

Synthesis, In Silico and Kinetics Evaluation of N-(β-d-glucopyranosyl)-2-arylimidazole-4(5)-carboxamides and N-(β-d-glucopyranosyl)-4(5)-arylimidazole-2-carboxamides as Glycogen Phosphorylase Inhibitors
 , , and
, , and
Abstract
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
| Submicromolar Inhibitors with Diverse Scaffolds | ||||
 |  |  | ||
| I (0.35) [15] | II X = S (0.16) [25] | IV X = N (0.41) [21] | ||
| III X = CH2 (0.63) [20] | V X = CH (0.031) [21] | |||
| Bioisosteric replacements in I and compounds studied | ||||
 | ||||
| Linker | Ar | |||
 | 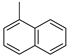 | 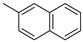 | ||
| a | b | c | ||
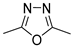 | 545 b | 172 b | 30 | |
| VI [26] | ||||
 | 136 b | 33 | no inh. | |
| VII [26] | ||||
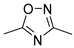 | 104 | 145 b | no inh. | |
| VIII [26] | ||||
 | 1 | not studied kinetically | 9.2 | |
| IX [24] | ||||
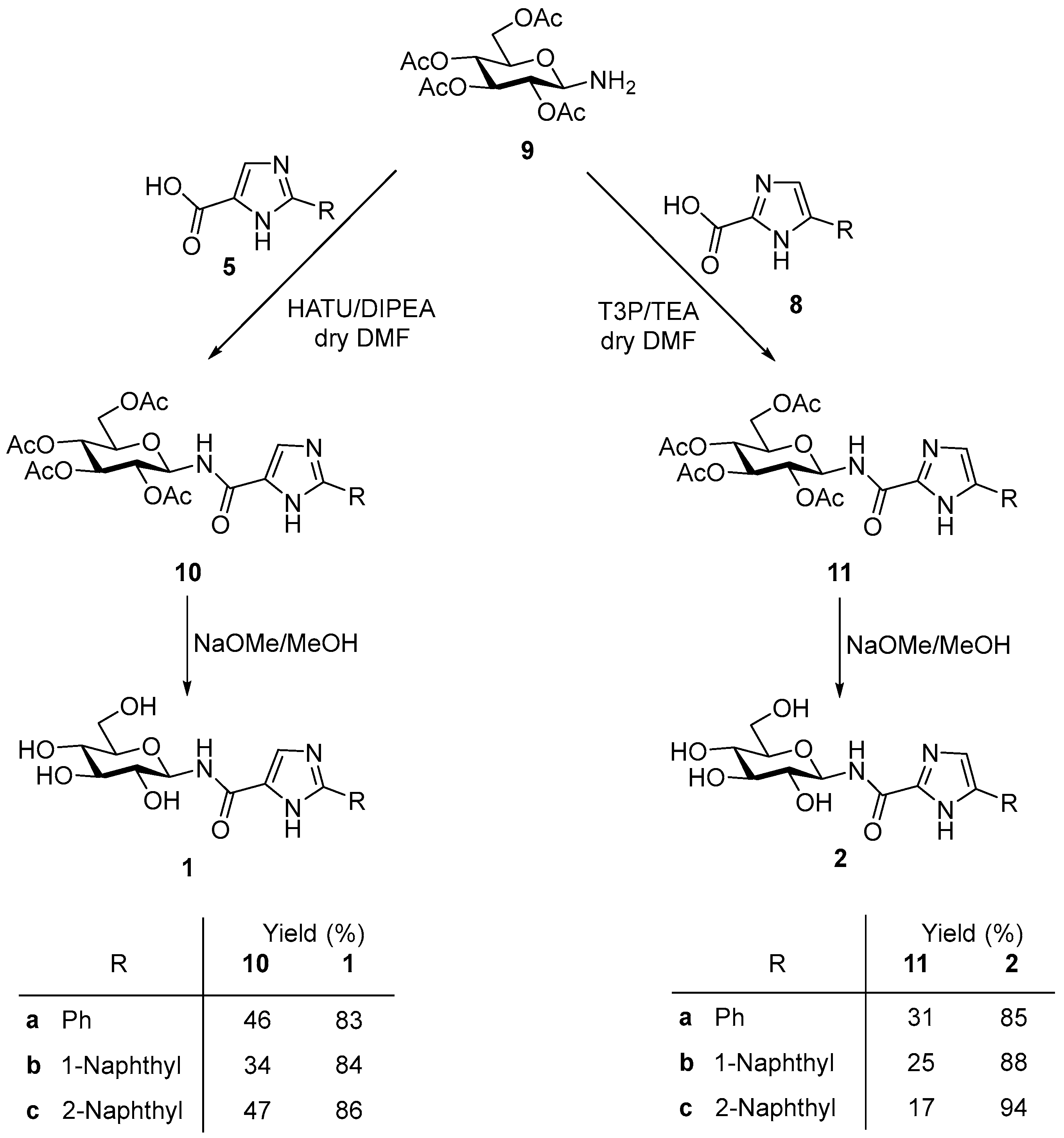
 | Target compounds in this study | |||
| 1 | ||||
 | ||||
| 2 | ||||
2. Results and Discussion
2.1. Computational
2.1.1. Unbound State Calculations
2.1.2. Bound State Calculations
2.2. Synthesis
2.3. Kinetics
3. Conclusions
4. Computational Details
4.1. Protein Preparation
4.2. Ligand Preparation
4.3. Docking Details
5. Experimental
5.1. General Methods
5.1.1. General Procedure A for the Synthesis of Ethyl 2-aryl-1H-imidazole-4(5)-carboxylate Derivatives
5.1.2. General Procedure B for the Synthesis of Ethyl 4(5)-aryl-1H-imidazole-2-carboxylate Derivatives
5.1.3. General Procedure C for the Ester Hydrolysis
5.1.4. General Procedure D for the Synthesis of N-(2,3,4,6-tetra-O-acetyl-β-d-glucopyranosyl)-2-aryl-1H-imidazole-4(5)-carboxamides
5.1.5. General Procedure E for the Synthesis of N-(2,3,4,6-tetra-O-acetyl-β-d-glucopyranosyl)-4(5)-aryl-1H-imidazole-2-carboxamides
5.1.6. General Procedure F for Zemplén Deacetylation
5.2. Synthesis and Characterisation of the Compounds
5.2.1. Ethyl 2-Phenyl-1H-imidazole-4(5)-carboxylate (4a) [36]
5.2.2. Ethyl 2-(1-Naphthyl)-1H-imidazole-4(5)-carboxylate (4b)
5.2.3. Ethyl 2-(2-Naphthyl)-1H-imidazole-4(5)-carboxylate (4c)
5.2.4. 2-Phenyl-1H-imidazole-4(5)-carboxylic Acid (5a) [34]
5.2.5. 2-(1-Naphthyl)-1H-imidazole-4(5)-carboxylic Acid (5b)
5.2.6. 2-(2-Naphthyl)-1H-imidazole-4(5)-carboxylic Acid (5c)
5.2.7. Ethyl 4(5)-Phenyl-1H-imidazole-2-carboxylate (7a) [32]
5.2.8. Ethyl 4(5)-(1-Naphthyl)-1H-imidazole-2-carboxylate (7b)
5.2.9. Ethyl 4(5)-(2-Naphthyl)-1H-imidazole-2-carboxylate (7c)
5.2.10. 4(5)-Phenyl-1H-imidazole-2-carboxylic Acid (8a) [35]
5.2.11. 4(5)-(1-Naphthyl)-1H-imidazole-2-carboxylic Acid (8b)
5.2.12. 4(5)-(2-Naphthyl)-1H-imidazole-2-carboxylic Acid (8c)
5.2.13. N-(2,3,4,6-Tetra-O-acetyl-β-d-glucopyranosyl)-2-phenyl-1H-imidazole-4(5)-carboxamide (10a)
5.2.14. N-(2,3,4,6-Tetra-O-acetyl-β-d-glucopyranosyl)-2-(1-naphthyl)-1H-imidazole-4(5)-carboxamide (10b)
