
Transcranial Magneto-Acoustic Stimulation Protects Synaptic Rehabilitation from Amyloid-Beta Plaques via Regulation of Microglial Functions

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
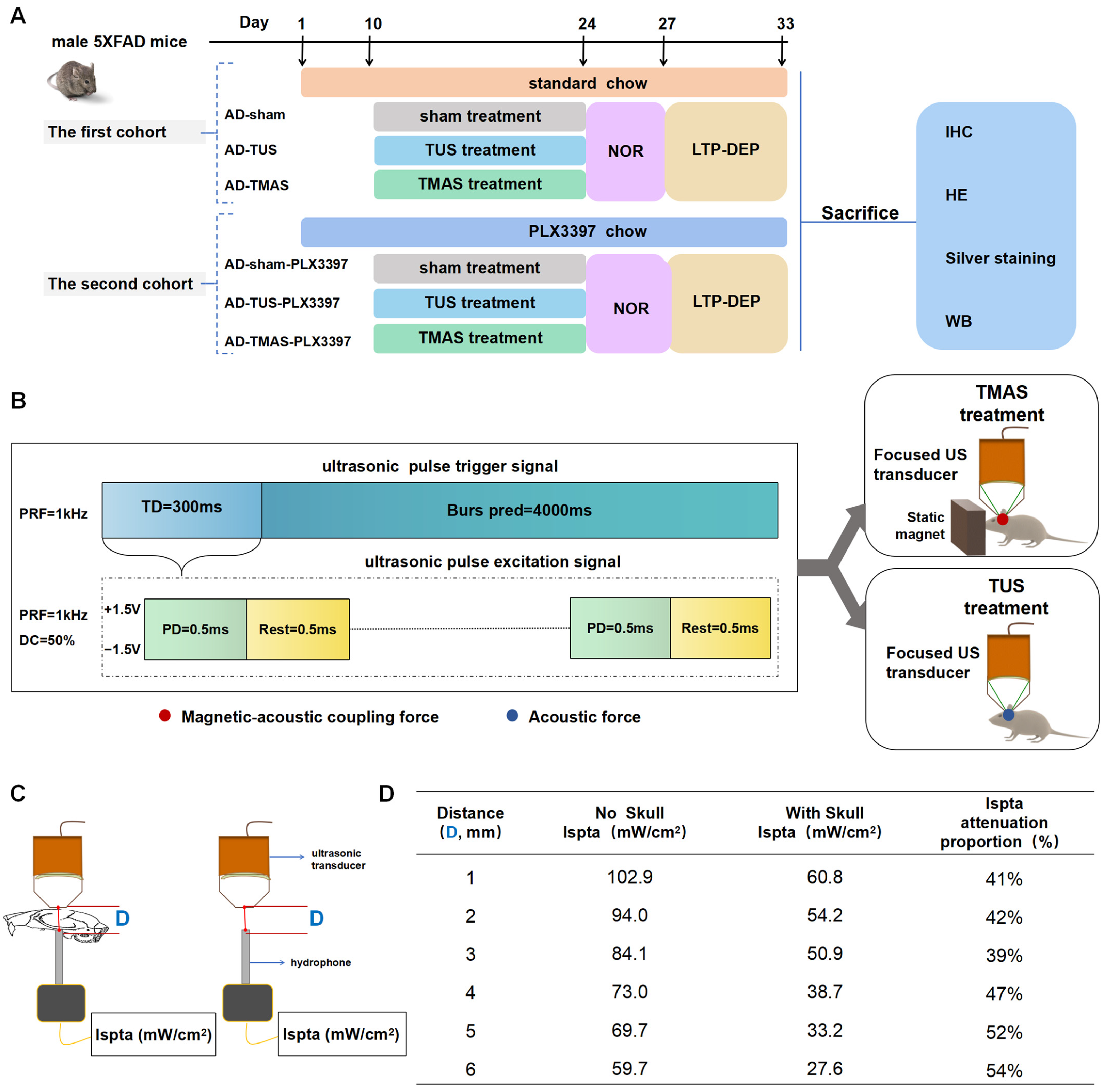
2.1. The Schedule for TUS and TMAS Treatments for 5xFAD Transgenic Mice
2.2. TMAS-TUS Treatment Promotes Activation of Microglia through the Cell Proliferation, Motility, and Phagocytic Capacities
2.3. TMAS-TUS Treatment Enhances Microglial Phagocytosis and Clearance of Aβ
2.4. TMAS-TUS Treatment Reduces Amyloid Burden and Related Toxicity
2.5. Microglia Are Necessary for TMAS-TUS Treatment-Induced Aβ Plaque Reduction
2.6. Microglia Are Necessary for TMAS-TUS Treatment-Induced Synaptic Rehabilitation
2.6.1. Effects of TMAS-TUS Treatment on NOR Tests
2.6.2. Effects of TMAS-TUS Treatment on Electrophysiological Variation in the Hippocampus of 5xFAD Mice
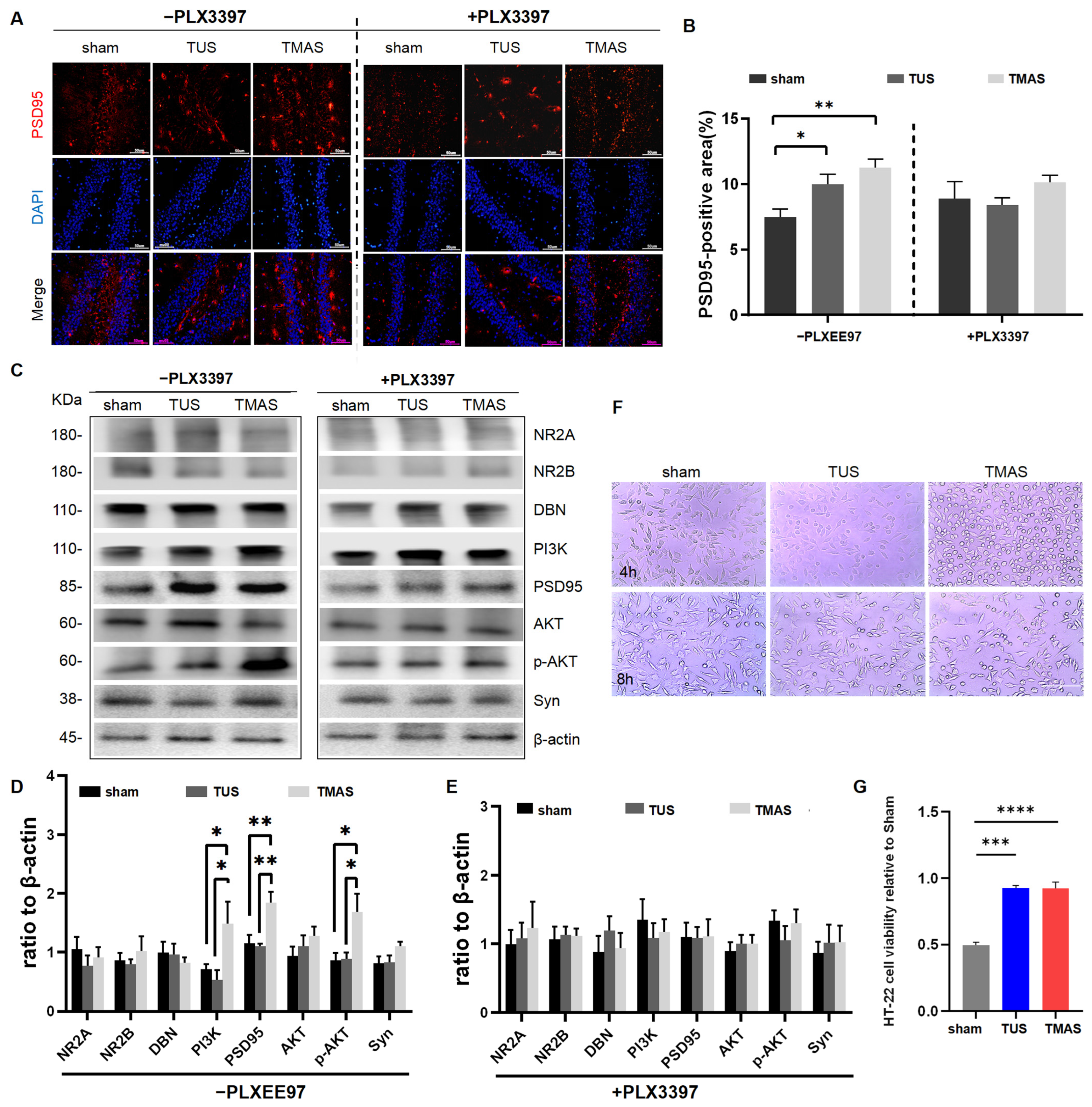
2.7. Effects of TMAS-TUS Treatment on Neuroplasticity-Associated Proteins and PI3K-AKT Signaling Protein Expressions in the Hippocampus of 5xFAD Mice
3. Discussion
4. Materials and Methods
4.1. Study Design
4.2. Animals
4.3. TMAS-TUS Experimental System and Treatment
4.4. Silver Staining
4.5. Cell Culture and Proliferation Assay
4.6. Aβ Phagocytosis Assays of BV2 Cells
4.7. Microglial Migration in Transwell Assays
4.8. Western Blotting
4.9. Immunohistochemistry and Microscopy
4.10. Novel Object Recognition (NOR) Test
4.11. In Vivo Electrophysiological Study
4.12. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gabrielli, M.; Prada, I.; Joshi, P.; Falcicchia, C.; D’Arrigo, G.; Rutigliano, G.; Battocchio, E.; Zenatelli, R.; Tozzi, F.; Radeghieri, A.; et al. Microglial large extracellular vesicles propagate early synaptic dysfunction in Alzheimer’s disease. Brain 2022, 145, 2849–2868. [Google Scholar] [CrossRef] [PubMed]
- Passeri, E.; Elkhoury, K.; Morsink, M.; Broersen, K.; Linder, M.; Tamayol, A.; Malaplate, C.; Yen, F.T.; Arab-Tehrany, E. Alzheimer’s Disease: Treatment Strategies and Their Limitations. Int. J. Mol. Sci. 2022, 23, 13954. [Google Scholar] [CrossRef] [PubMed]
- Karran, E.; Mercken, M.; De Strooper, B. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.S.; Jo, S.A. Mechanisms of Amyloid-beta Peptide Clearance: Potential Therapeutic Targets for Alzheimer’s Disease. Biomol. Ther. 2012, 20, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Gandy, S.; Heppner, F.L. Microglia as dynamic and essential components of the amyloid hypothesis. Neuron 2013, 78, 575–577. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Herron, S.; Silveira, S.; Kleemann, K.; Gauthier, C.; Mallah, D.; Cheng, Y.; Margeta, M.A.; Pitts, K.M.; Barry, J.L.; et al. Identification of a protective microglial state mediated by miR-155 and interferon-gamma signaling in a mouse model of Alzheimer’s disease. Nat. Neurosci. 2023, 26, 1196–1207. [Google Scholar] [CrossRef] [PubMed]
