located in the neurovascular unit.
5.1. Astrocyte Activation
Inactivation of astrocytic VEGFA expression reduces BBB leakage in inflammatory CNS diseases [143]. In the acute phase of stroke, excessive VEGF acts as a potent vascular permeability factor [17,141,144]. Ischemia/reperfusion-injury-mediated release of proinflammatory cytokines and other soluble mediators triggers paracellular permeability and tight junction disruption [145,146,147]. Tight junctions are disrupted during neuroinflammatory diseases, which results in the infiltration of monocytes into the brain parenchyma, where they become activated macrophages [147,148].
Cortical astrocytes located in the penumbra of an ischemic stroke rat model show enhanced levels of high mobility group box 1 (HMGB1) and its receptor TLR4 [149]. Administration of recombinant HMGB1 to the normal rat cortex triggers the expression of TLR4 and its downstream mediator, iNOS, in astrocytes [149]. HIF-1α-mediated human iNOS expression is seen in primary human astrocytes under cytokine-stimulated conditions [101]. HMGB1 is upregulated in human astrocytoma tissues. Moreover, hypoxia-induced HIF-1α is an upstream regulator of HMGB1 in human glioma stem cell lines [150]. iNOS-derived NO triggers the post-translational S-nitrosylation of HMGB1, leading to HMGB1 secretion and proinflammatory responses [151]. Secreted HMGB1 acts in a damage-associated molecular pattern (DAMP), activating the NLRP3 inflammasome [152,153]. Multiple inflammasome-related complications affect immune system homeostasis in patients with severe TBI [154].
5.2. Oligodendrocyte Activation
In the brain, oligodendrocytes produce myelin, which is a lipid-rich membrane. Oligodendrocytes in the white matter have a high metabolic demand that requires mitochondrial ATP production during remyelination processes [155]. White matter degeneration has been correlated with decreased cognitive function during normal brain aging [156]. Hypoxic oligodendrocyte precursor cell (OPC)-derived VEGF is associated with BBB impairment [157]. HIF-1α activates a unique set of genes in OPCs through interaction with the OPC-specific transcription factor OLIG2, which results in impaired oligodendrocyte formation [158]. The receptors for HMGB1 in OPCs include TLR2, TLR4, TLR9, and the receptor for advanced glycation end-product (RAGE) [159]. Treatment of OPCs with HMGB1 blocks OPC maturation into oligodendrocytes and triggers nuclear translocation of NF-κB through a TLR2-dependent pathway [159]. RAGE expression is influenced by hypoxia via nuclear translocation of NF-κB and HIF-1α [160].
5.3. Microglia/Macrophage Activation
The levels of mitochondrial DAMPs (i.e., mtDNA) in patients are often associated with the severity and prognosis of human diseases. Mitochondrial DAMPs are released into the extracellular space, causing immune responses [161]. Immune cells such as macrophages and microglia are activated under hypoxic conditions, leading to increased mobilization [162]. In human AD brains, endothelial cells upregulate genes involved in cytokine secretion and immune responses [29]. AD microglia downregulate homeostatic genes [29]. Inhibition of autophagy in microglia and macrophages exacerbates the innate immune responses and worsens brain injury outcomes [163]. Autophagic flux can be disrupted in brain cells following TBI in mice. Macrophage-/microglia-specific knockout of the essential autophagy gene beclin1 leads to an overall increase in neuroinflammation after TBI [163]. Increasing autophagy following rapamycin treatment decreases inflammation and improves the outcomes in wild-type mice after TBI [163].
In neuroinflammatory disease, increased HMGB1 expression can be detected in astrocytes, microglia, and infiltrating macrophages, which triggers the HMGB1–TLR4–NF-κB signaling pathway [164]. STAT3 activation in microglia may affect pericyte cell death [165]. STAT3 ablation in microglia induces pericyte survival in diabetic retinas via TNF-α–Akt–p70S6 kinase signaling [165]. Activated STAT3 increases HIF-1α protein stability and accelerates de novo synthesis of HIF-1α [118].
Depending on the disease stage and chronicity, microglia are stimulated differently, leading to particular activation states (M1 and M2), which correspond to altered microglial morphology, gene expression, and function [166]. Ramified microglial morphology (M2 phenotype) is associated with normal surveillance activity, while a more rounded phagocytic appearance (M1 phenotype) is observed in the damaged brain [166]. Macrophage-specific HIF-1α-deficient mice show suppressed wire-induced neointimal thickening and decreased infiltration of inflammatory cells as compared to wild-type mice [167]. This result implies that decreasing HIF-1α activity in macrophages may prevent the progression of vascular remodeling [167]. Additionally, HIF-1α-deficient macrophages are positively correlated with the phenotypic profile of M2 macrophages and negatively correlated with that of M1 macrophages [167].
5.4. Vascular Cells
When mice were exposed to chronic mild hypoxia (8% O2), leaky blood vessels were noted [168]. Prolonged hypoxia has deleterious effects on AD pathogenesis [1,14,169,170]. Microvessels obtained from the brains of patients with AD express higher levels of HIF-1α protein than do those in controls [171]. Plasma levels of HMGB1 increase within 30 min of severe trauma in humans, which correlates with tissue hypoperfusion [172].
In spinal cord injury models, primary mouse brain microvascular endothelial cells engulf myelin debris through immunoglobulin G opsonization [173]. Myelin debris in endothelial cells can then be delivered to the lysosomal degradation system via the autophagy pathway [173]. Autophagic degradation of myelin debris is required for endothelial cell proliferation, via VEGF [173]. The uptake of myelin debris by endothelial cells stimulates macrophage recruitment by upregulating monocyte chemoattractant protein-1 (MCP-1), inflammatory responses, and glial activation [173].
MCP-1 upregulates HIF-1α gene expression in human endothelial cells, resulting in VEGF induction [174]. In AD specimens, cerebral microvessels showed higher levels of MCP-1, IL-1β, IL-6, VEGF, and matrix metalloproteinase-9 (MMP-9) than those found in age-matched control brains [175,176,177]. MMPs are a family of zinc- and calcium-dependent endopeptidases degrading the extracellular matrix [175,178]. Among the MMPs, MMP-9 deficiency reduced BBB leakage following TBI in mice [179]. Animal models of TBI or stroke demonstrate higher levels of MMP-9 in the ipsilateral brain regions [9,179]. MMP-9 enables pericytes to detach from the basal lamina, migrate to the newly formed microvasculature, and balance the degradation and maturation of the vasculature after ischemic stroke [180,181]. HIF-1α induces vascular permeability during severe hypoxic conditions by increasing MMP-9 and VEGF expression [182].
6. Conclusions and Future Directions
This review has revealed the molecular mechanisms of a key molecule, HIF-1α, during severe hypoxic conditions, such as those in brain diseases. Severe and chronic hypoxia exacerbates inflammation, mitochondrial malfunction, excessive oxidative stress, and cell death, partly due to the disproportionate accumulation of HIF-1α.
Currently, most HIF-1α inhibitors have been tested in preclinical models of solid tumors [183]. There are currently no clinical trials using HIF-1α inhibitors in stroke, TBI, or AD (https://clinicaltrials.gov/ accessed on 8 April 2024). Instead, clinical trials using “hyperbaric oxygen treatment” are being carried out in mild TBI (NCT02089594, NCT01220713, NCT00594503), stroke (NCT04376359, NCT06148285, NCT04149379, NCT03431402), and mild cognitive impairment (NCT02085330). High O2 therapy can elevate cerebral blood flow and improve cognitive behavioral performances by diminishing hypoxia.
Developing techniques to diminish HIF-1α during severe hypoxia is valuable, creating a new direction for brain disease treatment. Proper inactivation of HIF-1α may contribute to the reduction in inflammasomes and cell damage and to enhanced mitochondrial function through the transcriptional regulations and post-modification of target molecules in neurodegenerative diseases, such as stroke, TBI, and AD.
Funding
This paper was supported by Konkuk University in 2023.