5.2.15. N-(2,3,4,6-Tetra-O-acetyl-β-d-glucopyranosyl)-2-(2-naphthyl)-1H-imidazole-4(5)-carboxamide (10c)
5.2.16. N-(2,3,4,6-Tetra-O-acetyl-β-d-glucopyranosyl)-4(5)-phenyl-1H-imidazole-2-carboxamide (11a)
5.2.17. N-(2,3,4,6-Tetra-O-acetyl-β-d-glucopyranosyl)-4(5)-(1-naphthyl)-1H-imidazole-2-carboxamide (11b)
5.2.18. N-(2,3,4,6-Tetra-O-acetyl-β-d-glucopyranosyl)-4(5)-(2-naphthyl)-1H-imidazole-2-carboxamide (11c)
5.2.19. N-(β-d-Glucopyranosyl)-2-phenyl-1H-imidazole-4(5)-carboxamide (1a)
5.2.20. N-(β-d-Glucopyranosyl)-2-(1-naphthyl)-1H-imidazole-4(5)-carboxamide (1b)
5.2.21. N-(β-d-Glucopyranosyl)-2-(2-naphthyl)-1H-imidazole-4(5)-carboxamide (1c)
5.2.22. N-(β-d-Glucopyranosyl)-4(5)-phenyl-1H-imidazole-2-carboxamide (2a)
5.2.23. N-(β-d-Glucopyranosyl)-4(5)-(1-naphthyl)-1H-imidazole-2-carboxamide (2b)
5.2.24. N-(β-d-Glucopyranosyl)-4(5)-(2-naphthyl)-1H-imidazole-2-carboxamide (2c)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Khan, M.A.B.; Hashim, M.J.; King, J.K.; Govender, R.D.; Mustafa, H.; Al Kaabi, J. Epidemiology of Type 2 Diabetes—Global Burden of Disease and Forecasted Trends. J. Epidemiol. Glob. Health 2020, 10, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Cavaghan, M.K.; Ehrmann, D.A.; Polonsky, K.S. Interactions between insulin resistance and insulin secretion in the development of glucose intolerance. J. Clin. Investig. 2000, 106, 329–333. [Google Scholar] [CrossRef]
- Israili, Z.H. Advances in the Treatment of Type 2 Diabetes Mellitus. Am. J. Ther. 2011, 18, 117–152. [Google Scholar] [CrossRef]
- Wagman, A.S.; Nuss, J.M. Current therapies and emerging targets for the treatment of diabetes. Curr. Pharm. Des. 2001, 7, 417–450. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Ding, Y.; Tanaka, Y.; Zhang, W. Risk factors contributing to type 2 diabetes and recent advances in the treatment and prevention. Int. J. Med. Sci. 2014, 11, 1185–1200. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.H.; Hoover, D.J.; Armento, S.J.; Stock, I.A.; McPherson, R.K.; Danley, D.E.; Stevenson, R.W.; Barrett, E.J.; Treadway, J.L. Discovery of a human liver glycogen phosphorylase inhibitor that lowers blood glucose in vivo. Proc. Natl. Acad. Sci. USA 1998, 95, 1776–1781. [Google Scholar] [CrossRef]
- Xu, L.; Sun, H. Pharmacological manipulation of brain glycogenolysis as a therapeutic approach to cerebral ischemia. Mini Rev. Med. Chem. 2010, 10, 1188–1193. [Google Scholar] [CrossRef]
- Guan, T.; Qian, Y.S.; Tang, X.Z.; Huang, M.H.; Huang, L.F.; Li, Y.M.; Sun, H.B. Maslinic Acid, a Natural Inhibitor of Glycogen Phosphorylase, Reduces Cerebral Ischemic Injury in Hyperglycemic Rats by GLT-1 Up-Regulation. J. Neurosci. Res. 2011, 89, 1829–1839. [Google Scholar] [CrossRef]
- Zois, C.E.; Harris, A.L. Glycogen metabolism has a key role in the cancer microenvironment and provides new targets for cancer therapy. J. Mol. Med. 2016, 94, 137–154. [Google Scholar] [CrossRef]
