
Enterotype-Dependent Probiotic-Mediated Changes in the Male Rat Intestinal Microbiome In Vivo and In Vitro
 , , , , , , ,
, , , , , , ,
Abstract
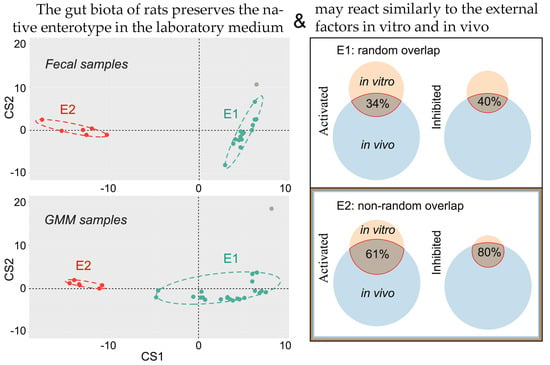
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Rat Intestinal Microbiomes of Two Enterotypes Responded Differently to Probiotic Administration In Vivo
2.2. Shaping Fecal Microbiomes, Probiotic Bacteria Were Counteracted by the Resident Biota
2.3. Multiple Probiotic Administration Can Promote Enterotype Shifts in Resident Microbiomes
2.4. Fecal Microbiomes of Two Enterotypes Transferred to GMM Responded Differently to the Introduction of Probiotic In Vitro
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains, Growth Conditions and Lyophilization
4.2. Animal Handling and Administration of Probiotics In Vivo
4.3. Revival of Fecal Bacteria
4.4. Probiotic Administration In Vitro
4.5. DNA Extraction
4.6. 16S Ribosomal RNA Gene Amplification
4.7. DNA Library Preparation and Sequencing
4.8. Identification of Taxa for Clustering
4.9. Enterotype Assessment and Visualization
4.10. Volcano Plots Visualization
4.11. Pie Charts Visualization
4.12. Biodiversity Assessment
4.13. Venn Diagrams
4.14. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, J.; Tan, Y.; Cheng, H.; Zhang, D.; Feng, W.; Peng, C. Functions of Gut Microbiota Metabolites, Current Status and Future Perspectives. Aging Dis. 2022, 13, 1106–1126. [Google Scholar] [CrossRef] [PubMed]
- Delgado, S.; Sánchez, B.; Margolles, A.; Ruas-Madiedo, P.; Ruiz, L. Molecules Produced by Probiotics and Intestinal Microorganisms with Immunomodulatory Activity. Nutrients 2020, 12, 391. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; O’Connor, A.L.; Becker, S.L.; Patel, R.K.; Martindale, R.G.; Tsikitis, V.L. Gut microbial metabolites and its impact on human health. Ann. Gastroenterol. 2023, 36, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, P.; Zhang, X. Probiotics Regulate Gut Microbiota: An Effective Method to Improve Immunity. Molecules 2021, 26, 6076. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.-J.; Wu, E. The role of gut microbiota in immune homeostasis and autoimmunity. Gut Microbes 2012, 3, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Jandhyala, S.M.; Talukdar, R.; Subramanyam, C.; Vuyyuru, H.; Sasikala, M.; Nageshwar Reddy, D. Role of the normal gut microbiota. World J. Gastroenterol. 2015, 21, 8787–8803. [Google Scholar] [CrossRef] [PubMed]
- Vemuri, R.; Gundamaraju, R.; Shastri, M.D.; Shukla, S.D.; Kalpurath, K.; Ball, M.; Tristram, S.; Shankar, E.M.; Ahuja, K.; Eri, R. Gut Microbial Changes, Interactions, and Their Implications on Human Lifecycle: An Ageing Perspective. Biomed. Res. Int. 2018, 2018, 4178607. [Google Scholar] [CrossRef] [PubMed]
- Nichols, R.G.; Davenport, E.R. The relationship between the gut microbiome and host gene expression: A review. Hum. Genet. 2021, 140, 747–760. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhernakova, D.V.; Kurilshikov, A.; Andreu-Sánchez, S.; Wang, D.; Augustijn, H.E.; Vich Vila, A.; Weersma, R.K.; Medema, M.H.; Netea, M.G.; et al. Influence of the microbiome, diet and genetics on inter-individual variation in the human plasma metabolome. Nat. Med. 2022, 28, 2333–2343. [Google Scholar] [CrossRef]