Conflicts of Interest
The author declares no conflict of interest.
References
- Zhang, F.; Niu, L.; Li, S.; Le, W. Pathological Impacts of Chronic Hypoxia on Alzheimer’s Disease. ACS Chem. Neurosci. 2019, 10, 902–909. [Google Scholar] [CrossRef] [PubMed]
- Shapira, R.; Gdalyahu, A.; Gottfried, I.; Sasson, E.; Hadanny, A.; Efrati, S.; Blinder, P.; Ashery, U. Hyperbaric oxygen therapy alleviates vascular dysfunction and amyloid burden in an Alzheimer’s disease mouse model and in elderly patients. Aging 2021, 13, 20935–20961. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Liu, G.; Zeng, X.; Xiang, Y.; Chen, X.; Le, W. Therapeutic effects of long-term HBOT on Alzheimer’s disease neuropathologies and cognitive impairment in APP(swe)/PS1(dE9) mice. Redox Biol. 2024, 70, 103006. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Life with oxygen. Science 2007, 318, 62–64. [Google Scholar] [CrossRef] [PubMed]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Epstein, A.C.; Gleadle, J.M.; McNeill, L.A.; Hewitson, K.S.; O’Rourke, J.; Mole, D.R.; Mukherji, M.; Metzen, E.; Wilson, M.I.; Dhanda, A.; et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 2001, 107, 43–54. [Google Scholar] [CrossRef]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Barteczek, P.; Li, L.; Ernst, A.S.; Bohler, L.I.; Marti, H.H.; Kunze, R. Neuronal HIF-1alpha and HIF-2alpha deficiency improves neuronal survival and sensorimotor function in the early acute phase after ischemic stroke. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2017, 37, 291–306. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.K.; Maki, T.; Mandeville, E.T.; Koh, S.H.; Hayakawa, K.; Arai, K.; Kim, Y.M.; Whalen, M.J.; Xing, C.; Wang, X.; et al. Dual effects of carbon monoxide on pericytes and neurogenesis in traumatic brain injury. Nat. Med. 2016, 22, 1335–1341. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.L.; Park, J.S.; Manzanero, S.; Choi, Y.; Baik, S.H.; Okun, E.; Gelderblom, M.; Fann, D.Y.; Magnus, T.; Launikonis, B.S.; et al. Evidence that collaboration between HIF-1alpha and Notch-1 promotes neuronal cell death in ischemic stroke. Neurobiol. Dis. 2014, 62, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Tao, T.; Xu, J.; Liu, Z.; Zou, Z.; Jin, M. HIF-1alpha attenuates neuronal apoptosis by upregulating EPO expression following cerebral ischemia-reperfusion injury in a rat MCAO model. Int. J. Mol. Med. 2020, 45, 1027–1036. [Google Scholar] [PubMed]
- Hassan, H.; Chen, R. Hypoxia in Alzheimer’s disease: Effects of hypoxia inducible factors. Neural Regen. Res. 2021, 16, 310–311. [Google Scholar] [PubMed]
- Amalia, L.; Sadeli, H.A.; Parwati, I.; Rizal, A.; Panigoro, R. Hypoxia-inducible factor-1alpha in acute ischemic stroke: Neuroprotection for better clinical outcome. Heliyon 2020, 6, e04286. [Google Scholar] [CrossRef] [PubMed]
- Jung, E.; Kim, Y.E.; Jeon, H.S.; Yoo, M.; Kim, M.; Kim, Y.M.; Koh, S.H.; Choi, Y.K. Chronic hypoxia of endothelial cells boosts HIF-1alpha-NLRP1 circuit in Alzheimer’s disease. Free Radic. Biol. Med. 2023, 204, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Mitroshina, E.V.; Savyuk, M.O.; Ponimaskin, E.; Vedunova, M.V. Hypoxia-Inducible Factor (HIF) in Ischemic Stroke and Neurodegenerative Disease. Front. Cell Dev. Biol. 2021, 9, 703084. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, M.; Choi, Y.K. The Role of a Neurovascular Signaling Pathway Involving Hypoxia-Inducible Factor and Notch in the Function of the Central Nervous System. Biomol. Ther. 2020, 28, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.; Chang, M.S.; Koh, S.H.; Choi, Y.K. Repair Mechanisms of the Neurovascular Unit after Ischemic Stroke with a Focus on VEGF. Int. J. Mol. Sci. 2021, 22, 8543. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, R.; Tyson, D.W.; Rosevear, H.M.; Brosius, F.C., 3rd. Hypoxia-inducible factor-1alpha is a critical mediator of hypoxia induced apoptosis in cardiac H9c2 and kidney epithelial HK-2 cells. BMC Cardiovasc. Disord. 2008, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Chen, Y.; Qin, L.; Xu, X.; Sun, Y.; Zhong, M.; Lu, Y.; Hu, K.; Wei, L.; Chen, J. Oxidative stress drives vascular smooth muscle cell damage in acute Stanford type A aortic dissection through HIF-1alpha/HO-1 mediated ferroptosis. Heliyon 2023, 9, e22857. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Zheng, Z.; Guo, Q.; Tian, M.; Yang, J.; Liu, X.; Zhu, X.; Liu, S. The role of HIF-1alpha/HO-1 pathway in hippocampal neuronal ferroptosis in epilepsy. iScience 2023, 26, 108098. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, J.; Zhao, T.; Chen, J.; Kang, L.; Wei, Y.; Han, L.; Shen, L.; Long, C.; Wu, S.; et al. Di-(2-ethylhexyl) phthalate exposure leads to ferroptosis via the HIF-1alpha/HO-1 signaling pathway in mouse testes. J. Hazard. Mater. 2022, 426, 127807. [Google Scholar] [CrossRef] [PubMed]
- Yuan, D.; Guan, S.; Wang, Z.; Ni, H.; Ding, D.; Xu, W.; Li, G. HIF-1alpha aggravated traumatic brain injury by NLRP3 inflammasome-mediated pyroptosis and activation of microglia. J. Chem. Neuroanat. 2021, 116, 101994. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.K.; Hong, J.; Lee, J.W.; Kim, S.S.; Sim, H.; Lee, J.C.; Kim, D.W.; Lim, S.S.; Kang, I.J.; Won, M.H. Ischemia-Induced Cognitive Impairment Is Improved via Remyelination and Restoration of Synaptic Density in the Hippocampus after Treatment with COG-Up((R)) in a Gerbil Model of Ischemic Stroke. Vet. Sci. 2021, 8, 321. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, T.; Chan, P.H. Reactive oxygen radicals and pathogenesis of neuronal death after cerebral ischemia. Antioxid. Redox Signal. 2003, 5, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Mahmood, A.; Chopp, M. Animal models of traumatic brain injury. Nat. Rev. Neurosci. 2013, 14, 128–142. [Google Scholar] [CrossRef] [PubMed]
- Ikonomovic, M.D.; Mi, Z.; Abrahamson, E.E. Disordered APP metabolism and neurovasculature in trauma and aging: Combined risks for chronic neurodegenerative disorders. Ageing Res. Rev. 2017, 34, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Desmond, D.W.; Moroney, J.T.; Sano, M.; Stern, Y. Incidence of dementia after ischemic stroke: Results of a longitudinal study. Stroke J. Cereb. Circ. 2002, 33, 2254–2260. [Google Scholar] [CrossRef] [PubMed]
