
Type II Interleukin-4 Receptor Activation in Basal Breast Cancer Cells Promotes Tumor Progression via Metabolic and Epigenetic Modulation
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Type II IL4R Is Associated with Reduced Survival of Basal Breast Cancer Patients
2.2. IL4 Increases Glucose Uptake and Metabolism
2.3. The pAKT/pACLY Signaling Axis Is Activated by IL4 Signaling
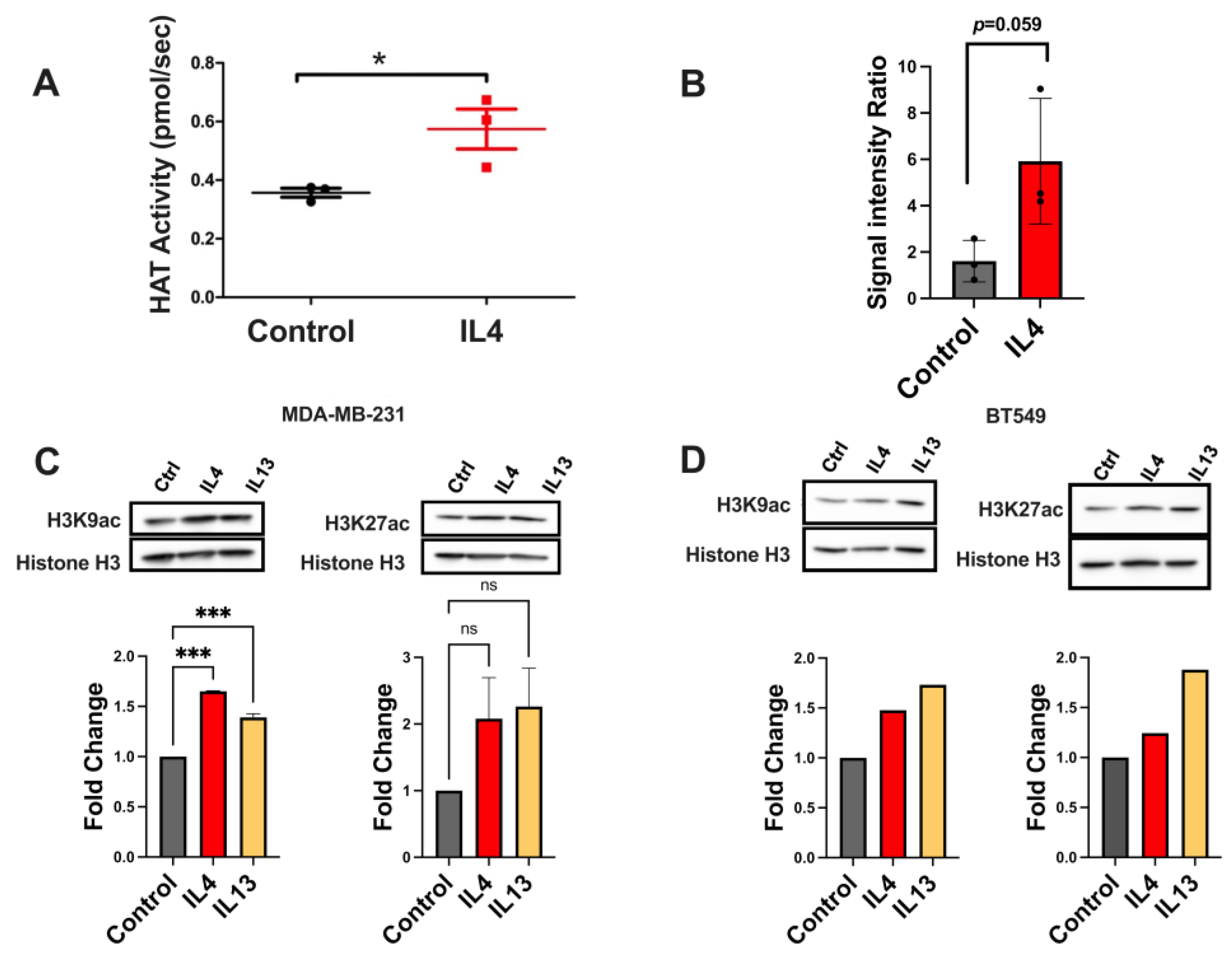