- Hasan, N.; Yang, H. Factors affecting the composition of the gut microbiota, and its modulation. PeerJ 2019, 7, e7502. [Google Scholar] [CrossRef]
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the gut microbiota in disease. Microb. Ecol. Health Dis. 2015, 26, 26191. [Google Scholar] [CrossRef] [PubMed]
- DeGruttola, A.K.; Low, D.; Mizoguchi, A.; Mizoguchi, E. Current Understanding of Dysbiosis in Disease in Human and Animal Models. Inflamm. Bowel Dis. 2016, 22, 1137–1150. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, L.J.; Monga, M.; Miller, A.W. Defining Dysbiosis for a Cluster of Chronic Diseases. Sci. Rep. 2019, 9, 12918. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Guevarra, R.B.; Kim, Y.T.; Kwon, J.; Kim, H.; Cho, J.H.; Kim, H.B.; Lee, J.H. Role of Probiotics in Human Gut Microbiome-Associated Diseases. J. Microbiol. Biotechnol. 2019, 29, 1335–1340. [Google Scholar] [CrossRef] [PubMed]
- Suk, K.T.; Kim, D.J. Gut microbiota: Novel therapeutic target for nonalcoholic fatty liver disease. Expert. Rev. Gastroenterol. Hepatol. 2019, 13, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Jung Lee, W.; Lattimer, L.D.; Stephen, S.; Borum, M.L.; Doman, D.B. Fecal Microbiota Transplantation: A Review of Emerging Indications Beyond Relapsing Clostridium difficile Toxin Colitis. Gastroenterol. Hepatol. (N. Y.) 2015, 11, 24–32. [Google Scholar] [PubMed]
- Sbahi, H.; Di Palma, J.A. Faecal microbiota transplantation: Applications and limitations in treating gastrointestinal disorders. BMJ Open Gastroenterol. 2016, 3, e000087. [Google Scholar] [CrossRef]
- Giles, E.M.; D’Adamo, G.L.; Forster, S.C. The future of faecal transplants. Nat. Rev. Microbiol. 2019, 17, 719. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.W.; Kuo, C.H.; Kuo, F.C.; Wang, Y.K.; Hsu, W.H.; Yu, F.J.; Hu, H.M.; Hsu, P.I.; Wang, J.Y.; Wu, D.C. Fecal microbiota transplantation: Review and update. J. Formos. Med. Assoc. 2019, 118 (Suppl. S1), S23–S31. [Google Scholar] [CrossRef]
- Choi, H.H.; Cho, Y.S. Fecal Microbiota Transplantation: Current Applications, Effectiveness, and Future Perspectives. Clin. Endosc. 2016, 49, 257–265. [Google Scholar] [CrossRef]
- Basson, A.R.; Zhou, Y.; Seo, B.; Rodriguez-Palacios, A.; Cominelli, F. Autologous fecal microbiota transplantation for the treatment of inflammatory bowel disease. Transl. Res. 2020, 226, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Allen-Vercoe, E.; Petrof, E.O. Fecal microbiota transplantation: In perspective. Therap. Adv. Gastroenterol. 2016, 9, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Moron, R.; Galvez, J.; Colmenero, M.; Anderson, P.; Cabeza, J.; Rodriguez-Cabezas, M.E. The Importance of the Microbiome in Critically Ill Patients: Role of Nutrition. Nutrients 2019, 11, 3002. [Google Scholar] [CrossRef] [PubMed]