- Snowdon, D.A.; Greiner, L.H.; Mortimer, J.A.; Riley, K.P.; Greiner, P.A.; Markesbery, W.R. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA 1997, 277, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Grubman, A.; Chew, G.; Ouyang, J.F.; Sun, G.; Choo, X.Y.; McLean, C.; Simmons, R.K.; Buckberry, S.; Vargas-Landin, D.B.; Poppe, D.; et al. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat. Neurosci. 2019, 22, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Diener, H.C.; Cunha, L.; Forbes, C.; Sivenius, J.; Smets, P.; Lowenthal, A. European Stroke Prevention Study. 2. Dipyridamole and acetylsalicylic acid in the secondary prevention of stroke. J. Neurol. Sci. 1996, 143, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Zhang, J.; Huang, G.; Yan, J.; Xu, C.; Dou, Z.; Sun, C.; Zhang, H. The crosstalk between HIFs and mitochondrial dysfunctions in cancer development. Cell Death Dis. 2021, 12, 215. [Google Scholar] [CrossRef] [PubMed]
- Renehan, A.G.; Booth, C.; Potten, C.S. What is apoptosis, and why is it important? BMJ 2001, 322, 1536–1538. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, L.; Liu, H.; Prabhakaran, K.; Zhang, X.; Borowitz, J.L.; Isom, G.E. HIF-1alpha activation by a redox-sensitive pathway mediates cyanide-induced BNIP3 upregulation and mitochondrial-dependent cell death. Free Radic. Biol. Med. 2007, 43, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhang, Z.; Sandhu, G.; Ma, X.; Yang, X.; Geiger, J.D.; Kong, J. Evidence of oxidative stress-induced BNIP3 expression in amyloid beta neurotoxicity. Brain Res. 2007, 1138, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Lu, J.; Wang, X.; Wu, H.; Mei, S.; Zheng, J.; Xu, S.; Lenahan, C.; Chen, S.; Zhang, J.; et al. HIF-1alpha Mediates TRAIL-Induced Neuronal Apoptosis via Regulating DcR1 Expression Following Traumatic Brain Injury. Front. Cell Neurosci. 2020, 14, 192. [Google Scholar] [CrossRef] [PubMed]
- McArthur, K.; Whitehead, L.W.; Heddleston, J.M.; Li, L.; Padman, B.S.; Oorschot, V.; Geoghegan, N.D.; Chappaz, S.; Davidson, S.; San Chin, H.; et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 2018, 359, eaao6047. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, K.; Hertlein, V.; Jenner, A.; Dellmann, T.; Gojkovic, M.; Pena-Blanco, A.; Dadsena, S.; Wajngarten, N.; Danial, J.S.H.; Thevathasan, J.V.; et al. The interplay between BAX and BAK tunes apoptotic pore growth to control mitochondrial-DNA-mediated inflammation. Mol. Cell 2022, 82, 933–949.e9. [Google Scholar] [CrossRef]
- Riley, J.S.; Quarato, G.; Cloix, C.; Lopez, J.; O’Prey, J.; Pearson, M.; Chapman, J.; Sesaki, H.; Carlin, L.M.; Passos, J.F.; et al. Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. EMBO J. 2018, 37, e99238. [Google Scholar] [CrossRef]
- Wincewicz, A.; Sulkowska, M.; Koda, M.; Sulkowski, S. Cumulative expression of HIF-1-alpha, Bax, Bcl-xL and P53 in human colorectal cancer. Pathology 2007, 39, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Wang, M.; Wu, Y.; Du, J.; Li, Y.; Su, A.; Zhong, L.; Xie, Z.; Gong, M.; Liang, J.; et al. The mechanisms of Huangqi Guizhi Wuwu decoction in treating ischaemic stroke based on network pharmacology and experiment verification. Pharm. Biol. 2023, 61, 1014–1029. [Google Scholar] [CrossRef]
- Guo, K.; Searfoss, G.; Krolikowski, D.; Pagnoni, M.; Franks, C.; Clark, K.; Yu, K.T.; Jaye, M.; Ivashchenko, Y. Hypoxia induces the expression of the pro-apoptotic gene BNIP3. Cell Death Differ. 2001, 8, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.J.; Jiang, B.H.; Chin, B.Y.; Iyer, N.V.; Alam, J.; Semenza, G.L.; Choi, A.M. Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia. J. Biol. Chem. 1997, 272, 5375–5381. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M. Heme oxygenase-1 in Alzheimer disease: A tribute to Moussa Youdim. J. Neural Transm. 2011, 118, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M. Glial HO-1 expression, iron deposition and oxidative stress in neurodegenerative diseases. Neurotox. Res. 1999, 1, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M.; Song, W.; Tavitian, A.; Cressatti, M. The sinister face of heme oxygenase-1 in brain aging and disease. Prog. Neurobiol. 2019, 172, 40–70. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.K.; Kim, Y.M. Beneficial and Detrimental Roles of Heme Oxygenase-1 in the Neurovascular System. Int. J. Mol. Sci. 2022, 23, 7041. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Cressatti, M.; Zukor, H.; Liberman, A.; Galindez, C.; Schipper, H.M. Parkinsonian features in aging GFAP.HMOX1 transgenic mice overexpressing human HO-1 in the astroglial compartment. Neurobiol. Aging 2017, 58, 163–179. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Ye, P.; Kong, C.; Chao, Y.; Yu, W.; Jiang, X.; Luo, J.; Gu, Y.; Chen, S.L. Mitoferrin 2 deficiency prevents mitochondrial iron overload-induced endothelial injury and alleviates atherosclerosis. Exp. Cell Res. 2021, 402, 112552. [Google Scholar] [CrossRef] [PubMed]
- Ayton, S.; Wang, Y.; Diouf, I.; Schneider, J.A.; Brockman, J.; Morris, M.C.; Bush, A.I. Brain iron is associated with accelerated cognitive decline in people with Alzheimer pathology. Mol. Psychiatry 2020, 25, 2932–2941. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.Y.; Park, E.; Lee, S.J.; Chung, S.W. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 2015, 6, 24393–24403. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.K.; Chen, S.E.; Chang, L.C. A Dual Role of Heme Oxygenase-1 in Cancer Cells. Int. J. Mol. Sci. 2018, 20, 39. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Wang, S.; Sun, Z.; Dong, H.; Yu, H.; Huang, M.; Gao, X. Ferroptosis Enhanced Diabetic Renal Tubular Injury via HIF-1alpha/HO-1 Pathway in db/db Mice. Front. Endocrinol. 2021, 12, 626390. [Google Scholar] [CrossRef] [PubMed]
- Henning, Y.; Blind, U.S.; Larafa, S.; Matschke, J.; Fandrey, J. Hypoxia aggravates ferroptosis in RPE cells by promoting the Fenton reaction. Cell Death Dis. 2022, 13, 662. [Google Scholar] [CrossRef] [PubMed]
- Vara-Perez, M.; Rossi, M.; Van den Haute, C.; Maes, H.; Sassano, M.L.; Venkataramani, V.; Michalke, B.; Romano, E.; Rillaerts, K.; Garg, A.D.; et al. BNIP3 promotes HIF-1alpha-driven melanoma growth by curbing intracellular iron homeostasis. EMBO J. 2021, 40, e106214. [Google Scholar] [CrossRef] [PubMed]
- Santana-Codina, N.; Mancias, J.D. The Role of NCOA4-Mediated Ferritinophagy in Health and Disease. Pharmaceuticals 2018, 11, 114. [Google Scholar] [CrossRef] [PubMed]