- Zois, C.E.; Hendriks, A.M.; Haider, S.; Pires, E.; Bridges, E.; Kalamida, D.; Voukantsis, D.; Lagerholm, B.C.; Fehrmann, R.S.N.; den Dunnen, W.F.A.; et al. Liver glycogen phosphorylase is upregulated in glioblastoma and provides a metabolic vulnerability to high dose radiation. Cell Death Dis. 2022, 13, 573. [Google Scholar] [CrossRef]
- Mathomes, R.T.; Koulas, S.M.; Tsialtas, I.; Stravodimos, G.; Welsby, P.J.; Psarra, A.M.G.; Stasik, I.; Leonidas, D.D.; Hayes, J.M. Multidisciplinary docking, kinetics and X-ray crystallography studies of baicalein acting as a glycogen phosphorylase inhibitor and determination of its’ potential against glioblastoma in cellular models. Chem.-Biol. Interact. 2023, 382, 110568. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.M.; Kantsadi, A.L.; Leonidas, D.D. Natural products and their derivatives as inhibitors of glycogen phosphorylase: Potential treatment for type 2 diabetes. Phytochem. Rev. 2014, 13, 471–498. [Google Scholar] [CrossRef]
- Somsák, L. Glucose derived inhibitors of glycogen phosphorylase. Compt. Rend. Chim. 2011, 14, 211–223. [Google Scholar] [CrossRef]
- Hayes, J.M. Computer-Aided Discovery of Glycogen Phosphorylase Inhibitors Exploiting Natural Products. In Discovery and Development of Antidiabetic Agents from Natural Products: Natural Product Drug Discovery, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 29–62. [Google Scholar]
- Somsák, L.; Czifrák, K.; Tóth, M.; Bokor, É.; Chrysina, E.D.; Alexacou, K.M.; Hayes, J.M.; Tiraidis, C.; Lazoura, E.; Leonidas, D.D.; et al. New inhibitors of glycogen phosphorylase as potential antidiabetic agents. Curr. Med. Chem. 2008, 15, 2933–2983. [Google Scholar] [CrossRef] [PubMed]
- Oikonomakos, N.G.; Kontou, M.; Zographos, S.E.; Tsitoura, H.S.; Johnson, L.N.; Watson, K.A.; Mitchell, E.P.; Fleet, G.W.; Son, J.C.; Bichard, C.J.; et al. The design of potential antidiabetic drugs: Experimental investigation of a number of β-d-glucose analogue inhibitors of glycogen phosphorylase. Eur. J. Drug Metab. Pharmacokinet. 1994, 19, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Watson, K.A.; Mitchell, E.P.; Johnson, L.N.; Son, J.C.; Bichard, C.J.; Orchard, M.G.; Fleet, G.W.; Oikonomakos, N.G.; Leonidas, D.D.; Kontou, M.; et al. Design of inhibitors of glycogen phosphorylase: A study of α- and β-C-glucosides and 1-thio-β-d-glucose compounds. Biochem. 1994, 33, 5745–5758. [Google Scholar] [CrossRef] [PubMed]
- Barr, D.; Szennyes, E.; Bokor, É.; Al-Oanzi, Z.H.; Moffatt, C.; Kun, S.; Docsa, T.; Sipos, A.; Davies, M.P.; Mathomes, R.T.; et al. Identification of C-β-d-Glucopyranosyl Azole-Type Inhibitors of Glycogen Phosphorylase That Reduce Glycogenolysis in Hepatocytes: In Silico Design, Synthesis, in Vitro Kinetics, and ex Vivo Studies. ACS Chem. Biol. 2019, 14, 1460–1470. [Google Scholar] [CrossRef] [PubMed]