- Fong, W.; Li, Q.; Yu, J. Gut microbiota modulation: A novel strategy for prevention and treatment of colorectal cancer. Oncogene 2020, 39, 4925–4943. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, L.; Wang, S.; Wei, J.; Qu, L.; Pan, L.; Xu, K. The role of the gut microbiota and probiotics associated with microbial metabolisms in cancer prevention and therapy. Front. Pharmacol. 2022, 13, 1025860. [Google Scholar] [CrossRef] [PubMed]
- Fijan, S. Microorganisms with Claimed Probiotic Properties: An Overview of Recent Literature. Int. J. Environ. Res. Public Health 2014, 11, 4745–4767. [Google Scholar] [CrossRef] [PubMed]
- Das, T.K.; Pradhan, S.; Chakrabarti, S.; Mondal, K.C.; Ghosh, K. Current status of probiotic and related health benefits. Appl. Food Res. 2022, 2, 100185. [Google Scholar] [CrossRef]
- Anjana; Tiwari, S.K. Bacteriocin-Producing Probiotic Lactic Acid Bacteria in Controlling Dysbiosis of the Gut Microbiota. Front. Cell. Infect. Microbiol. 2022, 12, 851140. [Google Scholar] [CrossRef]
- Bodke, H.; Jogdand, S. Role of Probiotics in Human Health. Cureus 2022, 14, e31313. [Google Scholar] [CrossRef]
- Ansari, F.; Neshat, M.; Pourjafar, H.; Jafari, S.M.; Samakkhah, S.A.; Mirzakhani, E. The role of probiotics and prebiotics in modulating of the gut-brain axis. Front. Nutr. 2023, 10, 1173660. [Google Scholar] [CrossRef]
- Roy, S.; Dhaneshwar, S. Role of prebiotics, probiotics, and synbiotics in management of inflammatory bowel disease: Current perspectives. World J. Gastroenterol. 2023, 29, 2078–2100. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Jin, W.; Liu, S.J.; Jiao, Z.; Li, X. Probiotics, prebiotics, and postbiotics in health and disease. MedComm 2023, 4, e420. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, J.; Wang, M.; Zheng, L.; Cen, Q.; Wang, F.; Zhu, L.; Pang, R.; Zhang, A. Bifidobacterium: A probiotic for the prevention and treatment of depression. Front. Microbiol. 2023, 14, 1174800. [Google Scholar] [CrossRef] [PubMed]
- Handajani, Y.S.; Hengky, A.; Schröder-Butterfill, E.; Hogervorst, E.; Turana, Y. Probiotic supplementation improved cognitive function in cognitively impaired and healthy older adults: A systematic review of recent trials. Neurol. Sci. 2023, 44, 1163–1169. [Google Scholar] [CrossRef] [PubMed]
- Cryan, J.F.; O’Riordan, K.J.; Cowan, C.S.M.; Sandhu, K.V.; Bastiaanssen, T.F.S.; Boehme, M.; Codagnone, M.G.; Cussotto, S.; Fulling, C.; Golubeva, A.V.; et al. The Microbiota-Gut-Brain Axis. Physiol. Rev. 2019, 99, 1877–2013. [Google Scholar] [CrossRef] [PubMed]
- Sudo, N.; Chida, Y.; Aiba, Y.; Sonoda, J.; Oyama, N.; Yu, X.N.; Kubo, C.; Koga, Y. Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice. J. Physiol. 2004, 558, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Zhou, S.; Chen, Q.; Liu, M.; Dong, M.; Hou, J.; Zhou, B. Tryptophan-5-HT pathway disorder was uncovered in the olfactory bulb of a depression mice model by metabolomic analysis. Front. Mol. Neurosci. 2022, 15, 965697. [Google Scholar] [CrossRef] [PubMed]