- Quiles Del Rey, M.; Mancias, J.D. NCOA4-Mediated Ferritinophagy: A Potential Link to Neurodegeneration. Front. Neurosci. 2019, 13, 238. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lozovatsky, L.; Sukumaran, A.; Gonzalez, L.; Jain, A.; Liu, D.; Ayala-Lopez, N.; Finberg, K.E. NCOA4 is regulated by HIF and mediates mobilization of murine hepatic iron stores after blood loss. Blood 2020, 136, 2691–2702. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; McManus, R.M.; Latz, E. Inflammasome signalling in brain function and neurodegenerative disease. Nat. Rev. Neurosci. 2018, 19, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Denes, A.; Coutts, G.; Lenart, N.; Cruickshank, S.M.; Pelegrin, P.; Skinner, J.; Rothwell, N.; Allan, S.M.; Brough, D. AIM2 and NLRC4 inflammasomes contribute with ASC to acute brain injury independently of NLRP3. Proc. Natl. Acad. Sci. USA 2015, 112, 4050–4055. [Google Scholar] [CrossRef] [PubMed]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Zhou, R.; Tschopp, J. The NLRP3 inflammasome: A sensor for metabolic danger? Science 2010, 327, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Mangan, M.S.J.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov. 2018, 17, 688. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Masters, S.L.; Gerlic, M.; Metcalf, D.; Preston, S.; Pellegrini, M.; O’Donnell, J.A.; McArthur, K.; Baldwin, T.M.; Chevrier, S.; Nowell, C.J.; et al. NLRP1 inflammasome activation induces pyroptosis of hematopoietic progenitor cells. Immunity 2012, 37, 1009–1023. [Google Scholar] [CrossRef] [PubMed]
- Faustin, B.; Lartigue, L.; Bruey, J.M.; Luciano, F.; Sergienko, E.; Bailly-Maitre, B.; Volkmann, N.; Hanein, D.; Rouiller, I.; Reed, J.C. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol. Cell 2007, 25, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.R.; Karki, R.; Kanneganti, T.D. Role of AIM2 inflammasome in inflammatory diseases, cancer and infection. Eur. J. Immunol. 2019, 49, 1998–2011. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Han, L.; Guo, J.; Wang, X.; Liu, D.; Tian, J.; Zhang, M.; An, F. AIM2 accelerates the atherosclerotic plaque progressions in ApoE-/- mice. Biochem. Biophys. Res. Commun. 2018, 498, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Guilbaud, E.; Tait, S.W.G.; Yamazaki, T.; Galluzzi, L. Mitochondrial control of inflammation. Nat. Rev. Immunol. 2023, 23, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, G.; Samal, D.; Khandayataray, P.; Murthy, M.K. A Review on Caspases: Key Regulators of Biological Activities and Apoptosis. Mol. Neurobiol. 2023, 60, 5805–5837. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Liu, L.; Li, R.; Wei, X.; Luan, W.; Liu, P.; Zhao, J. Hypoxia-Inducible Factor 1-alpha (HIF-1alpha) Induces Apoptosis of Human Uterosacral Ligament Fibroblasts through the Death Receptor and Mitochondrial Pathways. Med. Sci. Monit. 2018, 24, 8722–8733. [Google Scholar] [CrossRef] [PubMed]
- Chi, W.; Li, F.; Chen, H.; Wang, Y.; Zhu, Y.; Yang, X.; Zhu, J.; Wu, F.; Ouyang, H.; Ge, J.; et al. Caspase-8 promotes NLRP1/NLRP3 inflammasome activation and IL-1beta production in acute glaucoma. Proc. Natl. Acad. Sci. USA 2014, 111, 11181–11186. [Google Scholar] [CrossRef] [PubMed]
- Fink, S.L.; Cookson, B.T. Apoptosis, pyroptosis, and necrosis: Mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 2005, 73, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Stone, C.R.; Geng, X.; Ding, Y. Hypoxia-inducible factor-1 alpha and RIP3 triggers NLRP3 inflammasome in ischemic stroke. Brain Circ. 2018, 4, 191–192. [Google Scholar] [PubMed]
- Jiang, Q.; Geng, X.; Warren, J.; Eugene Paul Cosky, E.; Kaura, S.; Stone, C.; Li, F.; Ding, Y. Hypoxia Inducible Factor-1alpha (HIF-1alpha) Mediates NLRP3 Inflammasome-Dependent-Pyroptotic and Apoptotic Cell Death Following Ischemic Stroke. Neuroscience 2020, 448, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Mi, L.; Min, X.; Chai, Y.; Zhang, J.; Chen, X. NLRP1 Inflammasomes: A Potential Target for the Treatment of Several Types of Brain Injury. Front. Immunol. 2022, 13, 863774. [Google Scholar] [CrossRef]
- Fann, D.Y.; Lim, Y.A.; Cheng, Y.L.; Lok, K.Z.; Chunduri, P.; Baik, S.H.; Drummond, G.R.; Dheen, S.T.; Sobey, C.G.; Jo, D.G.; et al. Evidence that NF-kappaB and MAPK Signaling Promotes NLRP Inflammasome Activation in Neurons Following Ischemic Stroke. Mol. Neurobiol. 2018, 55, 1082–1096. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Zhang, H.; Lu, X.; Wang, J.; Zhang, X.; Sun, S.; Bao, Z.; Tian, W.; Ning, S.; Wang, L.; et al. Overexpression of MicroRNA-9a-5p Ameliorates NLRP1 Inflammasome-mediated Ischemic Injury in Rats Following Ischemic Stroke. Neuroscience 2020, 444, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, H.; Shindo, A.; Shimada, T.; Yata, K.; Wakita, H.; Takahashi, R.; Tomimoto, H. Chronic cerebral hypoperfusion activates AIM2 and NLRP3 inflammasome. Brain Res. 2020, 1736, 146779. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, R.; Tabassum, H.; Parvez, S. NLRP3 inflammasome in traumatic brain injury: Its implication in the disease pathophysiology and potential as a therapeutic target. Life Sci. 2023, 314, 121352. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Li, W.; Huang, S.; Yin, Z.; Xu, X.; Chen, F.; Kong, X.; Wang, H.; Zhang, J.; Lei, P. The pathological role of NLRs and AIM2 inflammasome-mediated pyroptosis in damaged blood-brain barrier after traumatic brain injury. Brain Res. 2018, 1697, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Brickler, T.; Gresham, K.; Meza, A.; Coutermarsh-Ott, S.; Williams, T.M.; Rothschild, D.E.; Allen, I.C.; Theus, M.H. Nonessential Role for the NLRP1 Inflammasome Complex in a Murine Model of Traumatic Brain Injury. Mediators Inflamm. 2016, 2016, 6373506. [Google Scholar] [CrossRef]
- Sun, X.; He, G.; Qing, H.; Zhou, W.; Dobie, F.; Cai, F.; Staufenbiel, M.; Huang, L.E.; Song, W. Hypoxia facilitates Alzheimer’s disease pathogenesis by up-regulating BACE1 gene expression. Proc. Natl. Acad. Sci. USA 2006, 103, 18727–18732. [Google Scholar] [CrossRef] [PubMed]
- Ashok, B.S.; Ajith, T.A.; Sivanesan, S. Hypoxia-inducible factors as neuroprotective agent in Alzheimer’s disease. Clin. Exp. Pharmacol. Physiol. 2017, 44, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhou, K.; Wang, R.; Cui, J.; Lipton, S.A.; Liao, F.F.; Xu, H.; Zhang, Y.W. Hypoxia-inducible factor 1alpha (HIF-1alpha)-mediated hypoxia increases BACE1 expression and beta-amyloid generation. J. Biol. Chem. 2007, 282, 10873–10880. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhang, Y.W.; Zhang, X.; Liu, R.; Zhang, X.; Hong, S.; Xia, K.; Xia, J.; Zhang, Z.; Xu, H. Transcriptional regulation of APH-1A and increased gamma-secretase cleavage of APP and Notch by HIF-1 and hypoxia. FASEB J. 2006, 20, 1275–1277. [Google Scholar] [CrossRef] [PubMed]
- Saresella, M.; La Rosa, F.; Piancone, F.; Zoppis, M.; Marventano, I.; Calabrese, E.; Rainone, V.; Nemni, R.; Mancuso, R.; Clerici, M. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol. Neurodegener. 2016, 11, 23. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Xu, T.; Fang, Q.; Zhang, H.; Yue, L.; Hu, G.; Sun, L. Quercetin hinders microglial activation to alleviate neurotoxicity via the interplay between NLRP3 inflammasome and mitophagy. Redox Biol. 2021, 44, 102010. [Google Scholar] [CrossRef] [PubMed]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Miao, Y.; Xiong, X.; Tan, J.; Han, Z.; Chen, F.; Lei, P.; Zhang, Q. Microglial exosomes alleviate intermittent hypoxia-induced cognitive deficits by suppressing NLRP3 inflammasome. Biol. Direct 2023, 18, 29. [Google Scholar] [CrossRef] [PubMed]