2.4. Histone Acetylation Leading to Gene Expression Changes Is Altered by IL4 Signaling
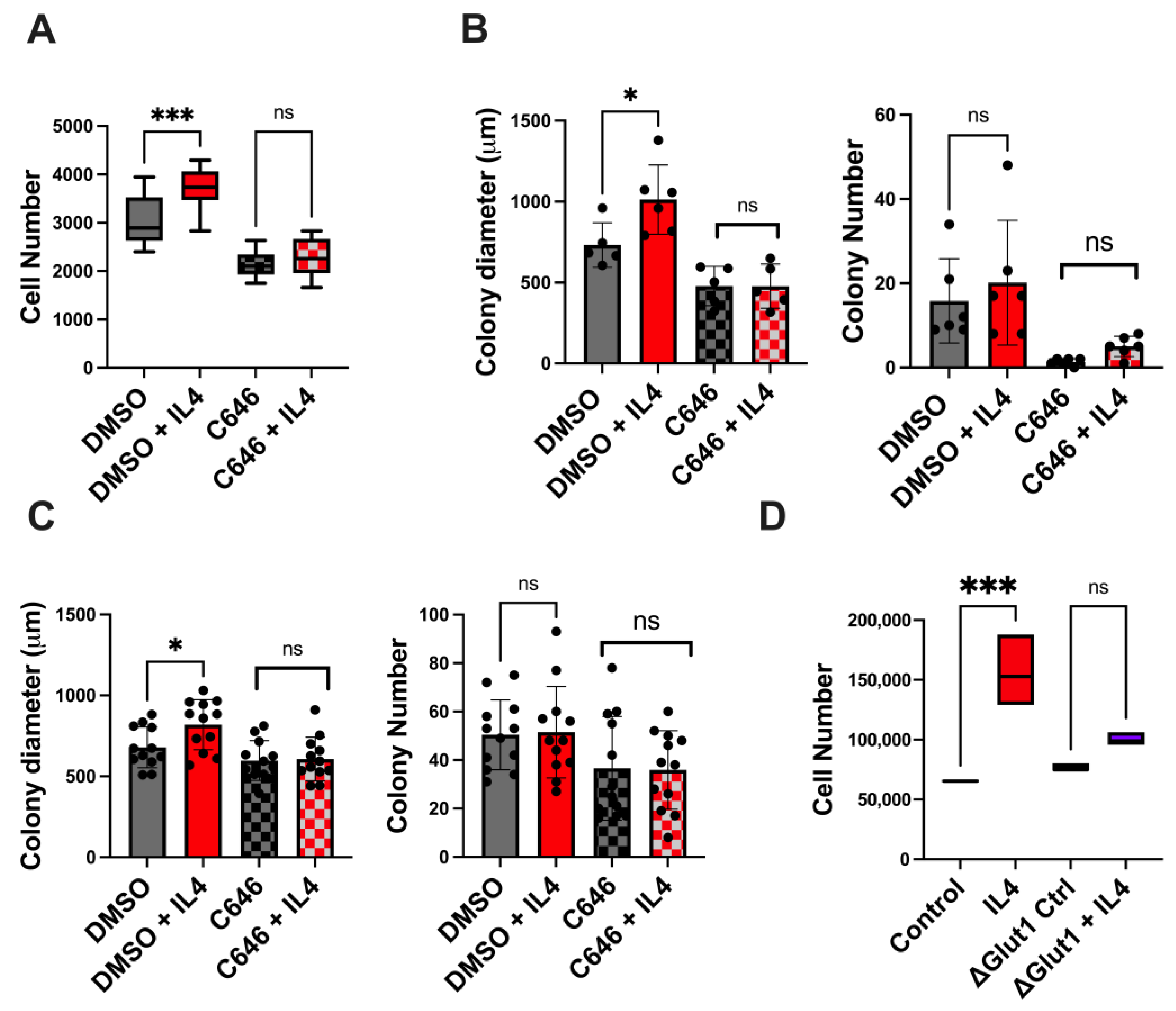
2.5. Both p300 and Glucose Uptake Contribute to Type II IL4R-Induced Proliferation and Survival
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- SEER*Explorer: An Interactive Website for SEER Cancer Statistics [Internet]. Surveillance Research Program, National Cancer Institute. Available online: https://seer.cancer.gov/statistics-network/ (accessed on 16 November 2023).