- Bravo, J.A.; Forsythe, P.; Chew, M.V.; Escaravage, E.; Savignac, H.M.; Dinan, T.G.; Bienenstock, J.; Cryan, J.F. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc. Natl. Acad. Sci. USA 2011, 108, 16050–16055. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Tian, P.; Zhu, H.; Zou, R.; Zhao, J.; Zhang, H.; Wang, G.; Chen, W. Lactobacillus paracasei CCFM1229 and Lactobacillus rhamnosus CCFM1228 Alleviated Depression- and Anxiety-Related Symptoms of Chronic Stress-Induced Depression in Mice by Regulating Xanthine Oxidase Activity in the Brain. Nutrients 2022, 14, 1294. [Google Scholar] [CrossRef]
- Messaoudi, M.; Lalonde, R.; Violle, N.; Javelot, H.; Desor, D.; Nejdi, A.; Bisson, J.F.; Rougeot, C.; Pichelin, M.; Cazaubiel, M.; et al. Assessment of psychotropic-like properties of a probiotic formulation (Lactobacillus helveticus R0052 and Bifidobacterium longum R0175) in rats and human subjects. Br. J. Nutr. 2011, 105, 755–764. [Google Scholar] [CrossRef]
- Kato-Kataoka, A.; Nishida, K.; Takada, M.; Kawai, M.; Kikuchi-Hayakawa, H.; Suda, K.; Ishikawa, H.; Gondo, Y.; Shimizu, K.; Matsuki, T.; et al. Fermented Milk Containing Lactobacillus casei Strain Shirota Preserves the Diversity of the Gut Microbiota and Relieves Abdominal Dysfunction in Healthy Medical Students Exposed to Academic Stress. Appl. Environ. Microbiol. 2016, 82, 3649–3658. [Google Scholar] [CrossRef]
- Möller, C.M.; Olsa, E.J.A.; Ginty, A.T.; Rapelje, A.L.; Tindall, C.L.; Holesh, L.A.; Petersen, K.L.; Conklin, S.M. Influence of Acute Multispecies and Multistrain Probiotic Supplementation on Cardiovascular Function and Reactivity to Psychological Stress in Young Adults: A Double-Blind, Randomized, Placebo-Controlled Trial. Psychosom. Med. 2017, 79, 914–919. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.R.; Allen, A.P.; Temko, A.; Hutch, W.; Kennedy, P.J.; Farid, N.; Murphy, E.; Boylan, G.; Bienenstock, J.; Cryan, J.F.; et al. Lost in translation? The potential psychobiotic Lactobacillus rhamnosus (JB-1) fails to modulate stress or cognitive performance in healthy male subjects. Brain Behav. Immun. 2017, 61, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.-M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Costea, P.I.; Hildebrand, F.; Arumugam, M.; Bäckhed, F.; Blaser, M.J.; Bushman, F.D.; de Vos, W.M.; Ehrlich, S.D.; Fraser, C.M.; Hattori, M.; et al. Enterotypes in the landscape of gut microbial community composition. Nat. Microbiol. 2018, 3, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.Y.; Rho, M.; Song, Y.-M.; Lee, K.; Sung, J.; Ko, G. Stability of Gut Enterotypes in Korean Monozygotic Twins and Their Association with Biomarkers and Diet. Sci. Rep. 2014, 4, 7348. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Mobeen, F.; Sharma, V.; Tulika, P. Enterotype Variations of the Healthy Human Gut Microbiome in Different Geographical Regions. Bioinformation 2018, 14, 560–573. [Google Scholar] [CrossRef] [PubMed]
- Azad, M.A.K.; Sarker, M.; Li, T.; Yin, J. Probiotic Species in the Modulation of Gut Microbiota: An Overview. Biomed. Res. Int. 2018, 2018, 9478630. [Google Scholar] [CrossRef]