- Rapino, C.; Bianchi, G.; Di Giulio, C.; Centurione, L.; Cacchio, M.; Antonucci, A.; Cataldi, A. HIF-1alpha cytoplasmic accumulation is associated with cell death in old rat cerebral cortex exposed to intermittent hypoxia. Aging Cell 2005, 4, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Lai, U.H.; Zhu, L.; Singh, A.; Ahmed, M.; Forsyth, N.R. Reactive Oxygen Species Formation in the Brain at Different Oxygen Levels: The Role of Hypoxia Inducible Factors. Front. Cell Dev. Biol. 2018, 6, 132. [Google Scholar] [CrossRef] [PubMed]
- Kung-Chun Chiu, D.; Pui-Wah Tse, A.; Law, C.T.; Ming-Jing Xu, I.; Lee, D.; Chen, M.; Kit-Ho Lai, R.; Wai-Hin Yuen, V.; Wing-Sum Cheu, J.; Wai-Hung Ho, D.; et al. Hypoxia regulates the mitochondrial activity of hepatocellular carcinoma cells through HIF/HEY1/PINK1 pathway. Cell Death Dis. 2019, 10, 934. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, T.U.; Yazihan, N.; Dalgic, A.; Kaya, E.E.; Salman, B.; Kocak, M.; Akcil, E. Role of ATP-dependent K channels in the effects of erythropoietin in renal ischaemia injury. Indian. J. Med. Res. 2015, 141, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhou, W.; Chen, W.; Wang, H.; Zhang, Y.; Yu, T. Mechanism of the hypoxia inducible factor 1/hypoxic response element pathway in rat myocardial ischemia/diazoxide post-conditioning. Mol. Med. Rep. 2020, 21, 1527–1536. [Google Scholar] [CrossRef] [PubMed]
- Gorlach, A.; Dimova, E.Y.; Petry, A.; Martinez-Ruiz, A.; Hernansanz-Agustin, P.; Rolo, A.P.; Palmeira, C.M.; Kietzmann, T. Reactive oxygen species, nutrition, hypoxia and diseases: Problems solved? Redox Biol. 2015, 6, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef]
- Wang, X.; Michaelis, E.K. Selective neuronal vulnerability to oxidative stress in the brain. Front. Aging Neurosci. 2010, 2, 12. [Google Scholar] [CrossRef] [PubMed]
- Garry, P.S.; Ezra, M.; Rowland, M.J.; Westbrook, J.; Pattinson, K.T. The role of the nitric oxide pathway in brain injury and its treatment--from bench to bedside. Exp. Neurol. 2015, 263, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Wang, C.; Jin, S.W.; Labrecque, M.P.; Beischlag, T.V.; Brockman, M.A.; Choy, J.C. Expression of human inducible nitric oxide synthase in response to cytokines is regulated by hypoxia-inducible factor-1. Free Radic. Biol. Med. 2019, 130, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Sri Swetha Victoria, V.; Praneeth Kumar, P.; Karmakar, S.; Swetha, M.; Reddy, A. Cross-talk between insulin resistance and nitrogen species in hypoxia leads to deterioration of tissue and homeostasis. Int. Immunopharmacol. 2023, 122, 110472. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Sun, Y.; Qi, Z.; Cao, L.; Ding, S. Mitochondrial transfer/transplantation: An emerging therapeutic approach for multiple diseases. Cell Biosci. 2022, 12, 66. [Google Scholar] [CrossRef] [PubMed]
- Norat, P.; Sokolowski, J.D.; Gorick, C.M.; Soldozy, S.; Kumar, J.S.; Chae, Y.; Yagmurlu, K.; Nilak, J.; Sharifi, K.A.; Walker, M.; et al. Intraarterial Transplantation of Mitochondria after Ischemic Stroke Reduces Cerebral Infarction. Stroke Vasc. Interv. Neurol. 2023, 3, e000644. [Google Scholar] [CrossRef] [PubMed]
- Nitzan, K.; Benhamron, S.; Valitsky, M.; Kesner, E.E.; Lichtenstein, M.; Ben-Zvi, A.; Ella, E.; Segalstein, Y.; Saada, A.; Lorberboum-Galski, H.; et al. Mitochondrial Transfer Ameliorates Cognitive Deficits, Neuronal Loss, and Gliosis in Alzheimer’s Disease Mice. J. Alzheimers Dis. 2019, 72, 587–604. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Qu, D.; Xi, Z.; Huan, Y.; Zhang, K.; Yu, C.; Yang, D.; Kang, J.; Lin, W.; Wu, S.; et al. Mitochondria transplantation protects traumatic brain injury via promoting neuronal survival and astrocytic BDNF. Transl. Res. 2021, 235, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Perez-Trevino, P.; Velasquez, M.; Garcia, N. Mechanisms of mitochondrial DNA escape and its relationship with different metabolic diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165761. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R.; Han, J. Mitochondrial Nucleoid: Shield and Switch of the Mitochondrial Genome. Oxid. Med. Cell Longev. 2017, 2017, 8060949. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef]
- Zhou, J.; Li, C.; Yao, W.; Alsiddig, M.C.; Huo, L.; Liu, H.; Miao, Y.L. Hypoxia-inducible factor-1alpha-dependent autophagy plays a role in glycolysis switch in mouse granulosa cells. Biol. Reprod. 2018, 99, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Lu, N.; Li, X.; Tan, R.; An, J.; Cai, Z.; Hu, X.; Wang, F.; Wang, H.; Lu, C.; Lu, H. HIF-1alpha/Beclin1-Mediated Autophagy Is Involved in Neuroprotection Induced by Hypoxic Preconditioning. J. Mol. Neurosci. 2018, 66, 238–250. [Google Scholar] [CrossRef] [PubMed]
- You, L.; Wang, Z.; Li, H.; Shou, J.; Jing, Z.; Xie, J.; Sui, X.; Pan, H.; Han, W. The role of STAT3 in autophagy. Autophagy 2015, 11, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Mylonis, I.; Kourti, M.; Samiotaki, M.; Panayotou, G.; Simos, G. Mortalin-mediated and ERK-controlled targeting of HIF-1alpha to mitochondria confers resistance to apoptosis under hypoxia. J. Cell Sci. 2017, 130, 466–479. [Google Scholar] [PubMed]
- Li, H.S.; Zhou, Y.N.; Li, L.; Li, S.F.; Long, D.; Chen, X.L.; Zhang, J.B.; Feng, L.; Li, Y.P. HIF-1alpha protects against oxidative stress by directly targeting mitochondria. Redox Biol. 2019, 25, 101109. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.J.; Wang, Z.Y.; Xu, L.; Chen, X.H.; Li, X.X.; Liao, W.T.; Ma, H.K.; Jiang, M.D.; Xu, T.T.; Xu, J.; et al. HIF-1alpha-BNIP3-mediated mitophagy in tubular cells protects against renal ischemia/reperfusion injury. Redox Biol. 2020, 36, 101671. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, A.; Betto, R.M.; Diamante, L.; Tesoriere, A.; Ghirardo, R.; Cioccarelli, C.; Meneghetti, G.; Peron, M.; Laquatra, C.; Tiso, N.; et al. STAT3 and HIF1alpha cooperatively mediate the transcriptional and physiological responses to hypoxia. Cell Death Discov. 2023, 9, 226. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.E.; Lee, H.G.; Cho, I.H.; Chung, D.H.; Yoon, S.H.; Yang, Y.M.; Lee, J.W.; Choi, S.; Park, J.W.; Ye, S.K.; et al. STAT3 is a potential modulator of HIF-1-mediated VEGF expression in human renal carcinoma cells. FASEB J. 2005, 19, 1296–1298. [Google Scholar] [CrossRef] [PubMed]