- Clayton, K.; Delpech, J.C.; Herron, S.; Iwahara, N.; Ericsson, M.; Saito, T.; Saido, T.C.; Ikezu, S.; Ikezu, T. Plaque associated microglia hyper-secrete extracellular vesicles and accelerate tau propagation in a humanized APP mouse model. Mol. Neurodegener. 2021, 16, 18. [Google Scholar] [CrossRef] [PubMed]
- Bobola, M.S.; Chen, L.; Ezeokeke, C.K.; Olmstead, T.A.; Nguyen, C.; Sahota, A.; Williams, R.G.; Mourad, P.D. Transcranial focused ultrasound, pulsed at 40 Hz, activates microglia acutely and reduces Abeta load chronically, as demonstrated in vivo. Brain Stimul. 2020, 13, 1014–1023. [Google Scholar] [CrossRef]
- Park, M.; Hoang, G.M.; Nguyen, T.; Lee, E.; Jung, H.J.; Choe, Y.; Lee, M.H.; Hwang, J.Y.; Kim, J.G.; Kim, T. Effects of transcranial ultrasound stimulation pulsed at 40 Hz on Abeta plaques and brain rhythms in 5xFAD mice. Transl. Neurodegener. 2021, 10, 48. [Google Scholar] [CrossRef]
- Zhou, X.; Liu, S.; Wang, Y.; Yin, T.; Yang, Z.; Liu, Z. High-Resolution Transcranial Electrical Simulation for Living Mice Based on Magneto-Acoustic Effect. Front. Neurosci. 2019, 13, 1342. [Google Scholar] [CrossRef]
- Wang, H.; Zhou, X.; Cui, D.; Liu, R.; Tan, R.; Wang, X.; Liu, Z.; Yin, T. Comparative Study of Transcranial Magneto-Acoustic Stimulation and Transcranial Ultrasound Stimulation of Motor Cortex. Front. Behav. Neurosci. 2019, 13, 241. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Feng, L.; Liu, S.; Zhou, X.; Yin, T.; Liu, Z.; Yang, Z. Transcranial Magneto-Acoustic Stimulation Improves Neuroplasticity in Hippocampus of Parkinson’s Disease Model Mice. Neurotherapeutics 2019, 16, 1210–1224. [Google Scholar] [CrossRef]
- Lam, S.; Herard, A.S.; Boluda, S.; Petit, F.; Eddarkaoui, S.; Cambon, K.; Picq, J.L.; Buee, L.; Duyckaerts, C.; Haik, S.; et al. Pathological changes induced by Alzheimer’s brain inoculation in amyloid-beta plaque-bearing mice. Acta Neuropathol. Com. 2022, 10, 112. [Google Scholar] [CrossRef] [PubMed]
- Horwood, J.M.; Dufour, F.; Laroche, S.; Davis, S. Signalling mechanisms mediated by the phosphoinositide 3-kinase/Akt cascade in synaptic plasticity and memory in the rat. Eur. J. Neurosci. 2006, 23, 3375–3384. [Google Scholar] [CrossRef]
- Razani, E.; Pourbagheri-Sigaroodi, A.; Safaroghli-Azar, A.; Zoghi, A.; Shanaki-Bavarsad, M.; Bashash, D. The PI3K/Akt signaling axis in Alzheimer’s disease: A valuable target to stimulate or suppress? Cell Stress Chaperones 2021, 26, 871–887. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Deficient brain insulin signalling pathway in Alzheimer’s disease and diabetes. J. Pathol. 2011, 225, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de la Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease—Is this type 3 diabetes? J. Alzheimers Dis. 2005, 7, 63–80. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Xiao, K.; Mi, X.; Li, N.; Guo, L.; Wang, X.; Sun, Y.; Li, G.D.; Zhou, Y. Ghrelin signaling in dCA1 suppresses neuronal excitability and impairs memory acquisition via PI3K/Akt/GSK-3beta cascades. Neuropharmacology 2022, 203, 108871. [Google Scholar] [CrossRef]