- Lee, S.; You, H.; Lee, M.; Kim, D.; Jung, S.; Park, Y.; Hyun, S. Different Reactions in Each Enterotype Depending on the Intake of Probiotic Yogurt Powder. Microorganisms 2021, 9, 1277. [Google Scholar] [CrossRef]
- Faith, J.J.; Guruge, J.L.; Charbonneau, M.; Subramanian, S.; Seedorf, H.; Goodman, A.L.; Clemente, J.C.; Knight, R.; Heath, A.C.; Leibel, R.L.; et al. The long-term stability of the human gut microbiota. Science 2013, 341, 1237439. [Google Scholar] [CrossRef]
- Haddad, E.N.; Sugino, K.Y.; Tucker, R.M.; Comstock, S.S. Gut enterotypes are stable during Bifidobacterium and Lactobacillus probiotic supplementation. J. Food Sci. 2020, 85, 1596–1604. [Google Scholar] [CrossRef] [PubMed]
- Roager, H.M.; Licht, T.R.; Poulsen, S.K.; Larsen, T.M.; Bahl, M.I. Microbial Enterotypes, Inferred by the Prevotella-to-Bacteroides Ratio, Remained Stable during a 6-Month Randomized Controlled Diet Intervention with the New Nordic Diet. Appl. Environ. Microbiol. 2014, 80, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.-J.; Wang, K.-C.; Yuan, L.-B.; Li, H.-F.; Xu, Y.-Y.; Wei, L.-Y.; Chen, L.; Jin, K.-K.; Lin, Q.-Q. Compositional and functional alterations of gut microbiota in patients with stroke. Nutr. Metab. Cardiovasc. Dis. 2021, 31, 3434–3448. [Google Scholar] [CrossRef]
- Cheng, M.; Ning, K. Stereotypes About Enterotype: The Old and New Ideas. Genom. Proteom. Bioinform. 2019, 17, 4–12. [Google Scholar] [CrossRef]
- Wang, D.; Tang, G.; Yu, J.; Li, Y.; Feng, L.; Liu, H.; Li, J.; Chen, L.; Cao, Y.; Yao, J. Microbial Enterotypes Shape the Divergence in Gut Fermentation, Host Metabolism, and Growth Rate of Young Goats. Microbiol. Spectr. 2023, 11, e04818-22. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, F.; Ye, H.; Lu, B. Integrative analysis of the gut microbiome and metabolome in a rat model with stress induced irritable bowel syndrome. Sci. Rep. 2021, 11, 17596. [Google Scholar] [CrossRef]
- Hansen, A.K.; Hansen, C.H.F. The microbiome and rodent models of immune mediated diseases. Mamm. Genome 2021, 32, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Landsiedel, R.; Hahn, D.; Ossig, R.; Ritz, S.; Sauer, L.; Buesen, R.; Rehm, S.; Wohlleben, W.; Groeters, S.; Strauss, V.; et al. Gut microbiome and plasma metabolome changes in rats after oral gavage of nanoparticles: Sensitive indicators of possible adverse health effects. Part. Fibre Toxicol. 2022, 19, 21. [Google Scholar] [CrossRef]
- Choi, S.I.; Kim, N.; Nam, R.H.; Park, J.H.; Nho, H.; Yu, J.E.; Song, C.H.; Lee, S.M.; Lee, D.H. Fecal Microbial Enterotypes Differentially Respond to a High-fat Diet Based on Sex in Fischer-344 Rats. J. Cancer Prev. 2021, 26, 277–288. [Google Scholar] [CrossRef]
- Fan, C.; Zhang, L.; Fu, H.; Liu, C.; Li, W.; Cheng, Q.; Zhang, H.; Jia, S.; Zhang, Y. Enterotypes of the Gut Microbial Community and Their Response to Plant Secondary Compounds in Plateau Pikas. Microorganisms 2020, 8, 1311. [Google Scholar] [CrossRef] [PubMed]