- Gao, A.; Jiang, J.; Xie, F.; Chen, L. Bnip3 in mitophagy: Novel insights and potential therapeutic target for diseases of secondary mitochondrial dysfunction. Clin. Chim. Acta 2020, 506, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Sun, X.; Zheng, C.; Xue, C.; Jin, Y.; Zhou, N.; Sun, S. The evolutionarily conserved hif-1/bnip3 pathway promotes mitophagy and mitochondrial fission in crustacean testes during hypoxia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2023, 324, R128–R142. [Google Scholar] [CrossRef] [PubMed]
- Burtscher, J.; Mallet, R.T.; Burtscher, M.; Millet, G.P. Hypoxia and brain aging: Neurodegeneration or neuroprotection? Ageing Res. Rev. 2021, 68, 101343. [Google Scholar] [CrossRef] [PubMed]
- Madhu, V.; Boneski, P.K.; Silagi, E.; Qiu, Y.; Kurland, I.; Guntur, A.R.; Shapiro, I.M.; Risbud, M.V. Hypoxic Regulation of Mitochondrial Metabolism and Mitophagy in Nucleus Pulposus Cells Is Dependent on HIF-1alpha-BNIP3 Axis. J. Bone Miner. Res. 2020, 35, 1504–1524. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhao, L.; Peng, R. Hypoxia-Inducible Factor 1 and Mitochondria: An Intimate Connection. Biomolecules 2022, 13, 50. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Lee, H.Y.; Hanna, R.A.; Gustafsson, A.B. Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1924–H1931. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ney, P.A. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009, 16, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Liu, Y.; Xia, X.; Sun, H.; Gao, J.; Ren, Q.; Zhou, T.; Ma, C.; Xia, J.; Yin, C. Activation of macrophage TBK1-HIF-1alpha-mediated IL-17/IL-10 signaling by hyperglycemia aggravates the complexity of coronary atherosclerosis: An in vivo and in vitro study. FASEB J. 2021, 35, e21609. [Google Scholar] [PubMed]
- Hu, H.; Guo, L.; Overholser, J.; Wang, X. Mitochondrial VDAC1: A Potential Therapeutic Target of Inflammation-Related Diseases and Clinical Opportunities. Cells 2022, 11, 3174. [Google Scholar] [CrossRef] [PubMed]
- Zinghirino, F.; Pappalardo, X.G.; Messina, A.; Nicosia, G.; De Pinto, V.; Guarino, F. VDAC Genes Expression and Regulation in Mammals. Front. Physiol. 2021, 12, 708695. [Google Scholar] [CrossRef] [PubMed]
- Guarino, F.; Zinghirino, F.; Mela, L.; Pappalardo, X.G.; Ichas, F.; De Pinto, V.; Messina, A. NRF-1 and HIF-1alpha contribute to modulation of human VDAC1 gene promoter during starvation and hypoxia in HeLa cells. Biochim. Biophys. Acta Bioenerg. 2020, 1861, 148289. [Google Scholar] [CrossRef] [PubMed]
- Keinan, N.; Tyomkin, D.; Shoshan-Barmatz, V. Oligomerization of the mitochondrial protein voltage-dependent anion channel is coupled to the induction of apoptosis. Mol. Cell Biol. 2010, 30, 5698–5709. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Gupta, R.; Blanco, L.P.; Yang, S.; Shteinfer-Kuzmine, A.; Wang, K.; Zhu, J.; Yoon, H.E.; Wang, X.; Kerkhofs, M.; et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science 2019, 366, 1531–1536. [Google Scholar] [CrossRef] [PubMed]
- Ben-Hail, D.; Begas-Shvartz, R.; Shalev, M.; Shteinfer-Kuzmine, A.; Gruzman, A.; Reina, S.; De Pinto, V.; Shoshan-Barmatz, V. Novel Compounds Targeting the Mitochondrial Protein VDAC1 Inhibit Apoptosis and Protect against Mitochondrial Dysfunction. J. Biol. Chem. 2016, 291, 24986–25003. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Mizrachi, D.; Keinan, N. Oligomerization of the mitochondrial protein VDAC1: From structure to function and cancer therapy. Prog. Mol. Biol. Transl. Sci. 2013, 117, 303–334. [Google Scholar]
- Riddle, S.R.; Ahmad, A.; Ahmad, S.; Deeb, S.S.; Malkki, M.; Schneider, B.K.; Allen, C.B.; White, C.W. Hypoxia induces hexokinase II gene expression in human lung cell line A549. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 278, L407–L416. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hydroxylation of HIF-1: Oxygen sensing at the molecular level. Physiology 2004, 19, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, S.; Arii, S.; Mori, A.; Isobe, N.; Yang, W.; Oe, H.; Fujimoto, A.; Yonenaga, Y.; Sakashita, H.; Imamura, M. Hexokinase II and VEGF expression in liver tumors: Correlation with hypoxia-inducible factor 1 alpha and its significance. J. Hepatol. 2004, 40, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Pastorino, J.G.; Hoek, J.B. Regulation of hexokinase binding to VDAC. J. Bioenerg. Biomembr. 2008, 40, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Baik, S.H.; Ramanujan, V.K.; Becker, C.; Fett, S.; Underhill, D.M.; Wolf, A.J. Hexokinase dissociation from mitochondria promotes oligomerization of VDAC that facilitates NLRP3 inflammasome assembly and activation. Sci. Immunol. 2023, 8, eade7652. [Google Scholar] [CrossRef] [PubMed]
- Smilansky, A.; Dangoor, L.; Nakdimon, I.; Ben-Hail, D.; Mizrachi, D.; Shoshan-Barmatz, V. The Voltage-dependent Anion Channel 1 Mediates Amyloid beta Toxicity and Represents a Potential Target for Alzheimer Disease Therapy. J. Biol. Chem. 2015, 290, 30670–30683. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Reddy, P.H. RNA silencing of genes involved in Alzheimer’s disease enhances mitochondrial function and synaptic activity. Biochim. Biophys. Acta 2013, 1832, 2368–2378. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.G.; Zhang, L.; Jiang, Q.; Zhang, R.; Davies, K.; Powers, C.; Bruggen, N.; Chopp, M. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J. Clin. Investig. 2000, 106, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Kim, W.J.; Choi, Y.K.; Song, H.S.; Son, M.J.; Gelman, I.H.; Kim, Y.J.; Kim, K.W. SSeCKS regulates angiogenesis and tight junction formation in blood-brain barrier. Nat. Med. 2003, 9, 900–906. [Google Scholar] [CrossRef] [PubMed]
- Argaw, A.T.; Asp, L.; Zhang, J.; Navrazhina, K.; Pham, T.; Mariani, J.N.; Mahase, S.; Dutta, D.J.; Seto, J.; Kramer, E.G.; et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J. Clin. Investig. 2012, 122, 2454–2468. [Google Scholar] [CrossRef] [PubMed]
- Pedram, A.; Razandi, M.; Levin, E.R. Deciphering vascular endothelial cell growth factor/vascular permeability factor signaling to vascular permeability. Inhibition by atrial natriuretic peptide. J. Biol. Chem. 2002, 277, 44385–44398. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.O.; Harper, S.J. Regulation of vascular permeability by vascular endothelial growth factors. Vascul Pharmacol. 2002, 39, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, K.E.; Witt, K.A. Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol. Dis. 2008, 32, 200–219. [Google Scholar] [CrossRef] [PubMed]
- Knowland, D.; Arac, A.; Sekiguchi, K.J.; Hsu, M.; Lutz, S.E.; Perrino, J.; Steinberg, G.K.; Barres, B.A.; Nimmerjahn, A.; Agalliu, D. Stepwise recruitment of transcellular and paracellular pathways underlies blood-brain barrier breakdown in stroke. Neuron 2014, 82, 603–617. [Google Scholar] [CrossRef] [PubMed]