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-Negative Breast Cancer: Clinical Features and Patterns of Recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef] [PubMed]
- Voduc, K.D.; Cheang, M.C.U.; Tyldesley, S.; Gelmon, K.; Nielsen, T.O.; Kennecke, H. Breast Cancer Subtypes and the Risk of Local and Regional Relapse. J. Clin. Oncol. 2010, 28, 1684–1691. [Google Scholar] [CrossRef] [PubMed]
- Kennecke, H.; Yerushalmi, R.; Woods, R.; Cheang, M.C.U.; Voduc, D.; Speers, C.H.; Nielsen, T.O.; Gelmon, K. Metastatic Behavior of Breast Cancer Subtypes. J. Clin. Oncol. 2010, 28, 3271–3277. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Dewanjee, S.; Li, Y.; Jha, N.K.; Chen, Z.-S.; Kumar, A.; Vishakha; Behl, T.; Jha, S.K.; Tang, H. Advancements in Clinical Aspects of Targeted Therapy and Immunotherapy in Breast Cancer. Mol. Cancer 2023, 22, 105. [Google Scholar] [CrossRef] [PubMed]
- Fasoulakis, Z.; Kolios, G.; Papamanolis, V.; Kontomanolis, E.N. Interleukins Associated with Breast Cancer. Cureus 2018, 10, e3549. [Google Scholar] [CrossRef] [PubMed]
- Briukhovetska, D.; Dörr, J.; Endres, S.; Libby, P.; Dinarello, C.A.; Kobold, S. Interleukins in Cancer: From Biology to Therapy. Nat. Rev. Cancer 2021, 21, 481–499. [Google Scholar] [CrossRef] [PubMed]
- Todaro, M.; Lombardo, Y.; Francipane, M.G.; Alea, M.P.; Cammareri, P.; Iovino, F.; Di Stefano, A.B.; Di Bernardo, C.; Agrusa, A.; Condorelli, G.; et al. Apoptosis Resistance in Epithelial Tumors Is Mediated by Tumor-Cell-Derived Interleukin-4. Cell Death Differ. 2008, 15, 762–772. [Google Scholar] [CrossRef] [PubMed]
- Venmar, K.T.; Carter, K.J.; Hwang, D.G.; Dozier, E.A.; Fingleton, B. IL4 Receptor ILR4α Regulates Metastatic Colonization by Mammary Tumors through Multiple Signaling Pathways. Cancer Res. 2014, 74, 4329–4340. [Google Scholar] [CrossRef] [PubMed]
- Erb, H.H.H.; Guggenberger, F.; Santer, F.R.; Culig, Z. Interleukin-4 Induces a CD44high/CD49bhigh PC3 Subpopulation with Tumor-initiating Characteristics. J. Cell. Biochem. 2018, 119, 4103–4112. [Google Scholar] [CrossRef]
- Chen, J.; Gong, C.; Mao, H.; Li, Z.; Fang, Z.; Chen, Q.; Lin, M.; Jiang, X.; Hu, Y.; Wang, W.; et al. E2F1/SP3/STAT6 Axis Is Required for IL-4-Induced Epithelial-Mesenchymal Transition of Colorectal Cancer Cells. Int. J. Oncol. 2018, 53, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Ouyang, Y.; Cai, J.; Xiong, L.; Chen, Y.; Zeng, X.; Liu, A. High Expression of IL-4R Enhances Proliferation and Invasion of Hepatocellular Carcinoma Cells. Int. J. Biol. Markers 2017, 32, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Keegan, A.D.; Leonard, W.J.; Zhu, J. Recent Advances in Understanding the Role of IL-4 Signaling. Fac. Rev. 2021, 10, 71. [Google Scholar] [CrossRef] [PubMed]