- Bokor, É.; Kun, S.; Docsa, T.; Gergely, P.; Somsák, L. 4(5)-Aryl-2-C-glucopyranosyl-imidazoles as New Nanomolar Glucose Analogue Inhibitors of Glycogen Phosphorylase. ACS Med. Chem. Lett. 2015, 6, 1215–1219. [Google Scholar] [CrossRef] [PubMed]
- Goyard, D.; Kónya, B.; Chajistamatiou, A.S.; Chrysina, E.D.; Leroy, J.; Balzarin, S.; Tournier, M.; Tousch, D.; Petit, P.; Duret, C.; et al. Glucose-derived spiro-isoxazolines are anti-hyperglycemic agents against type 2 diabetes through glycogen phosphorylase inhibition. Eur. J. Med. Chem. 2016, 108, 444–454. [Google Scholar] [CrossRef]
- Bokor, É. N- and C-Glycopyranosyl heterocycles as glycogen phosphorylase inhibitors. In Recent Trends in Carbohydrate Chemistry—Synthesis, Structure and Function of Carbohydrates; Rauter, A.P., Christensen, B.E., Somsák, L., Kosma, P., Adamo, R., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; Volume 1, pp. 253–300. [Google Scholar]
- Meanwell, N.A. Synopsis of Some Recent Tactical Application of Bioisosteres in Drug Design. J. Med. Chem. 2011, 54, 2529–2591. [Google Scholar] [CrossRef]
- Lima, L.M.A.; Barreiro, E.J. Bioisosterism: A useful strategy for molecular modification and drug design. Curr. Med. Chem. 2005, 12, 23–49. [Google Scholar] [CrossRef]
- Begum, J.; Varga, G.; Docsa, T.; Gergely, P.; Hayes, J.M.; Juhász, L.; Somsák, L. Computationally motivated synthesis and enzyme kinetic evaluation of N-(β-d-glucopyranosyl)-1,2,4-triazolecarboxamides as glycogen phosphorylase inhibitors. Medchemcomm 2015, 6, 80–89. [Google Scholar] [CrossRef]
- Goyard, D.; Kónya, B.; Czifrák, K.; Larini, P.; Demontrond, F.; Leroy, J.; Balzarin, S.; Tournier, M.; Tousch, D.; Petit, P.; et al. Glucose-based spiro-oxathiazoles as in vivo anti-hyperglycemic agents through glycogen phosphorylase inhibition. Org. Biomol. Chem. 2020, 18, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Polyák, M.; Varga, G.; Szilágyi, B.; Juhász, L.; Docsa, T.; Gergely, P.; Begum, J.; Hayes, J.M.; Somsák, L. Synthesis, enzyme kinetics and computational evaluation of N-(β-d-glucopyranosyl) oxadiazolecarboxamides as glycogen phosphorylase inhibitors. Bioorg. Med. Chem. 2013, 21, 5738–5747. [Google Scholar] [CrossRef] [PubMed]
- Kun, S.; Mathomes, R.T.; Docsa, T.; Somsák, L.; Hayes, J.M. Design and Synthesis of 3-(β-d-Glucopyranosyl)-4-amino/4-guanidino Pyrazole Derivatives and Analysis of Their Glycogen Phosphorylase Inhibitory Potential. Molecules 2023, 28, 3005. [Google Scholar] [CrossRef]
- Milletti, F.; Vulpetti, A. Tautomer Preference in PDB Complexes and its Impact on Structure-Based Drug Discovery. J. Chem. Inf. Model. 2010, 50, 1062–1074. [Google Scholar] [CrossRef] [PubMed]
- Onufriev, A.V.; Alexov, E. Protonation and pK changes in protein-ligand binding. Q. Rev. Biophys. 2013, 46, 181–209. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Software Suite; Schrödinger LLC.: New York, NY, USA, 2020.