- Gabbouj, S.; Ryhanen, S.; Marttinen, M.; Wittrahm, R.; Takalo, M.; Kemppainen, S.; Martiskainen, H.; Tanila, H.; Haapasalo, A.; Hiltunen, M.; et al. Altered Insulin Signaling in Alzheimer’s Disease Brain—Special Emphasis on PI3K-Akt Pathway. Front. Neurosci. 2019, 13, 629. [Google Scholar] [CrossRef]
- Jung, S.Y.; Kim, D.Y. Treadmill exercise improves motor and memory functions in cerebral palsy rats through activation of PI3K-Akt pathway. J. Exerc. Rehabil. 2017, 13, 136–142. [Google Scholar] [CrossRef]
- Weinhard, L.; di Bartolomei, G.; Bolasco, G.; Machado, P.; Schieber, N.L.; Neniskyte, U.; Exiga, M.; Vadisiute, A.; Raggioli, A.; Schertel, A.; et al. Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat. Commun. 2018, 9, 1228. [Google Scholar] [CrossRef]
- Huang, Y.; Happonen, K.E.; Burrola, P.G.; O’Connor, C.; Hah, N.; Huang, L.; Nimmerjahn, A.; Lemke, G. Microglia use TAM receptors to detect and engulf amyloid beta plaques. Nat. Immunol. 2021, 22, 586–594. [Google Scholar] [CrossRef]
- Qiu, Y.; Shen, X.; Ravid, O.; Atrakchi, D.; Rand, D.; Wight, A.E.; Kim, H.J.; Liraz-Zaltsman, S.; Cooper, I.; Schnaider, B.M.; et al. Definition of the contribution of an Osteopontin-producing CD11c(+) microglial subset to Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2023, 120, e2076052176. [Google Scholar] [CrossRef] [PubMed]
- Wlodarczyk, A.; Lobner, M.; Cedile, O.; Owens, T. Comparison of microglia and infiltrating CD11c+ cells as antigen presenting cells for T cell proliferation and cytokine response. J. Neuroinflamm. 2014, 11, 57. [Google Scholar] [CrossRef]
- Blasi, E.; Barluzzi, R.; Bocchini, V.; Mazzolla, R.; Bistoni, F. Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J. Neuroimmunol. 1990, 27, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, O.; Coleman, M. Alzheimer’s Disease: Etiology, Neuropathology and Pathogenesis; Exon Publications: Brisbane, Australia, 2020. [Google Scholar]
- Jo, K.W.; Lee, D.; Cha, D.G.; Oh, E.; Choi, Y.H.; Kim, S.; Park, E.S.; Kim, J.K.; Kim, K.T. Gossypetin ameliorates 5xFAD spatial learning and memory through enhanced phagocytosis against Abeta. Alzheimers Res. Ther. 2022, 14, 158. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Jantti, H.; Sitnikova, V.; Ishchenko, Y.; Shakirzyanova, A.; Giudice, L.; Ugidos, I.F.; Gomez-Budia, M.; Korvenlaita, N.; Ohtonen, S.; Belaya, I.; et al. Microglial amyloid beta clearance is driven by PIEZO1 channels. J. Neuroinflamm. 2022, 19, 147. [Google Scholar] [CrossRef]
- Maccioni, R.B.; Munoz, J.P.; Barbeito, L. The molecular bases of Alzheimer’s disease and other neurodegenerative disorders. Arch. Med. Res. 2001, 32, 367–381. [Google Scholar] [CrossRef]
- Sharoar, M.G.; Palko, S.; Ge, Y.; Saido, T.C.; Yan, R. Accumulation of saposin in dystrophic neurites is linked to impaired lysosomal functions in Alzheimer’s disease brains. Mol. Neurodegener. 2021, 16, 45. [Google Scholar] [CrossRef]