- Venmar, K.T.; Kimmel, D.W.; Cliffel, D.E.; Fingleton, B. IL4 Receptor α Mediates Enhanced Glucose and Glutamine Metabolism to Support Breast Cancer Growth. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2015, 1853, 1219–1228. [Google Scholar] [CrossRef] [PubMed]
- Dey, P.; Li, J.; Zhang, J.; Chaurasiya, S.; Strom, A.; Wang, H.; Liao, W.-T.; Cavallaro, F.; Denz, P.; Bernard, V.; et al. Oncogenic KRAS-Driven Metabolic Reprogramming in Pancreatic Cancer Cells Utilizes Cytokines from the Tumor Microenvironment. Cancer Discov. 2020, 10, 608–625. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Aksoylar, H.I.; Yu, J.; Snyder, N.W.; Worth, A.J.; Iyer, S.S.; Wang, J.; Ben-Sahra, I.; Byles, V.; Polynne-Stapornkul, T.; et al. Akt-mTORC1 Signaling Regulates Acly to Integrate Metabolic Input to Control of Macrophage Activation. eLife 2016, 5, e11612. [Google Scholar] [CrossRef] [PubMed]
- Győrffy, B. Survival Analysis across the Entire Transcriptome Identifies Biomarkers with the Highest Prognostic Power in Breast Cancer. Comput. Struct. Biotechnol. J. 2021, 19, 4101–4109. [Google Scholar] [CrossRef] [PubMed]
- Nelms, K.; Keegan, A.D.; Zamorano, J.; Ryan, J.J.; Paul, W.E. The IL-4 Receptor: Signaling Mechanisms and Biologic Functions. Annu. Rev. Immunol. 1999, 17, 701–738. [Google Scholar] [CrossRef] [PubMed]
- Cluntun, A.A.; Huang, H.; Dai, L.; Liu, X.; Zhao, Y.; Locasale, J.W. The Rate of Glycolysis Quantitatively Mediates Specific Histone Acetylation Sites. Cancer Metab. 2015, 3, 10. [Google Scholar] [CrossRef]
- Sivanand, S.; Viney, I.; Wellen, K.E. Spatiotemporal Control of Acetyl-CoA Metabolism in Chromatin Regulation. Trends Biochem. Sci. 2018, 43, 61–74. [Google Scholar] [CrossRef]
- Noe, J.T.; Rendon, B.E.; Geller, A.E.; Conroy, L.R.; Morrissey, S.M.; Young, L.E.A.; Bruntz, R.C.; Kim, E.J.; Wise-Mitchell, A.; Barbosa de Souza Rizzo, M.; et al. Lactate Supports a Metabolic-Epigenetic Link in Macrophage Polarization. Sci. Adv. 2021, 7, eabi8602. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Wang, X.; Duan, Z.; Zhang, C.; Fu, X.; Yang, J.; Liu, X.; He, J. Histone H3k9 and H3k27 Acetylation Regulates IL-4/STAT6-Mediated Igε Transcription in B Lymphocytes. Anat. Rec. 2015, 298, 1431–1439. [Google Scholar] [CrossRef]
- Rudolph, A.-K.; Walter, T.; Erkel, G. The Fungal Metabolite Cyclonerodiol Inhibits IL-4/IL-13 Induced Stat6-Signaling through Blocking the Association of Stat6 with P38, ERK1/2 and P300. Int. Immunopharmacol. 2018, 65, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Bowers, E.M.; Yan, G.; Mukherjee, C.; Orry, A.; Wang, L.; Holbert, M.A.; Crump, N.T.; Hazzalin, C.A.; Liszczak, G.; Yuan, H.; et al. Virtual Ligand Screening of the P300/CBP Histone Acetyltransferase: Identification of a Selective Small Molecule Inhibitor. Chem. Biol. 2010, 17, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-Y.; Lee, C.-J.; Choi, J.-H.; Kim, J.-H.; Kim, J.-W.; Kim, J.-Y.; Nam, J.-S. The JAK2/STAT3/CCND2 Axis Promotes Colorectal Cancer Stem Cell Persistence and Radioresistance. J. Exp. Clin. Cancer Res. 2019, 38, 399. [Google Scholar] [CrossRef] [PubMed]