- Dale, S.E.P.M.; Kingston, C.; Morris, J.A.; Sikervar, V.; Watkin, S.V. Herbicidal Imidazole-Containing Compounds. Worldwide Applications WO2023247358A1, 28 December 2023. p. WO2023247358A2023247351. [Google Scholar]
- Thurkauf, A.H.R.F.; Yuan, J.; Peterson, J.M. Preparation of 4-Aminomethyl-2-Substituted Imidazole Derivatives and 2-Aminomethyl-4-Substituted Imidazole Derivatives; New Classes of Dopamine Receptor Subtype Specific Ligands. Worldwide Applications US20030018025, 23 January 2003. [Google Scholar]
- Kantsadi, A.L.; Bokor, É.; Kun, S.; Stravodimos, G.A.; Chatzileontiadou, D.S.M.; Leonidas, D.D.; Juhász-Tóth, E.; Szakács, A.; Batta, G.; Docsa, T.; et al. Synthetic, enzyme kinetic, and protein crystallographic studies of C-β-d-glucopyranosyl pyrroles and imidazoles reveal and explain low nanomolar inhibition of human liver glycogen phosphorylase. Eur. J. Med. Chem. 2016, 123, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Sívek, R.; Bures, F.; Pytela, O.; Kulhánek, J. Imidazole-based potential bi- and tridentate nitrogen ligands: Synthesis, characterization and application in asymmetric catalysis. Molecules 2008, 13, 2326–2339. [Google Scholar] [CrossRef]
- Lont, P.J.; Van der Plas, H.C. Ring Transformations in Reactions of Heterocyclic Halogeno Compounds with Nucleophiles (XXX) Reaction of Chlorophenyl-Pyrazines and Chlorodiphenyl-Pyrazines with Potassium Amide in Liquid-Ammonia. Recl. Trav. Chim. Pays-Bas 1973, 92, 449–459. [Google Scholar] [CrossRef]
- Huang, Y.; Zu, X.D.; Wu, F.; Xu, J.Y.; Wu, X.M.; Yao, H.Q. Highly efficient oxidation of 2-imidazoline-5-carboxylic derivatives to imidazole-5-carboxylic derivatives by dioxygen. Tetrahedron 2012, 68, 3123–3128. [Google Scholar] [CrossRef]