- Desai, B.S.; Monahan, A.J.; Carvey, P.M.; Hendey, B. Blood-brain barrier pathology in Alzheimer’s and Parkinson’s disease: Implications for drug therapy. Cell Transplant. 2007, 16, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Famakin, B.M.; Tsymbalyuk, O.; Tsymbalyuk, N.; Ivanova, S.; Woo, S.K.; Kwon, M.S.; Gerzanich, V.; Simard, J.M. HMGB1 is a Potential Mediator of Astrocytic TLR4 Signaling Activation following Acute and Chronic Focal Cerebral Ischemia. Neurol. Res. Int. 2020, 2020, 3929438. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Li, H.; Li, Y.; Zhang, Y.; Liu, G.; Mi, H.; Li, H.; Xiao, Q.; Niu, L.; Yu, X. Hypoxia-induced HMGB1 promotes glioma stem cells self-renewal and tumorigenicity via RAGE. iScience 2022, 25, 104872. [Google Scholar] [CrossRef]
- Yang, R.; Gao, Y.; Li, H.; Huang, W.; Tu, D.; Yang, M.; Liu, X.; Hong, J.S.; Gao, H.M. Posttranslational S-nitrosylation modification regulates HMGB1 secretion and promotes its proinflammatory and neurodegenerative effects. Cell Rep. 2022, 40, 111330. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Wu, M.; Lu, Y.; Wu, X.; Yu, B.; Chen, R.; Lu, J.; Tong, H. HMGB1-activatied NLRP3 inflammasome induces thrombocytopenia in heatstroke rat. PeerJ 2022, 10, e13799. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Kang, R.; Tang, D. The mechanism of HMGB1 secretion and release. Exp. Mol. Med. 2022, 54, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Relja, B.; Land, W.G. Damage-associated molecular patterns in trauma. Eur. J. Trauma. Emerg. Surg. 2020, 46, 751–775. [Google Scholar] [CrossRef] [PubMed]
- Meyer, N.; Rinholm, J.E. Mitochondria in Myelinating Oligodendrocytes: Slow and Out of Breath? Metabolites 2021, 11, 359. [Google Scholar] [CrossRef] [PubMed]
- Sams, E.C. Oligodendrocytes in the aging brain. Neuronal Signal 2021, 5, NS20210008. [Google Scholar] [CrossRef] [PubMed]
- Manukjan, N.; Majcher, D.; Leenders, P.; Caiment, F.; van Herwijnen, M.; Smeets, H.J.; Suidgeest, E.; van der Weerd, L.; Vanmierlo, T.; Jansen, J.F.A.; et al. Hypoxic oligodendrocyte precursor cell-derived VEGFA is associated with blood-brain barrier impairment. Acta Neuropathol. Commun. 2023, 11, 128. [Google Scholar] [CrossRef] [PubMed]
- Allan, K.C.; Hu, L.R.; Scavuzzo, M.A.; Morton, A.R.; Gevorgyan, A.S.; Cohn, E.F.; Clayton, B.L.L.; Bederman, I.R.; Hung, S.; Bartels, C.F.; et al. Non-canonical Targets of HIF1a Impair Oligodendrocyte Progenitor Cell Function. Cell Stem Cell 2021, 28, 257–272 e11. [Google Scholar] [CrossRef] [PubMed]
- Rouillard, M.E.; Hu, J.; Sutter, P.A.; Kim, H.W.; Huang, J.K.; Crocker, S.J. The Cellular Senescence Factor Extracellular HMGB1 Directly Inhibits Oligodendrocyte Progenitor Cell Differentiation and Impairs CNS Remyelination. Front. Cell Neurosci. 2022, 16, 833186. [Google Scholar] [CrossRef]
- Bai, W.; Zhou, J.; Zhou, N.; Liu, Q.; Cui, J.; Zou, W.; Zhang, W. Hypoxia-increased RAGE expression regulates chemotaxis and pro-inflammatory cytokines release through nuclear translocation of NF-kappa B and HIF1alpha in THP-1 cells. Biochem. Biophys. Res. Commun. 2018, 495, 2282–2288. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Hisata, S.; Choi, A.M. The Roles of Mitochondrial Damage-Associated Molecular Patterns in Diseases. Antioxid. Redox Signal. 2015, 23, 1329–1350. [Google Scholar] [CrossRef] [PubMed]
- Amersfoort, J.; Eelen, G.; Carmeliet, P. Immunomodulation by endothelial cells—Partnering up with the immune system? Nat. Rev. Immunol. 2022, 22, 576–588. [Google Scholar] [CrossRef] [PubMed]
- Hegdekar, N.; Sarkar, C.; Bustos, S.; Ritzel, R.M.; Hanscom, M.; Ravishankar, P.; Philkana, D.; Wu, J.; Loane, D.J.; Lipinski, M.M. Inhibition of autophagy in microglia and macrophages exacerbates innate immune responses and worsens brain injury outcomes. Autophagy 2023, 19, 2026–2044. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Jing, Y.; Zhao, X.; Wang, M.; Zhang, M.; Ma, R.; Ma, W.; Lv, Y.; Zhu, L. Modulation of the HMGB1/TLR4/NF-kappaB signaling pathway in the CNS by matrine in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2021, 352, 577480. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.H.; Lee, D.H.; Jeong, H.S.; Kim, S.H.; Ye, S.K.; Cho, C.H. STAT3 activation in microglia increases pericyte apoptosis in diabetic retinas through TNF-a/AKT/p70S6 kinase signaling. Biochem. Biophys. Res. Commun. 2022, 613, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Donat, C.K.; Scott, G.; Gentleman, S.M.; Sastre, M. Microglial Activation in Traumatic Brain Injury. Front. Aging Neurosci. 2017, 9, 208. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, T.; Kurobe, H.; Sugasawa, N.; Kinoshita, H.; Higashida, M.; Matsuoka, Y.; Yoshida, Y.; Hirata, Y.; Sakata, M.; Maxfield, M.W.; et al. Role of macrophage-derived hypoxia-inducible factor (HIF)-1alpha as a mediator of vascular remodelling. Cardiovasc. Res. 2013, 99, 705–715. [Google Scholar] [CrossRef]
- Halder, S.K.; Milner, R. Mild hypoxia triggers transient blood-brain barrier disruption: A fundamental protective role for microglia. Acta Neuropathol. Commun. 2020, 8, 175. [Google Scholar] [CrossRef]
- Iadecola, C.; Gottesman, R.F. Cerebrovascular Alterations in Alzheimer Disease. Circ. Res. 2018, 123, 406–408. [Google Scholar] [CrossRef] [PubMed]
- Grammas, P.; Tripathy, D.; Sanchez, A.; Yin, X.; Luo, J. Brain microvasculature and hypoxia-related proteins in Alzheimer’s disease. Int. J. Clin. Exp. Pathol. 2011, 4, 616–627. [Google Scholar] [PubMed]
- Grammas, P.; Samany, P.G.; Thirumangalakudi, L. Thrombin and inflammatory proteins are elevated in Alzheimer’s disease microvessels: Implications for disease pathogenesis. J. Alzheimers Dis. 2006, 9, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.J.; Brohi, K.; Calfee, C.S.; Rahn, P.; Chesebro, B.B.; Christiaans, S.C.; Carles, M.; Howard, M.; Pittet, J.F. Early release of high mobility group box nuclear protein 1 after severe trauma in humans: Role of injury severity and tissue hypoperfusion. Crit. Care 2009, 13, R174. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Zheng, Y.; Sun, L.; Badea, S.R.; Jin, Y.; Liu, Y.; Rolfe, A.J.; Sun, H.; Wang, X.; Cheng, Z.; et al. Microvascular endothelial cells engulf myelin debris and promote macrophage recruitment and fibrosis after neural injury. Nat. Neurosci. 2019, 22, 421–435. [Google Scholar] [CrossRef]
- Hong, K.H.; Ryu, J.; Han, K.H. Monocyte chemoattractant protein-1-induced angiogenesis is mediated by vascular endothelial growth factor-A. Blood 2005, 105, 1405–1407. [Google Scholar] [CrossRef] [PubMed]
- Sandsmark, D.K.; Bashir, A.; Wellington, C.L.; Diaz-Arrastia, R. Cerebral Microvascular Injury: A Potentially Treatable Endophenotype of Traumatic Brain Injury-Induced Neurodegeneration. Neuron 2019, 103, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Grammas, P.; Ovase, R. Inflammatory factors are elevated in brain microvessels in Alzheimer’s disease. Neurobiol. Aging 2001, 22, 837–842. [Google Scholar] [CrossRef]