- Aronica, M.A.; Goenka, S.; Boothby, M. IL-4-Dependent Induction of BCL-2 and BCL-X(L)IN Activated T Lymphocytes through a STAT6- and Pi 3-Kinase-Independent Pathway. Cytokine 2000, 12, 578–587. [Google Scholar] [CrossRef] [PubMed]
- Kaloni, D.; Diepstraten, S.T.; Strasser, A.; Kelly, G.L. BCL-2 Protein Family: Attractive Targets for Cancer Therapy. Apoptosis 2023, 28, 20–38. [Google Scholar] [CrossRef] [PubMed]
- Dang, B.; Gao, Q.; Zhang, L.; Zhang, J.; Cai, H.; Zhu, Y.; Zhong, Q.; Liu, J.; Niu, Y.; Mao, K.; et al. The Glycolysis/HIF-1α Axis Defines the Inflammatory Role of IL-4-Primed Macrophages. Cell Rep. 2023, 42, 112471. [Google Scholar] [CrossRef]
- Zhao, Q.; Chu, Z.; Zhu, L.; Yang, T.; Wang, P.; Liu, F.; Huang, Y.; Zhang, F.; Zhang, X.; Ding, W.; et al. 2-Deoxy-d-Glucose Treatment Decreases Anti-Inflammatory M2 Macrophage Polarization in Mice with Tumor and Allergic Airway Inflammation. Front. Immunol. 2017, 8, 637. [Google Scholar] [CrossRef]
- Ishida, K.; Nagatake, T.; Saika, A.; Kawai, S.; Node, E.; Hosomi, K.; Kunisawa, J. Induction of Unique Macrophage Subset by Simultaneous Stimulation with LPS and IL-4. Front. Immunol. 2023, 14, 1111729. [Google Scholar] [CrossRef]
- Dufort, F.J.; Bleiman, B.F.; Gumina, M.R.; Blair, D.; Wagner, D.J.; Roberts, M.F.; Abu-Amer, Y.; Chiles, T.C. Cutting Edge: IL-4-Mediated Protection of Primary B Lymphocytes from Apoptosis via Stat6-Dependent Regulation of Glycolytic Metabolism. J. Immunol. 2007, 179, 4953–4957. [Google Scholar] [CrossRef] [PubMed]
- Crofts, K.F.; Holbrook, B.C.; Soto-Pantoja, D.R.; Ornelles, D.A.; Alexander-Miller, M.A. TCR Dependent Metabolic Programming Regulates Autocrine IL-4 Production Resulting in Self-Tuning of the CD8+ T Cell Activation Setpoint. Front. Immunol. 2020, 11, 540. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhang, S.; Vuckovic, I.; Jeon, R.; Lerman, A.; Folmes, C.D.; Dzeja, P.P.; Herrmann, J. Glycolytic Stimulation Is Not a Requirement for M2 Macrophage Differentiation. Cell Metab. 2018, 28, 463–475.e4. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Li, W.; Hager, S.; Wilson, J.L.; Afjehi-Sadat, L.; Heiss, E.H.; Weichhart, T.; Heffeter, P.; Weckwerth, W. Targeting PHGDH Reverses the Immunosuppressive Phenotype of Tumor-Associated Macrophages through α-Ketoglutarate and mTORC1 Signaling. Cell Mol. Immunol. 2024. [Google Scholar] [CrossRef] [PubMed]
- Namgaladze, D.; Zukunft, S.; Schnütgen, F.; Kurrle, N.; Fleming, I.; Fuhrmann, D.; Brüne, B. Polarization of Human Macrophages by Interleukin-4 Does Not Require ATP-Citrate Lyase. Front. Immunol. 2018, 9, 2858. [Google Scholar] [CrossRef] [PubMed]