- Carpino, L.A.; Imazumi, H.; El-Faham, A.; Ferrer, F.J.; Zhang, C.W.; Lee, Y.S.; Foxman, B.M.; Henklein, P.; Hanay, C.; Mügge, C.; et al. The uronium/guanidinium peptide coupling reagents: Finally the true uronium salts. Angew. Chem. Int. Ed. 2002, 41, 442–445. [Google Scholar] [CrossRef]
- Al Musaimi, O.; Wisdom, R.; Talbiersky, P.; De La Torre, B.G.; Albericio, F. Propylphosphonic Anhydride (T3P®) as Coupling Reagent for Solid-Phase Peptide Synthesis. Chemistryselect 2021, 6, 2649–2657. [Google Scholar] [CrossRef]
- Sipos, A.; Szennyes, E.; Hajnal, N.E.; Kun, S.; Szabó, K.E.; Uray, K.; Somsák, L.; Docsa, T.; Bokor, É. Dual-Target Compounds against Type 2 Diabetes Mellitus: Proof of Concept for Sodium Dependent Glucose Transporter (SGLT) and Glycogen Phosphorylase (GP) Inhibitors. Pharmaceuticals 2021, 14, 364. [Google Scholar] [CrossRef] [PubMed]
- Ősz, E.; Somsák, L.; Szilágyi, L.; Kovács, L.; Docsa, T.; Tóth, B.; Gergely, P. Efficient inhibition of muscle and liver glycogen phosphorylases by a new glucopyranosylidene-spiro-thiohydantoin. Bioorg. Med. Chem. Lett. 1999, 9, 1385–1390. [Google Scholar] [CrossRef] [PubMed]
- Kun, S.; Begum, J.; Kyriakis, E.; Stamati, E.C.V.; Barkas, T.A.; Szennyes, E.; Bokor, É.; Szabó, K.E.; Stravodimos, G.A.; Sipos, A.; et al. A multidisciplinary study of 3-(β-d-glucopyranosyl)-5-substituted-1,2,4-triazole derivatives as glycogen phosphorylase inhibitors: Computation, synthesis, crystallography and kinetics reveal new potent inhibitors. Eur. J. Med. Chem. 2018, 147, 266–278. [Google Scholar] [CrossRef]