- Di Meco, A.; Kemal, S.; Popovic, J.; Chandra, S.; Sadleir, K.; Vassar, R. Poloxamer-188 Exacerbates Brain Amyloidosis, Presynaptic Dystrophies, and Pathogenic Microglial Activation in 5XFAD Mice. Curr. Alzheimer Res. 2022, 19, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Yu, K.; He, B. Transcranial focused ultrasound induces sustained synaptic plasticity in rat hippocampus. Brain Stimul. 2022, 15, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Phan, T.T.; Lee, K.; Kim, J.S.; Lee, S.Y.; Lee, J.M.; Do, J.; Lee, D.; Kim, S.P.; Lee, K.P.; et al. Long-lasting forms of plasticity through patterned ultrasound-induced brainwave entrainment. Sci. Adv. 2024, 10, eadk3198. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Lin, Z.; Wang, K.; Liu, X.; Zhou, W.; Meng, L.; Huang, J.; Yuan, K.; Niu, L.; Zheng, H. Transcranial Low-Intensity Pulsed Ultrasound Modulates Structural and Functional Synaptic Plasticity in Rat Hippocampus. IEEE Trans. Ultrason. Ferroelectr. Freq. Control 2019, 66, 930–938. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, S.; Niu, X.; Yu, K.; He, B. Transcranial ultrasound neuromodulation induces neuronal correlation change in the rat somatosensory cortex. J. Neural Eng. 2022, 19, 056002. [Google Scholar] [CrossRef]
- Kong, C.; Ahn, J.W.; Kim, S.; Park, J.Y.; Na, Y.C.; Chang, J.W.; Chung, S.; Chang, W.S. Long-lasting restoration of memory function and hippocampal synaptic plasticity by focused ultrasound in Alzheimer’s disease. Brain Stimul. 2023, 16, 857–866. [Google Scholar] [CrossRef]
- Leinenga, G.; Gotz, J. Scanning ultrasound removes amyloid-beta and restores memory in an Alzheimer’s disease mouse model. Sci. Transl. Med. 2015, 7, 278ra33. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Kong, C.; Lee, J.; Choi, B.Y.; Sim, J.; Koh, C.S.; Park, M.; Na, Y.C.; Suh, S.W.; Chang, W.S.; et al. Focused ultrasound-induced blood-brain barrier opening improves adult hippocampal neurogenesis and cognitive function in a cholinergic degeneration dementia rat model. Alzheimers Res. Ther. 2019, 11, 110. [Google Scholar] [CrossRef]
- Lee, C.H.; Chen, H.M.; Yeh, L.K.; Hong, M.Y.; Huang, G.S. Dosage-dependent induction of behavioral decline in Caenorhabditis elegans by long-term treatment of static magnetic fields. J. Radiat. Res. 2012, 53, 24–32. [Google Scholar] [CrossRef]
- Hung, Y.C.; Lee, J.H.; Chen, H.M.; Huang, G.S. Effects of static magnetic fields on the development and aging of Caenorhabditis elegans. J. Exp. Biol. 2010, 213, 2079–2085. [Google Scholar] [CrossRef]
- Peng, Y.; Lu, K.; Li, Z.; Zhao, Y.; Wang, Y.; Hu, B.; Xu, P.; Shi, X.; Zhou, B.; Pennington, M.; et al. Blockade of Kv1.3 channels ameliorates radiation-induced brain injury. Neuro-Oncology 2014, 16, 528–539. [Google Scholar] [CrossRef] [PubMed]
- El-Sherbeeny, N.A.; Ibrahiem, A.T.; Ali, H.S.; Farag, N.E.; Toraih, E.A.; Zaitone, S.A. Carbamazepine conquers spinal GAP43 deficiency and sciatic Nav1.5 upregulation in diabetic mice: Novel mechanisms in alleviating allodynia and hyperalgesia. Arch. Pharm. Res. 2020, 43, 724–734. [Google Scholar] [CrossRef]