- Sivanand, S.; Rhoades, S.; Jiang, Q.; Lee, J.V.; Benci, J.; Zhang, J.; Yuan, S.; Viney, I.; Zhao, S.; Carrer, A.; et al. Nuclear Acetyl-CoA Production by ACLY Promotes Homologous Recombination. Mol. Cell 2017, 67, 252–265.e6. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Kang, H.; Wang, J.; Dong, Y.; Peng, R.; Zhao, H.; Wu, W.; Guan, H.; Li, F. Inhibition of ACLY Leads to Suppression of Osteoclast Differentiation and Function via Regulation of Histone Acetylation. J. Bone Miner. Res. 2021, 36, 2065–2080. [Google Scholar] [CrossRef]
- Czimmerer, Z.; Halasz, L.; Daniel, B.; Varga, Z.; Bene, K.; Domokos, A.; Hoeksema, M.; Shen, Z.; Berger, W.K.; Cseh, T.; et al. The Epigenetic State of IL-4-Polarized Macrophages Enables Inflammatory Cistromic Expansion and Extended Synergistic Response to TLR Ligands. Immunity 2022, 55, 2006–2026.e6. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Shu, D.; Zheng, M.; Wang, J.; Luo, C.; Wang, Y.; Guo, F.; Zou, X.; Lv, X.; Li, Y.; et al. Microbial Metabolite Butyrate Facilitates M2 Macrophage Polarization and Function. Sci. Rep. 2016, 6, 24838. [Google Scholar] [CrossRef]
- Gingras, S.; Simard, J.; Groner, B.; Pfitzner, E. P300/CBP Is Required for Transcriptional Induction by Interleukin-4 and Interacts with Stat6. Nucleic Acids Res. 1999, 27, 2722–2729. [Google Scholar] [CrossRef]
- Shankaranarayanan, P.; Chaitidis, P.; Kühn, H.; Nigam, S. Acetylation by Histone Acetyltransferase CREB-Binding Protein/P300 of STAT6 Is Required for Transcriptional Activation of the 15-Lipoxygenase-1 Gene. J. Biol. Chem. 2001, 276, 42753–42760. [Google Scholar] [CrossRef]
- Zhou, X.; Chen, H.; Shi, Y.; Li, J.; Ma, X.; Du, L.; Hu, Y.; Tao, M.; Zhong, Q.; Yan, D.; et al. Histone Deacetylase 8 Inhibition Prevents the Progression of Peritoneal Fibrosis by Counteracting the Epithelial-Mesenchymal Transition and Blockade of M2 Macrophage Polarization. Front. Immunol. 2023, 14, 1137332. [Google Scholar] [CrossRef] [PubMed]
- López-Bravo, M.; Minguito De La Escalera, M.; Domínguez, P.M.; González-Cintado, L.; Del Fresno, C.; Martín, P.; Martínez Del Hoyo, G.; Ardavín, C. IL-4 Blocks TH1-Polarizing/Inflammatory Cytokine Gene Expression during Monocyte-Derived Dendritic Cell Differentiation through Histone Hypoacetylation. J. Allergy Clin. Immunol. 2013, 132, 1409–1419.e13. [Google Scholar] [CrossRef] [PubMed]
- Yıldırım-Buharalıoğlu, G.; Bond, M.; Sala-Newby, G.B.; Hindmarch, C.C.T.; Newby, A.C. Regulation of Epigenetic Modifiers, Including KDM6B, by Interferon-γ and Interleukin-4 in Human Macrophages. Front. Immunol. 2017, 8, 92. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Efthymiadou, A.; Liang, N.; Fan, R.; Treuter, E. Antagonistic Action of GPS2 and KDM1A at Enhancers Governs Alternative Macrophage Activation by Interleukin 4. Nucleic Acids Res. 2023, 51, 1067–1086. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, Y.; Tano, Y.; Fujii, M.; Imai, Y.; Norimatsu, Y.; Yasukawa, M.; Watanabe, M.; Yamada, T. The H3K9 Demethylase Plant Homeodomain Finger Protein 2 Regulates Interleukin 4 Production in CD4+ T Cells. Cytokine 2024, 175, 156506. [Google Scholar] [CrossRef] [PubMed]