- Thirumangalakudi, L.; Samany, P.G.; Owoso, A.; Wiskar, B.; Grammas, P. Angiogenic proteins are expressed by brain blood vessels in Alzheimer’s disease. J. Alzheimers Dis. 2006, 10, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Alluri, H.; Wilson, R.L.; Anasooya Shaji, C.; Wiggins-Dohlvik, K.; Patel, S.; Liu, Y.; Peng, X.; Beeram, M.R.; Davis, M.L.; Huang, J.H.; et al. Melatonin Preserves Blood-Brain Barrier Integrity and Permeability via Matrix Metalloproteinase-9 Inhibition. PLoS ONE 2016, 11, e0154427. [Google Scholar] [CrossRef] [PubMed]
- Muradashvili, N.; Benton, R.L.; Saatman, K.E.; Tyagi, S.C.; Lominadze, D. Ablation of matrix metalloproteinase-9 gene decreases cerebrovascular permeability and fibrinogen deposition post traumatic brain injury in mice. Metab. Brain Dis. 2015, 30, 411–426. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.Y.; Guo, Z.N.; Zhang, D.H.; Qu, Y.; Jin, H. The Role of Pericytes in Ischemic Stroke: Fom Cellular Functions to Therapeutic Targets. Front. Mol. Neurosci. 2022, 15, 866700. [Google Scholar] [CrossRef] [PubMed]
- Zozulya, A.; Weidenfeller, C.; Galla, H.J. Pericyte-endothelial cell interaction increases MMP-9 secretion at the blood-brain barrier in vitro. Brain Res. 2008, 1189, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Guan, C.; Liu, C.; Li, H.; Wu, J.; Sun, C. Targeting hypoxia-inducible factor-1alpha: A new strategy for triple-negative breast cancer therapy. Biomed. Pharmacother. 2022, 156, 113861. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Wang, F.; Yang, H.; Wang, Z. Action Sites and Clinical Application of HIF-1alpha Inhibitors. Molecules 2022, 27, 3426. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The roles of hypoxia-inducible factor (HIF)-1α in different oxygen tension conditions in the central nervous system (CNS). In severe hypoxic regions of the brain, HIF-1α can induce sustained inflammation and mitochondrial dysfunction, consequently leading to cell death in the form of apoptosis, pyroptosis, and ferroptosis. HIF-1α stabilization during mild hypoxia may enhance cell regeneration (i.e., angiogenesis and neurogenesis), mitochondrial biogenesis, and cell survival in the brain. During normoxia, HIF-1α can be degraded in the CNS.
Figure 1.
The roles of hypoxia-inducible factor (HIF)-1α in different oxygen tension conditions in the central nervous system (CNS). In severe hypoxic regions of the brain, HIF-1α can induce sustained inflammation and mitochondrial dysfunction, consequently leading to cell death in the form of apoptosis, pyroptosis, and ferroptosis. HIF-1α stabilization during mild hypoxia may enhance cell regeneration (i.e., angiogenesis and neurogenesis), mitochondrial biogenesis, and cell survival in the brain. During normoxia, HIF-1α can be degraded in the CNS.

Figure 2.
Involvement of HIF-1α in mitochondrial-mediated cell damage during severe hypoxia in the CNS. Mitochondria act as multifaceted regulators of cell death, and HIF-1α mediates MPTP, apoptosis, ferroptosis, inflammasome formation, and pyroptosis. Abbreviations: AIM2, absent in melanoma 2; BAX, BCL-2-associated X, apoptosis regulator; BAK, BCL-2 antagonist/killer 1; BNIP3, BCL-2/adenovirus E1B 19 kDa-interacting protein 3; cGAS, cytosolic cyclic GMP–AMP synthase; CNS, central nervous system; HIF-1α, hypoxia-inducible factor-1α; HO-1, heme oxygenase-1; IRF3, interferon regulatory factor; Mfrn2, mitoferrin 2; mtDNA, mitochondrial DNA; MPTP, mitochondrial permeability transition pore; NLRP1, NLR family pyrin domain containing 1; STING, stimulator of interferon genes; TBK1, TANK-binding kinase 1; ROS, reactive oxygen species; VDAC1; voltage-dependent anion channel 1.
Figure 2.
Involvement of HIF-1α in mitochondrial-mediated cell damage during severe hypoxia in the CNS. Mitochondria act as multifaceted regulators of cell death, and HIF-1α mediates MPTP, apoptosis, ferroptosis, inflammasome formation, and pyroptosis. Abbreviations: AIM2, absent in melanoma 2; BAX, BCL-2-associated X, apoptosis regulator; BAK, BCL-2 antagonist/killer 1; BNIP3, BCL-2/adenovirus E1B 19 kDa-interacting protein 3; cGAS, cytosolic cyclic GMP–AMP synthase; CNS, central nervous system; HIF-1α, hypoxia-inducible factor-1α; HO-1, heme oxygenase-1; IRF3, interferon regulatory factor; Mfrn2, mitoferrin 2; mtDNA, mitochondrial DNA; MPTP, mitochondrial permeability transition pore; NLRP1, NLR family pyrin domain containing 1; STING, stimulator of interferon genes; TBK1, TANK-binding kinase 1; ROS, reactive oxygen species; VDAC1; voltage-dependent anion channel 1.

Figure 3.
HIF-1α-mediated glial activation and BBB leakage during severe hypoxia in the CNS. In severe hypoxia, HIF-1α can regulate the release of cytokines and production of inflammatory mediators in endothelial cells and activated glial cells (i.e., astrocytes, oligodendrocytes, microglia), leading to enhanced BBB leakage. Abbreviations: BBB, blood–brain barrier; CNS, central nervous system; HMGB1, high-mobility group box 1; IL-1β, interleukin-1β; iNOS, inducible nitric oxide synthase; MMP-9, matrix metalloproteinase; NF-κB, nuclear factor-κB; NLRP3, NLR family pyrin domain-containing 3; ROS, reactive oxygen species; TNF-α, tumor necrosis factor-α; VEGF, vascular endothelial growth factor.
Figure 3.
HIF-1α-mediated glial activation and BBB leakage during severe hypoxia in the CNS. In severe hypoxia, HIF-1α can regulate the release of cytokines and production of inflammatory mediators in endothelial cells and activated glial cells (i.e., astrocytes, oligodendrocytes, microglia), leading to enhanced BBB leakage. Abbreviations: BBB, blood–brain barrier; CNS, central nervous system; HMGB1, high-mobility group box 1; IL-1β, interleukin-1β; iNOS, inducible nitric oxide synthase; MMP-9, matrix metalloproteinase; NF-κB, nuclear factor-κB; NLRP3, NLR family pyrin domain-containing 3; ROS, reactive oxygen species; TNF-α, tumor necrosis factor-α; VEGF, vascular endothelial growth factor.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Choi, Y.K. Detrimental Roles of Hypoxia-Inducible Factor-1α in Severe Hypoxic Brain Diseases. Int. J. Mol. Sci. 2024, 25, 4465. https://doi.org/10.3390/ijms25084465
AMA Style
Choi YK. Detrimental Roles of Hypoxia-Inducible Factor-1α in Severe Hypoxic Brain Diseases. International Journal of Molecular Sciences. 2024; 25(8):4465. https://doi.org/10.3390/ijms25084465
Chicago/Turabian StyleChoi, Yoon Kyung. 2024. "Detrimental Roles of Hypoxia-Inducible Factor-1α in Severe Hypoxic Brain Diseases" International Journal of Molecular Sciences 25, no. 8: 4465. https://doi.org/10.3390/ijms25084465
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.