- Wu, T.; Wang, X.; Cheng, J.; Liang, X.; Li, Y.; Chen, M.; Kong, L.; Tang, M. Nitrogen-doped graphene quantum dots induce ferroptosis through disrupting calcium homeostasis in microglia. Part. Fibre Toxicol. 2022, 19, 22. [Google Scholar] [CrossRef]
- Eder, C.; Klee, R.; Heinemann, U. Involvement of stretch-activated Cl- channels in ramification of murine microglia. J. Neurosci. 1998, 18, 7127–7137. [Google Scholar] [CrossRef]
- Demir, I.E.; Tieftrunk, E.; Schorn, S.; Saricaoglu, O.C.; Pfitzinger, P.L.; Teller, S.; Wang, K.; Waldbaur, C.; Kurkowski, M.U.; Wormann, S.M.; et al. Activated Schwann cells in pancreatic cancer are linked to analgesia via suppression of spinal astroglia and microglia. Gut 2016, 65, 1001–1014. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Chen, Q.; Zhu, H.; Hou, L.; Liu, W.; Yang, Q.; Shen, H.; Chai, G.; Zhang, B.; Chen, S.; et al. Microglial Piezo1 senses Abeta fibril stiffness to restrict Alzheimer’s disease. Neuron 2023, 111, 15–29. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, M.; Jia, P.; Liu, F.F.; Chen, K.; Meng, F.Y.; Hong, J.H.; Zhang, T.; Jin, X.H.; Shi, J. The analgesic action of larixyl acetate, a potent TRPC6 inhibitor, in rat neuropathic pain model induced by spared nerve injury. J. Neuroinflam. 2020, 17, 118. [Google Scholar] [CrossRef]
- Wang, W.; Li, Y.; Ma, F.; Sheng, X.; Chen, K.; Zhuo, R.; Wang, C.; Zheng, H.; Zhang, Y.W.; Bu, G.; et al. Microglial repopulation reverses cognitive and synaptic deficits in an Alzheimer’s disease model by restoring BDNF signaling. Brain Behav. Immun. 2023, 113, 275–288. [Google Scholar] [CrossRef]
- Kohman, R.A.; DeYoung, E.K.; Bhattacharya, T.K.; Peterson, L.N.; Rhodes, J.S. Wheel running attenuates microglia proliferation and increases expression of a proneurogenic phenotype in the hippocampus of aged mice. Brain Behav. Immun. 2012, 26, 803–810. [Google Scholar] [CrossRef]
- Nguyen, P.T.; Dorman, L.C.; Pan, S.; Vainchtein, I.D.; Han, R.T.; Nakao-Inoue, H.; Taloma, S.E.; Barron, J.J.; Molofsky, A.B.; Kheirbek, M.A.; et al. Microglial Remodeling of the Extracellular Matrix Promotes Synapse Plasticity. Cell 2020, 182, 388–403. [Google Scholar] [CrossRef]
- Valentinova, K.; Tchenio, A.; Trusel, M.; Clerke, J.A.; Lalive, A.L.; Tzanoulinou, S.; Matera, A.; Moutkine, I.; Maroteaux, L.; Paolicelli, R.C.; et al. Morphine withdrawal recruits lateral habenula cytokine signaling to reduce synaptic excitation and sociability. Nat. Neurosci. 2019, 22, 1053–1056. [Google Scholar] [CrossRef]
- Picard, K.; Corsi, G.; Decoeur, F.; Di Castro, M.A.; Bordeleau, M.; Persillet, M.; Laye, S.; Limatola, C.; Tremblay, M.E.; Nadjar, A. Microglial homeostasis disruption modulates non-rapid eye movement sleep duration and neuronal activity in adult female mice. Brain Behav. Immun. 2023, 107, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Gmitrov, J. Static Magnetic Field Versus Systemic Calcium Channel Blockade Effect on Microcirculation: Possible Mechanisms and Clinical Implementation. Bioelectromagnetics 2020, 41, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Liao, Z.; Gu, L.; Zhang, L.; Zhang, L.; Tian, X.; Li, J.; Fang, Z.; Zhang, X. Moderate Intensity Static Magnetic Fields Prevent Thrombus Formation in Rats and Mice. Bioelectromagnetics 2020, 41, 52–62. [Google Scholar] [CrossRef]