- Vento-Tormo, R.; Company, C.; Rodríguez-Ubreva, J.; de la Rica, L.; Urquiza, J.M.; Javierre, B.M.; Sabarinathan, R.; Luque, A.; Esteller, M.; Aran, J.M.; et al. IL-4 Orchestrates STAT6-Mediated DNA Demethylation Leading to Dendritic Cell Differentiation. Genome Biol. 2016, 17, 4. [Google Scholar] [CrossRef] [PubMed]
- Ono, H.; Kato, T.; Murase, Y.; Nakamura, Y.; Ishikawa, Y.; Watanabe, S.; Akahoshi, K.; Ogura, T.; Ogawa, K.; Ban, D.; et al. C646 Inhibits G2/M Cell Cycle-Related Proteins and Potentiates Anti-Tumor Effects in Pancreatic Cancer. Sci. Rep. 2021, 11, 10078. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, Y.; Shiraki, K.; Sugimoto, K.; Yada, T.; Tameda, M.; Ogura, S.; Yamamoto, N.; Takei, Y.; Ito, M. Epigenetic Regulation of Proliferation and Invasion in Hepatocellular Carcinoma Cells by CBP/P300 Histone Acetyltransferase Activity. Int. J. Oncol. 2016, 48, 533–540. [Google Scholar] [CrossRef]
- Santer, F.R.; Höschele, P.P.S.; Oh, S.J.; Erb, H.H.H.; Bouchal, J.; Cavarretta, I.T.; Parson, W.; Meyers, D.J.; Cole, P.A.; Culig, Z. Inhibition of the Acetyltransferases P300 and CBP Reveals a Targetable Function for P300 in the Survival and Invasion Pathways of Prostate Cancer Cell Lines. Mol. Cancer Ther. 2011, 10, 1644–1655. [Google Scholar] [CrossRef]
- Wawrzyniak, D.; Grabowska, M.; Głodowicz, P.; Kuczyński, K.; Kuczyńska, B.; Fedoruk-Wyszomirska, A.; Rolle, K. Down-Regulation of Tenascin-C Inhibits Breast Cancer Cells Development by Cell Growth, Migration, and Adhesion Impairment. PLoS ONE 2020, 15, e0237889. [Google Scholar] [CrossRef]
- Huang, T.; Lu, C.; Zhang, Y.; Lin, B.; Zhang, Z.; Zhu, D.; Wang, L.; Lu, Y. Effect of Activating Cancer-Associated Fibroblasts Biomarker TNC on Immune Cell Infiltration and Prognosis in Breast Cancer. Ann. Med. 2023, 55, 2250987. [Google Scholar] [CrossRef]
- Wan, H.; Li, Z.; Wang, H.; Cai, F.; Wang, L. ST8SIA1 Inhibition Sensitizes Triple Negative Breast Cancer to Chemotherapy via Suppressing Wnt/β-Catenin and FAK/Akt/mTOR. Clin. Transl. Oncol. 2021, 23, 902–910. [Google Scholar] [CrossRef]
- Ring, A.; Kaur, P.; Lang, J.E. EP300 Knockdown Reduces Cancer Stem Cell Phenotype, Tumor Growth and Metastasis in Triple Negative Breast Cancer. BMC Cancer 2020, 20, 1076. [Google Scholar] [CrossRef]
- Shiao, S.L.; Ruffell, B.; DeNardo, D.G.; Faddegon, B.A.; Park, C.C.; Coussens, L.M. TH2-Polarized CD4+ T Cells and Macrophages Limit Efficacy of Radiotherapy. Cancer Immunol. Res. 2015, 3, 518–525. [Google Scholar] [CrossRef]
