
ONT in Clinical Diagnostics of Repeat Expansion Disorders: Detection and Reporting Challenges


Abstract
1. Introduction
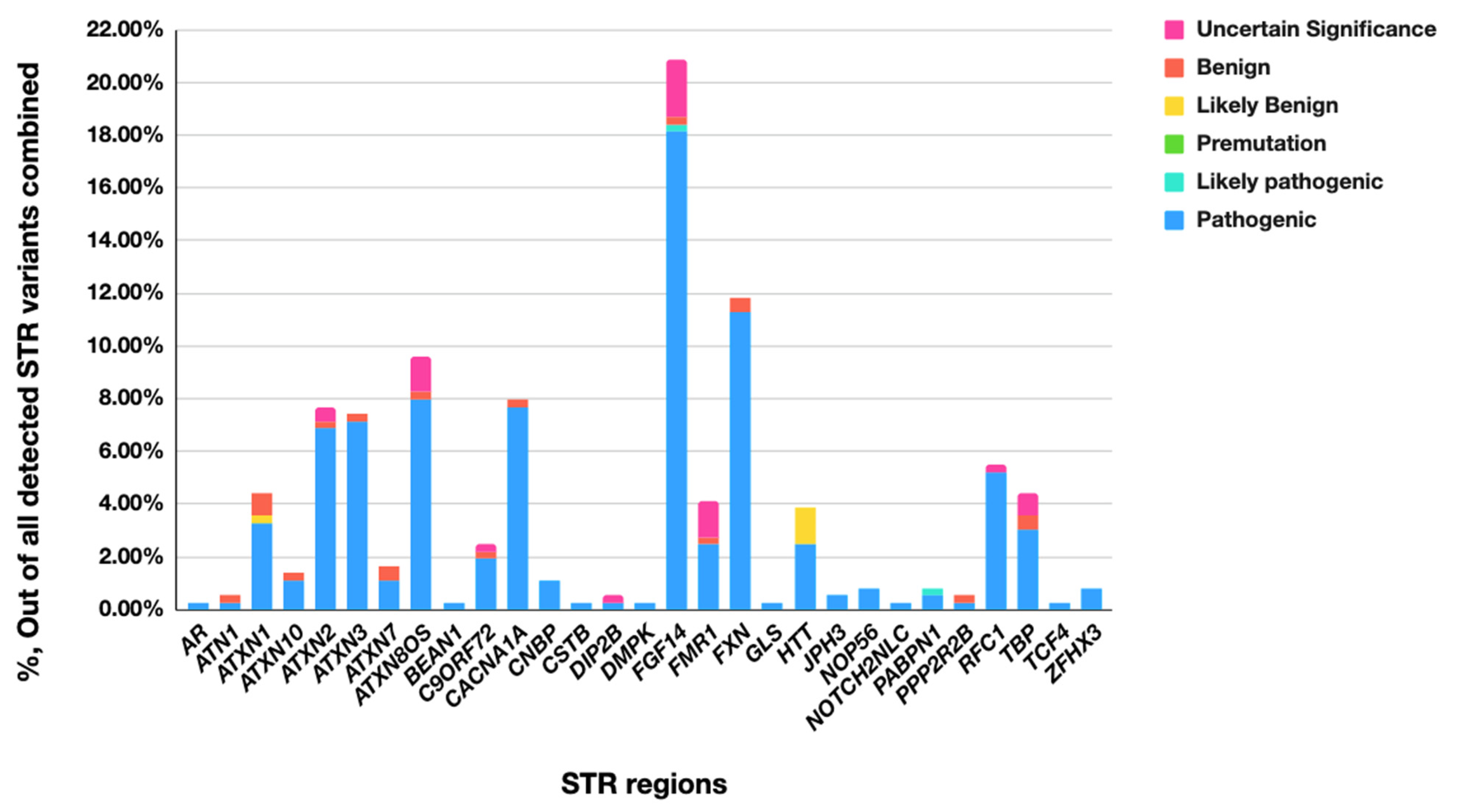
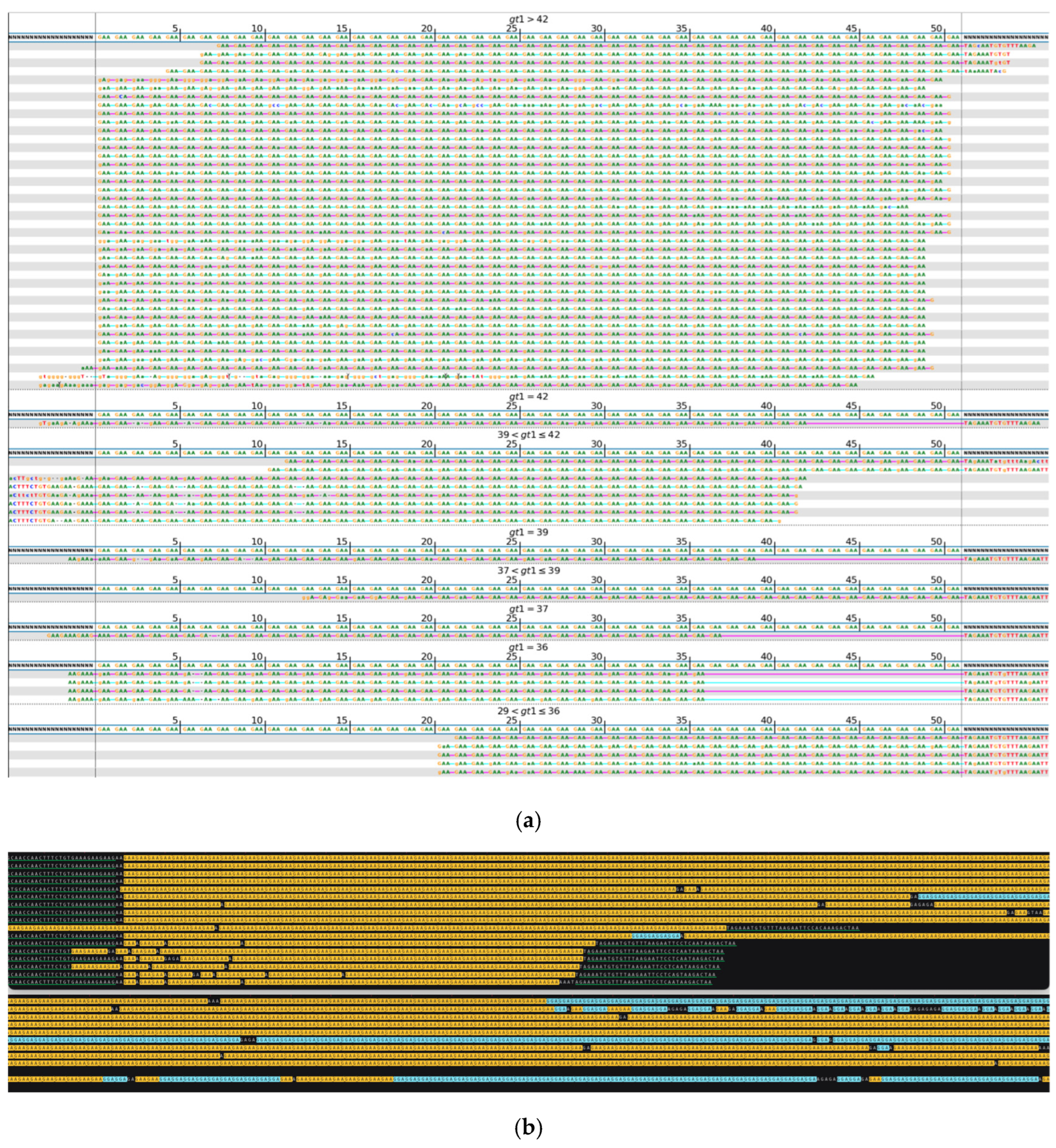
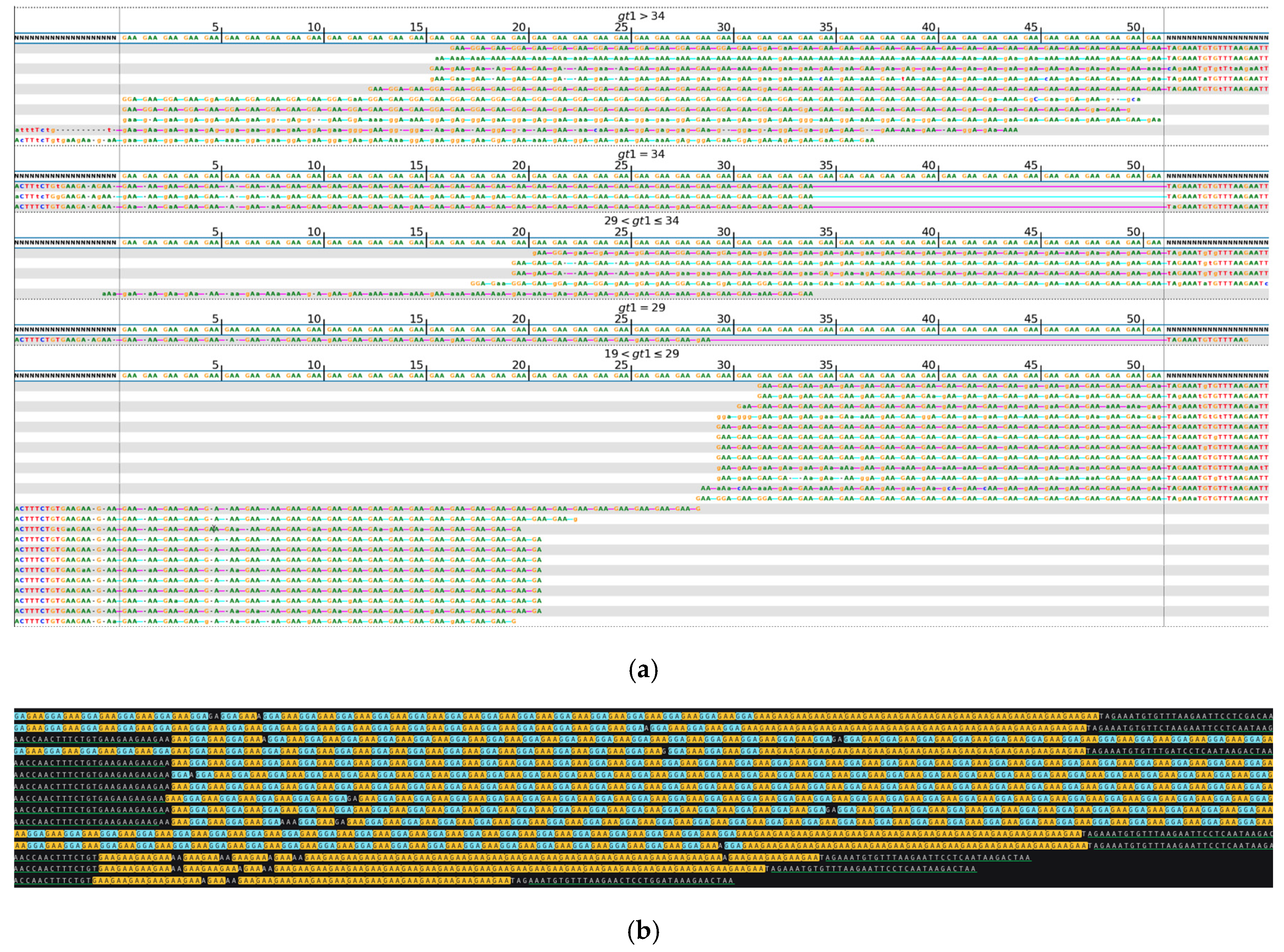
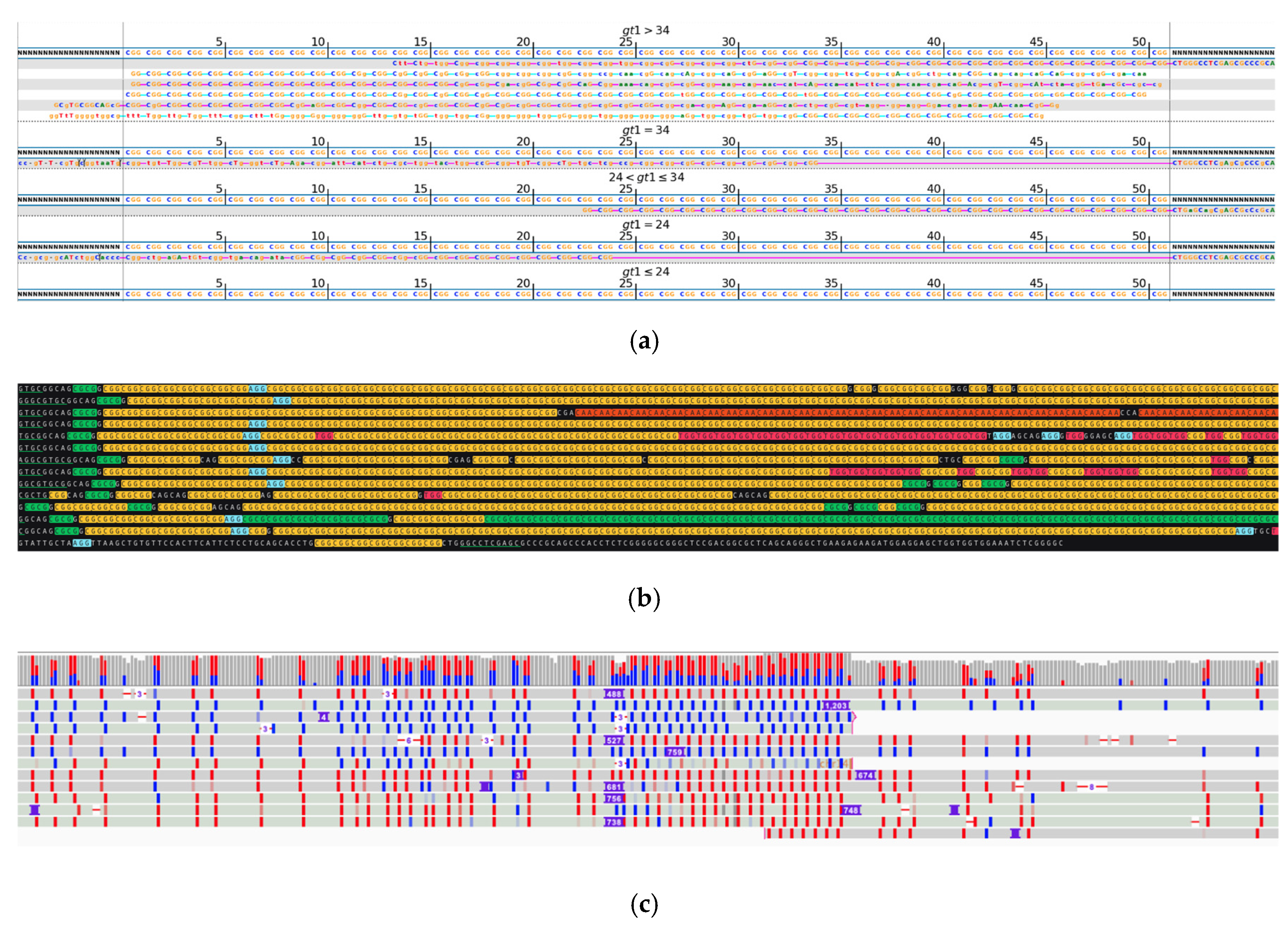
2. Results
3. Discussion
4. Materials and Methods
4.1. Samples Selection
4.2. Library Preparation and Illumina Short-Read Sequencing
4.3. Bioinformatics and Analytical Approach Illumina Short Reads
4.4. Library Preparation and ONT Long-Read Sequencing
4.5. Bioinformatics and Analytical Approach to ONT Long-Reads
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NGS | Next-Generation Sequencing |
| WGS | Whole-Genome Sequencing |
| STR | Short Tandem Repeat |
| VNTR | Variable Number Tandem Repeat |
| ONT | Oxford Nanopore Technologies |
| HPO | Human Phenotype Ontology |