- Gmitrov, J. Geomagnetic field modulates artificial static magnetic field effect on arterial baroreflex and on microcirculation. Int. J. Biometeorol. 2007, 51, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Okano, H.; Masuda, H.; Ohkubo, C. Decreased plasma levels of nitric oxide metabolites, angiotensin II, and aldosterone in spontaneously hypertensive rats exposed to 5 mT static magnetic field. Bioelectromagnetics 2005, 26, 161–172. [Google Scholar] [CrossRef]
- Cheng, L.; Yang, B.; Du, H.; Zhou, T.; Li, Y.; Wu, J.; Cao, Z.; Xu, A. Moderate intensity of static magnetic fields can alter the avoidance behavior and fat storage of Caenorhabditis elegans via serotonin. Environ. Sci. Pollut. R. 2022, 29, 43102–43113. [Google Scholar] [CrossRef]
- Sinha, A.S.; Shibata, S.; Takamatsu, Y.; Akita, T.; Fukuda, A.; Mima, T. Static Magnetic Field Stimulation Enhances Shunting Inhibition via a SLC26 Family Cl(-) Channel, Inducing Intrinsic Plasticity. J. Neurosci. 2024, 44, e1324222024. [Google Scholar] [CrossRef]
- Choi, W.; Cho, H.; Kim, G.; Youn, I.; Key, J.; Han, S. Targeted thrombolysis by magnetoacoustic particles in photothrombotic stroke model. Biomater. Res. 2022, 26, 58. [Google Scholar] [CrossRef]
- Kim, T.; Kim, H.J.; Choi, W.; Lee, Y.M.; Pyo, J.H.; Lee, J.; Kim, J.; Kim, J.; Kim, J.H.; Kim, C.; et al. Deep brain stimulation by blood-brain-barrier-crossing piezoelectric nanoparticles generating current and nitric oxide under focused ultrasound. Nat. Biomed. Eng. 2023, 7, 149–163. [Google Scholar] [CrossRef]
- Singer, A.; Dutta, S.; Lewis, E.; Chen, Z.; Chen, J.C.; Verma, N.; Avants, B.; Feldman, A.K.; O’Malley, J.; Beierlein, M.; et al. Magnetoelectric Materials for Miniature, Wireless Neural Stimulation at Therapeutic Frequencies. Neuron 2020, 107, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, Y.; Dong, S.; Wang, P.; Chen, W.; Lu, Z.; Ye, D.; Pan, B.; Wu, D.; Vecitis, C.D.; et al. Ultrasonic activation of inert poly(tetrafluoroethylene) enables piezocatalytic generation of reactive oxygen species. Nat. Commun. 2021, 12, 3508. [Google Scholar] [CrossRef] [PubMed]
- Sood, A.; Desseigne, M.; Dev, A.; Maurizi, L.; Kumar, A.; Millot, N.; Han, S.S. A Comprehensive Review on Barium Titanate Nanoparticles as a Persuasive Piezoelectric Material for Biomedical Applications: Prospects and Challenges. Small 2023, 19, e2206401. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Chen, Y.; Shi, J. Piezocatalytic Tumor Therapy by Ultrasound-Triggered and BaTiO(3) -Mediated Piezoelectricity. Adv. Mater. 2020, 32, e2001976. [Google Scholar] [CrossRef]
- Chen, S.; Zhu, P.; Mao, L.; Wu, W.; Lin, H.; Xu, D.; Lu, X.; Shi, J. Piezocatalytic Medicine: An Emerging Frontier using Piezoelectric Materials for Biomedical Applications. Adv. Mater. 2023, 35, e2208256. [Google Scholar] [CrossRef] [PubMed]