- Sondergaard, C.R.; Olsson, M.H.M.; Rostkowski, M.; Jensen, J.H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pK(a) Values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef] [PubMed]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.J.; Reboul, M.; Xiang, J.Y.; Wang, L.L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular-Orbital Studies of Organic-Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; Defrees, D.J.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. XXIII. A Polarization-Type Basis Set for 2nd-Row Elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef]
- Marenich, A.V.; Olson, R.M.; Kelly, C.P.; Cramer, C.J.; Truhlar, D.G. Self-consistent reaction field model for aqueous and nonaqueous solutions based on accurate polarized partial charges. J. Chem. Theory Comput. 2007, 3, 2011–2033. [Google Scholar] [CrossRef] [PubMed]
- Helferich, B.M.A. Über N-Glykoside. Chem. Berichte 1951, 85, 1–8. [Google Scholar] [CrossRef]
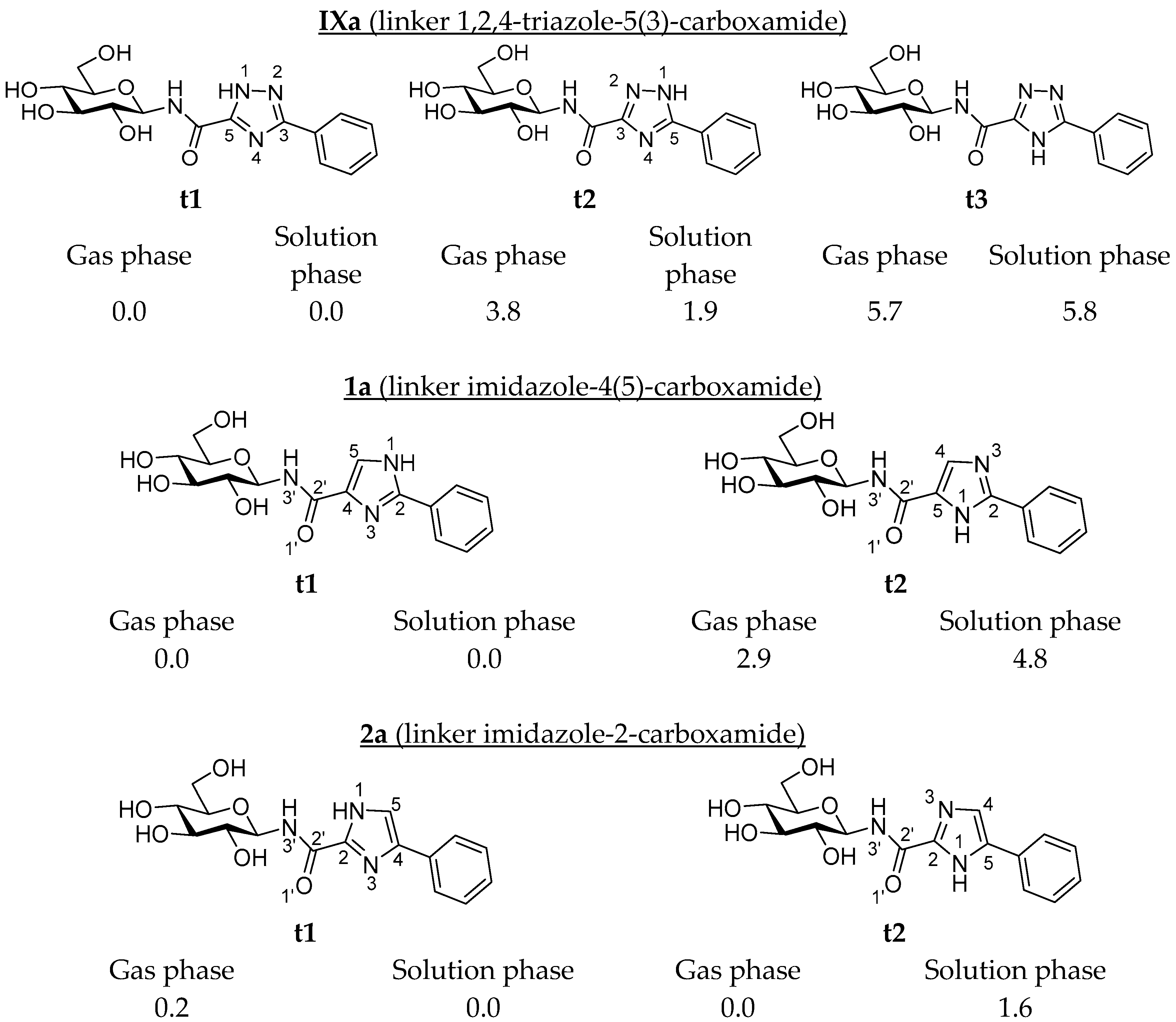

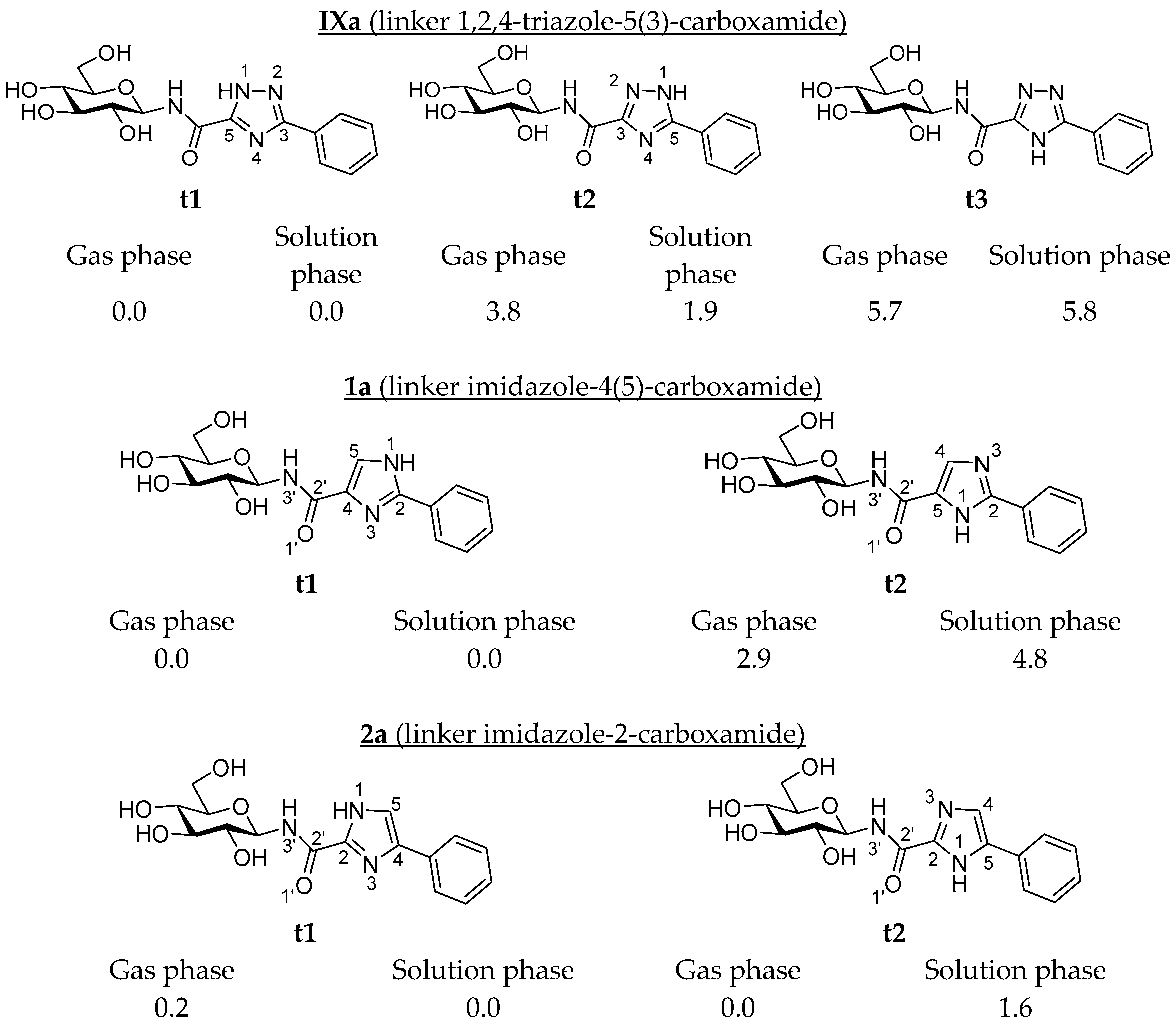
| Ligand | Tautomer a | Phenyl (a) | 1-Naphthyl (b) | 2-Naphthyl (c) |
| IX | t1 | −9.52 (−9.42) | −9.77 (−9.69) | −10.80 (−10.71) |
| t2 | −9.49 (−7.99) | −9.92 (−8.42) | −10.97 (−9.47) | |
| t3 | −9.33 (−7.51) | −9.79 (−7.97) | −10.67 (−8.85) | |
| 1 | t1 | −9.81 (−9.17) | −10.27 (−9.63) | −10.97 (−10.33) |
| t2 | −9.55 (−9.30) | −9.89 (−9.64) | −10.97 (−10.72) | |
| 2 | t1 | −9.70 (−9.29) | −9.93 (−9.53) | −11.00 (−10.59) |
| t2 | −9.61 (−9.18) | −9.99 (−9.56) | −10.99 (−10.55) |
 (i) R-B(OH)2/[Pd]/dry TEA/dry 1,4-dioxane/reflux/Ar; (ii) LiOH/EtOH-H2O/50 °C | ||||
| Yields (%) | ||||
| Entry | R | 4 | 5 | |
| 1 | a | 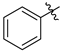 | 31 | 68 |
| 2 | b |  | 25 | 63 |
| 3 | c | 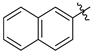 | 19 | 61 |