- Palleja, A.; Mikkelsen, K.H.; Forslund, S.K.; Kashani, A.; Allin, K.H.; Nielsen, T.; Hansen, T.H.; Liang, S.; Feng, Q.; Zhang, C.; et al. Recovery of gut microbiota of healthy adults following antibiotic exposure. Nat. Microbiol. 2018, 3, 1255–1265. [Google Scholar] [CrossRef]
- Fassarella, M.; Blaak, E.E.; Penders, J.; Nauta, A.; Smidt, H.; Zoetendal, E.G. Gut microbiome stability and resilience: Elucidating the response to perturbations in order to modulate gut health. Gut 2021, 70, 595–605. [Google Scholar] [CrossRef]
- Cuesta-Zuluaga, J.d.l.; Kelley, S.T.; Chen, Y.; Escobar, J.S.; Mueller, N.T.; Ley, R.E.; McDonald, D.; Huang, S.; Swafford, A.D.; Knight, R.; et al. Age- and Sex-Dependent Patterns of Gut Microbial Diversity in Human Adults. mSystems 2019, 4, e00261-19. [Google Scholar] [CrossRef]
- Bolsega, S.; Bleich, A.; Basic, M. Synthetic Microbiomes on the Rise—Application in Deciphering the Role of Microbes in Host Health and Disease. Nutrients 2021, 13, 4173. [Google Scholar] [CrossRef] [PubMed]
- Mabwi, H.A.; Kim, E.; Song, D.-G.; Yoon, H.S.; Pan, C.-H.; Komba, E.V.G.; Ko, G.; Cha, K.H. Synthetic gut microbiome: Advances and challenges. Comput. Struct. Biotechnol. J. 2021, 19, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.G.; Ho, P.-Y.; Aranda-Díaz, A.; Jain, S.; Yu, F.B.; Meng, X.; Wang, M.; Iakiviak, M.; Nagashima, K.; Zhao, A.; et al. Design, construction, and in vivo augmentation of a complex gut microbiome. Cell 2022, 185, 3617–3636.e9. [Google Scholar] [CrossRef] [PubMed]
- Rackayová, V.; Flatt, E.; Braissant, O.; Grosse, J.; Capobianco, D.; Mastromarino, P.; McMillin, M.; DeMorrow, S.; McLin, V.A.; Cudalbu, C. Probiotics improve the neurometabolic profile of rats with chronic cholestatic liver disease. Sci. Rep. 2021, 11, 2269. [Google Scholar] [CrossRef]
- Inatomi, T.; Honma, M. Ameliorating effect of probiotics in a rat model of chronic kidney disease. PLoS ONE 2023, 18, e0281745. [Google Scholar] [CrossRef]
- Parra, I.; Martínez, I.; Vásquez, L.; Gongora-Alfaro, J.; Tizabi, Y.; Mendieta, L. Neuroprotective and Immunomodulatory Effects of Probiotics in a Rat Model of Parkinson’s Disease. Neurotox. Res. 2023, 41, 187–200. [Google Scholar] [CrossRef]
- Nguyen, T.L.A.; Vieira-Silva, S.; Liston, A.; Raes, J. How informative is the mouse for human gut microbiota research? Dis. Models Mech. 2015, 8, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.L.; Kallstrom, G.; Faith, J.J.; Reyes, A.; Moore, A.; Dantas, G.; Gordon, J.I. Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice. Proc. Natl. Acad. Sci. USA 2011, 108, 6252–6257. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Kim, N.; Nam, R.H.; Park, J.H.; Choi, S.I.; Park, Y.-T.; Kim, Y.-R.; Seok, Y.-J.; Shin, C.M.; Lee, D.H. Gut microbiota and butyrate level changes associated with the long-term administration of proton pump inhibitors to old rats. Sci. Rep. 2019, 9, 6626. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, R.; Wang, S.; Solberg Woods, L.C.; Seshie, O.; Chung, S.T.; Shively, C.A.; Register, T.C.; Craft, S.; McClain, D.A.; Yadav, H. Comparative Microbiome Signatures and Short-Chain Fatty Acids in Mouse, Rat, Non-human Primate, and Human Feces. Front. Microbiol. 2018, 9, 2897. [Google Scholar] [CrossRef] [PubMed]