- Ge, M.; Xu, D.; Chen, Z.; Wei, C.; Zhang, Y.; Yang, C.; Chen, Y.; Lin, H.; Shi, J. Magnetostrictive-Piezoelectric-Triggered Nanocatalytic Tumor Therapy. Nano Lett. 2021, 21, 6764–6772. [Google Scholar] [CrossRef]
- Di Ianni, T.; Morrison, K.P.; Yu, B.; Murphy, K.R.; de Lecea, L.; Airan, R.D. High-throughput ultrasound neuromodulation in awake and freely behaving rats. Brain Stimul. 2023, 16, 1743–1752. [Google Scholar] [CrossRef]
- He, J.; Zhu, Y.; Wu, C.; Wu, J.; Chen, Y.; Yuan, M.; Cheng, Z.; Zeng, L.; Ji, X. Simultaneous multi-target ultrasound neuromodulation in freely-moving mice based on a single-element ultrasound transducer. J. Neural Eng. 2023, 20, 016021. [Google Scholar] [CrossRef]
- Song, C.; Knopfel, T. Optogenetics enlightens neuroscience drug discovery. Nat. Rev. Drug Discov. 2016, 15, 97–109. [Google Scholar] [CrossRef]
- Yuan, Y.; Yan, J.; Ma, Z.; Li, X. Noninvasive Focused Ultrasound Stimulation Can Modulate Phase-Amplitude Coupling between Neuronal Oscillations in the Rat Hippocampus. Front. Neurosci. 2016, 10, 348. [Google Scholar] [CrossRef]
- Yuan, Y.; Yan, J.; Ma, Z.; Li, X. Effect of noninvasive focused ultrasound stimulation on gamma oscillations in rat hippocampus. Neuroreport 2016, 27, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Nyborg, W.L. Biological effects of ultrasound: Development of safety guidelines. Part II: General review. Ultrasound Med. Biol. 2001, 27, 301–333. [Google Scholar] [CrossRef]
- Duck, F.A. Medical and non-medical protection standards for ultrasound and infrasound. Prog. Biophys. Mol. Bio. 2007, 93, 176–191. [Google Scholar] [CrossRef]
- Kim, E.D.; Won, Y.H.; Park, S.H.; Seo, J.H.; Kim, D.S.; Ko, M.H.; Kim, G.W. Efficacy and Safety of a Stimulator Using Low-Intensity Pulsed Ultrasound Combined with Transcutaneous Electrical Nerve Stimulation in Patients with Painful Knee Osteoarthritis. Pain. Res. Manag. 2019, 2019, 7964897. [Google Scholar] [CrossRef]
- Rabut, C.; Yoo, S.; Hurt, R.C.; Jin, Z.; Li, H.; Guo, H.; Ling, B.; Shapiro, M.G. Ultrasound Technologies for Imaging and Modulating Neural Activity. Neuron 2020, 108, 93–110. [Google Scholar] [CrossRef]
- Schafer, M.E.; Spivak, N.M.; Korb, A.S.; Bystritsky, A. Design, Development, and Operation of a Low-Intensity Focused Ultrasound Pulsation (LIFUP) System for Clinical Use. IEEE Trans. Ultrason. Ferroelectr. Freq. Control 2021, 68, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Ignell, R.; Hill, S.R. The Golgi Method. Cold Spring Harb. Protoc. 2022, 2022, Pdb.top107695. [Google Scholar] [CrossRef] [PubMed]
- Lavenir, I.; Passarella, D.; Masuda-Suzukake, M.; Curry, A.; Holton, J.L.; Ghetti, B.; Goedert, M. Silver staining (Campbell-Switzer) of neuronal alpha-synuclein assemblies induced by multiple system atrophy and Parkinson’s disease brain extracts in transgenic mice. Acta Neuropathol. Com. 2019, 7, 148. [Google Scholar] [CrossRef]