| gDNA | Genomic DNA |
| SSC | Small Sequence Change |
| SV | Structural Variant |
| CNV | Copy Number Variant |
| UPD | Uniparental Disomy |
| HAC | High-Accuracy Settings |
| IGV | Integrative Genomics Viewer |
| VUS | Variant of Uncertain Significance |
| SCA | Spinocerebellar Ataxia |
| FXTAS | Fragile X-Associated Tremor/Ataxia Syndrome |
| FXPOI | Fragile X-Associated Primary Ovarian Insufficiency |
References
- Wigby, K.M.; Brockman, D.; Costain, G.; Hale, C.; Taylor, S.L.; Belmont, J.; Bick, D.; Dimmock, D.; Fernbach, S.; Greally, J.; et al. Evidence Review and Considerations for Use of First Line Genome Sequencing to Diagnose Rare Genetic Disorders. NPJ Genom. Med. 2024, 9, 15. [Google Scholar] [CrossRef]
- van der Sanden, B.P.G.H.; Schobers, G.; Corominas Galbany, J.; Koolen, D.A.; Sinnema, M.; van Reeuwijk, J.; Stumpel, C.T.R.M.; Kleefstra, T.; de Vries, B.B.A.; Ruiterkamp-Versteeg, M.; et al. The Performance of Genome Sequencing as a First-Tier Test for Neurodevelopmental Disorders. Eur. J. Human. Genet. 2022, 31, 81–88. [Google Scholar] [CrossRef]
- Rajan-Babu, I.-S.; Peng, J.J.; Chiu, R.; Birch, P.; Couse, M.; Guimond, C.; Lehman, A.; Mwenifumbo, J.; van Karnebeek, C.; Friedman, J.; et al. Genome-Wide Sequencing as a First-Tier Screening Test for Short Tandem Repeat Expansions. Genome Med. 2021, 13, 126. [Google Scholar] [CrossRef] [PubMed]
- Billingsley, K.J.; Meredith, M.; Daida, K.; Alvarez Jerez, P.; Negi, S.; Malik, L.; Genner, R.M.; Moller, A.; Zheng, X.; Gibson, S.B.; et al. Long-Read Sequencing of Hundreds of Diverse Brains Provides Insight into the Impact of Structural Variation on Gene Expression and DNA Methylation. Preprint. bioRxiv 2024. [Google Scholar] [CrossRef]