- Couch, C.E.; Stagaman, K.; Spaan, R.S.; Combrink, H.J.; Sharpton, T.J.; Beechler, B.R.; Jolles, A.E. Diet and gut microbiome enterotype are associated at the population level in African buffalo. Nat. Commun. 2021, 12, 2267. [Google Scholar] [CrossRef] [PubMed]
- Song, E.J.; Shin, J.H. Personalized Diets based on the Gut Microbiome as a Target for Health Maintenance: From Current Evidence to Future Possibilities. J. Microbiol. Biotechnol. 2022, 32, 1497–1505. [Google Scholar] [CrossRef] [PubMed]
- Raymond, F.; Ouameur, A.A.; Déraspe, M.; Iqbal, N.; Gingras, H.; Dridi, B.; Leprohon, P.; Plante, P.-L.; Giroux, R.; Bérubé, È.; et al. The initial state of the human gut microbiome determines its reshaping by antibiotics. ISME J. 2016, 10, 707–720. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Du, M.X.; Abuduaini, R.; Yu, H.Y.; Li, D.H.; Wang, Y.J.; Zhou, N.; Jiang, M.Z.; Niu, P.X.; Han, S.S.; et al. Enlightening the taxonomy darkness of human gut microbiomes with a cultured biobank. Microbiome 2021, 9, 119. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Huang, X.; Chen, J.; Han, B. Formation of a Mixed-Species Biofilm Is a Survival Strategy for Unculturable Lactic Acid Bacteria and Saccharomyces cerevisiae in Daqu, a Chinese Traditional Fermentation Starter. Front. Microbiol. 2020, 11, 138. [Google Scholar] [CrossRef]
- Vartoukian, S.R. Cultivation strategies for growth of uncultivated bacteria. J. Oral. Biosci. 2016, 58, 142–149. [Google Scholar] [CrossRef]
- Bodor, A.; Bounedjoum, N.; Vincze, G.; Kis, Á.; Laczi, K.; Bende, G.; Szilágyi, Á.; Kovács, T.; Perei, K.; Rákhely, G. Challenges of unculturable bacteria: Environmental perspectives. Rev. Environ. Sci. Bio/Technol. 2020, 19, 1–22. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolzhetsov, N.; Markelova, N.; Frolova, M.; Alikina, O.; Glazunova, O.; Safonova, L.; Kalashnikova, I.; Yudin, V.; Makarov, V.; Keskinov, A.; et al. Enterotype-Dependent Probiotic-Mediated Changes in the Male Rat Intestinal Microbiome In Vivo and In Vitro. Int. J. Mol. Sci. 2024, 25, 4558. https://doi.org/10.3390/ijms25084558
Kolzhetsov N, Markelova N, Frolova M, Alikina O, Glazunova O, Safonova L, Kalashnikova I, Yudin V, Makarov V, Keskinov A, et al. Enterotype-Dependent Probiotic-Mediated Changes in the Male Rat Intestinal Microbiome In Vivo and In Vitro. International Journal of Molecular Sciences. 2024; 25(8):4558. https://doi.org/10.3390/ijms25084558
Chicago/Turabian StyleKolzhetsov, Nikolay, Natalia Markelova, Maria Frolova, Olga Alikina, Olga Glazunova, Lubov Safonova, Irina Kalashnikova, Vladimir Yudin, Valentin Makarov, Anton Keskinov, and et al. 2024. "Enterotype-Dependent Probiotic-Mediated Changes in the Male Rat Intestinal Microbiome In Vivo and In Vitro" International Journal of Molecular Sciences 25, no. 8: 4558. https://doi.org/10.3390/ijms25084558