- Li, Y.; Xu, B.; Ren, X.; Wang, L.; Xu, Y.; Zhao, Y.; Yang, C.; Yuan, C.; Li, H.; Tong, X.; et al. Inhibition of CISD2 promotes ferroptosis through ferritinophagy-mediated ferritin turnover and regulation of p62-Keap1-NRF2 pathway. Cell Mol. Biol. Lett. 2022, 27, 81. [Google Scholar] [CrossRef]
- Martinez, G.; Cappetta, D.; Telesca, M.; Urbanek, K.; Castaldo, G.; Dhellemmes, M.; Mele, V.G.; Chioccarelli, T.; Porreca, V.; Barbotin, A.L.; et al. Cytochalasin D restores nuclear size acting on F-actin and IZUMO1 localization in low-quality spermatozoa. Int. J. Biol. Sci. 2023, 19, 2234–2255. [Google Scholar] [CrossRef]
- Sun, Y.; Sommerville, N.R.; Liu, J.; Ngan, M.P.; Poon, D.; Ponomarev, E.D.; Lu, Z.; Kung, J.; Rudd, J.A. Intra-gastrointestinal amyloid-beta1-42 oligomers perturb enteric function and induce Alzheimer’s disease pathology. J. Physiol. 2020, 598, 4209–4223. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, H.J.; Park, S.K.; Park, J.H.; Jeong, H.R.; Lee, S.; Lee, H.; Seol, E.; Hoe, H.S. Donepezil Regulates LPS and Abeta-Stimulated Neuroinflammation through MAPK/NLRP3 Inflammasome/STAT3 Signaling. Int. J. Mol. Sci. 2021, 22, 10637. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, M.; Catto, M.; Sabate, R.; Francesconi, V.; Laurini, E.; Pricl, S.; Pisani, L.; Miniero, D.V.; Liuzzi, G.M.; Gatta, E.; et al. Thioxanthenone-based derivatives as multitarget therapeutic leads for Alzheimer’s disease. Eur. J. Med. Chem. 2023, 250, 115169. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Li, K.; Li, X.; Xu, L.; Yang, Z. Sodium butyrate ameliorates the impairment of synaptic plasticity by inhibiting the neuroinflammation in 5XFAD mice. Chem.-Biol. Interact. 2021, 341, 109452. [Google Scholar] [CrossRef]
- Chu, F.; Li, K.; Li, X.; Xu, L.; Huang, J.; Yang, Z. Graphene Oxide Ameliorates the Cognitive Impairment through Inhibiting PI3K/Akt/mTOR Pathway to Induce Autophagy in AD Mouse Model. Neurochem. Res. 2021, 46, 309–325. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, C.; Tan, R.; Zhou, X.; Wang, R.; Wang, X.; Ma, R.; Chu, F.; Li, Y.; Yin, T.; Liu, Z. Transcranial Magneto-Acoustic Stimulation Protects Synaptic Rehabilitation from Amyloid-Beta Plaques via Regulation of Microglial Functions. Int. J. Mol. Sci. 2024, 25, 4651. https://doi.org/10.3390/ijms25094651
Zhang C, Tan R, Zhou X, Wang R, Wang X, Ma R, Chu F, Li Y, Yin T, Liu Z. Transcranial Magneto-Acoustic Stimulation Protects Synaptic Rehabilitation from Amyloid-Beta Plaques via Regulation of Microglial Functions. International Journal of Molecular Sciences. 2024; 25(9):4651. https://doi.org/10.3390/ijms25094651
Chicago/Turabian StyleZhang, Chunlan, Ruxin Tan, Xiaoqing Zhou, Ruru Wang, Xin Wang, Ren Ma, Fangxuan Chu, Ying Li, Tao Yin, and Zhipeng Liu. 2024. "Transcranial Magneto-Acoustic Stimulation Protects Synaptic Rehabilitation from Amyloid-Beta Plaques via Regulation of Microglial Functions" International Journal of Molecular Sciences 25, no. 9: 4651. https://doi.org/10.3390/ijms25094651