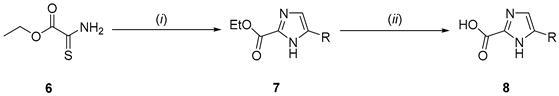 (i) Et3OBF4/dry DCM, then 2-amino-1-arylethan-1-one/NaOAc/AcOH/70 °C; (ii) LiOH/EtOH-H2O/50 °C | ||||
| Yields (%) | ||||
| Entry | R | 7 | 8 | |
| 1 | a | 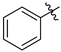 | 57 | 71 |
| 2 | b |  | 29 | 75 |
| 3 | c | 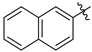 | 75 | 82 |
| Compound | Ar | ||
 |  |  | |
| a | b | c | |
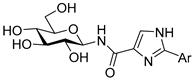 | 10.4 ± 6.2 | 16.2 ± 8.1 | 12.8 ± 3.0 |
| 1 | |||
 | 67.4 ± 9.7 | 4.1 ± 0.8 | 3.3 ± 0.1 |
| 2 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Homolya, L.; Mathomes, R.T.; Varga, L.; Docsa, T.; Juhász, L.; Hayes, J.M.; Somsák, L. Synthesis, In Silico and Kinetics Evaluation of N-(β-d-glucopyranosyl)-2-arylimidazole-4(5)-carboxamides and N-(β-d-glucopyranosyl)-4(5)-arylimidazole-2-carboxamides as Glycogen Phosphorylase Inhibitors. Int. J. Mol. Sci. 2024, 25, 4591. https://doi.org/10.3390/ijms25094591
Homolya L, Mathomes RT, Varga L, Docsa T, Juhász L, Hayes JM, Somsák L. Synthesis, In Silico and Kinetics Evaluation of N-(β-d-glucopyranosyl)-2-arylimidazole-4(5)-carboxamides and N-(β-d-glucopyranosyl)-4(5)-arylimidazole-2-carboxamides as Glycogen Phosphorylase Inhibitors. International Journal of Molecular Sciences. 2024; 25(9):4591. https://doi.org/10.3390/ijms25094591
Chicago/Turabian StyleHomolya, Levente, Rachel T. Mathomes, Luca Varga, Tibor Docsa, László Juhász, Joseph M. Hayes, and László Somsák. 2024. "Synthesis, In Silico and Kinetics Evaluation of N-(β-d-glucopyranosyl)-2-arylimidazole-4(5)-carboxamides and N-(β-d-glucopyranosyl)-4(5)-arylimidazole-2-carboxamides as Glycogen Phosphorylase Inhibitors" International Journal of Molecular Sciences 25, no. 9: 4591. https://doi.org/10.3390/ijms25094591