- Ruggiu, M.; Guérin, M.V.; Corre, B.; Bardou, M.; Alonso, R.; Russo, E.; Garcia, Z.; Feldmann, L.; Lemaître, F.; Dusseaux, M.; et al. Anti-PD-1 Therapy Triggers Tfh Cell–Dependent IL-4 Release to Boost CD8 T Cell Responses in Tumor-Draining Lymph Nodes. J. Exp. Med. 2024, 221, e20232104. [Google Scholar] [CrossRef]
- Moraga, I.; Richter, D.; Wilmes, S.; Winkelmann, H.; Jude, K.; Thomas, C.; Suhoski, M.M.; Engleman, E.G.; Piehler, J.; Garcia, K.C. Instructive Roles for Cytokine-Receptor Binding Parameters in Determining Signaling and Functional Potency. Sci. Signal. 2015, 8, ra114. [Google Scholar] [CrossRef]
- Little, A.C.; Pathanjeli, P.; Wu, Z.; Bao, L.; Goo, L.E.; Yates, J.A.; Oliver, C.R.; Soellner, M.B.; Merajver, S.D. IL-4/IL-13 Stimulated Macrophages Enhance Breast Cancer Invasion Via Rho-GTPase Regulation of Synergistic VEGF/CCL-18 Signaling. Front. Oncol. 2019, 9, 456. [Google Scholar] [CrossRef]
- LaMarche, N.M.; Hegde, S.; Park, M.D.; Maier, B.B.; Troncoso, L.; Le Berichel, J.; Hamon, P.; Belabed, M.; Mattiuz, R.; Hennequin, C.; et al. An IL-4 Signalling Axis in Bone Marrow Drives pro-Tumorigenic Myelopoiesis. Nature 2024, 625, 166–174. [Google Scholar] [CrossRef]
- Shechter, D.; Dormann, H.L.; Allis, C.D.; Hake, S.B. Extraction, Purification and Analysis of Histones. Nat. Protoc. 2007, 2, 1445–1457. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-Based Analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.J.; Gazin, C.; Lawson, N.D.; Pagès, H.; Lin, S.M.; Lapointe, D.S.; Green, M.R. ChIPpeakAnno: A Bioconductor Package to Annotate ChIP-Seq and ChIP-Chip Data. BMC Bioinform. 2010, 11, 237. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, F.; Ryan, D.P.; Grüning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dündar, F.; Manke, T. deepTools2: A next Generation Web Server for Deep-Sequencing Data Analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Williams, D.; Hargrove-Wiley, E.; Bindeman, W.; Valent, D.; Miranda, A.X.; Beckstead, J.; Fingleton, B. Type II Interleukin-4 Receptor Activation in Basal Breast Cancer Cells Promotes Tumor Progression via Metabolic and Epigenetic Modulation. Int. J. Mol. Sci. 2024, 25, 4647. https://doi.org/10.3390/ijms25094647
Williams D, Hargrove-Wiley E, Bindeman W, Valent D, Miranda AX, Beckstead J, Fingleton B. Type II Interleukin-4 Receptor Activation in Basal Breast Cancer Cells Promotes Tumor Progression via Metabolic and Epigenetic Modulation. International Journal of Molecular Sciences. 2024; 25(9):4647. https://doi.org/10.3390/ijms25094647
Chicago/Turabian StyleWilliams, Demond, Ebony Hargrove-Wiley, Wendy Bindeman, Daniel Valent, Adam X. Miranda, Jacob Beckstead, and Barbara Fingleton. 2024. "Type II Interleukin-4 Receptor Activation in Basal Breast Cancer Cells Promotes Tumor Progression via Metabolic and Epigenetic Modulation" International Journal of Molecular Sciences 25, no. 9: 4647. https://doi.org/10.3390/ijms25094647