- Gustafson, J.A.; Gibson, S.B.; Damaraju, N.; Zalusky, M.P.; Hoekzema, K.; Twesigomwe, D.; Yang, L.; Snead, A.A.; Richmond, P.A.; De Coster, W.; et al. High-Coverage Nanopore Sequencing of Samples from the 1000 Genomes Project to Build a Comprehensive Catalog of Human Genetic Variation. Genome Res. 2024, 34, 2061. [Google Scholar] [CrossRef] [PubMed]
- Gymrek, M.; Willems, T.; Reich, D.; Erlich, Y. Interpreting Short Tandem Repeat Variations in Humans Using Mutational Constraint. Nat. Genet. 2017, 49, 1495. [Google Scholar] [CrossRef]
- Mahmoud, M.; Gobet, N.; Cruz-Dávalos, D.I.; Mounier, N.; Dessimoz, C.; Sedlazeck, F.J. Structural Variant Calling: The Long and the Short of It. Genome Biol. 2019, 20, 1–14. [Google Scholar] [CrossRef]
- Pellerin, D.; Danzi, M.; Renaud, M.; Houlden, H.; Synofzik, M.; Zuchner, S.; Brais, B. GAA-FGF14-Related Ataxia. In GeneReviews®; University of Washington: Seattle, WA, USA, 2024. Available online: https://www.ncbi.nlm.nih.gov/books/NBK599589/ (accessed on 10 February 2025).
- Chiu, R.; Rajan-Babu, I.-S.; Friedman, J.M.; Birol, I. A Comprehensive Tandem Repeat Catalog of the Human Genome. medRxiv 2024. [Google Scholar] [CrossRef]
- Hiatt, L.; Weisburd, B.; Dolzhenko, E.; VanNoy, G.E.; Kurtas, E.N.; Rehm, H.L.; Quinlan, A.; Dashnow, H. STRchive: A Dynamic Resource Detailing Population-Level and Locus-Specific Insights at Tandem Repeat Disease Loci. medRxiv 2024. [Google Scholar] [CrossRef]
- Dolzhenko, E.; English, A.; Dashnow, H.; De Sena Brandine, G.; Mokveld, T.; Rowell, W.J.; Karniski, C.; Kronenberg, Z.; Danzi, M.C.; Cheung, W.A.; et al. Characterization and Visualization of Tandem Repeats at Genome Scale. Nat. Biotechnol. 2024, 42, 1606–1614. [Google Scholar] [CrossRef]
- Chintalaphani, S.R.; Pineda, S.S.; Deveson, I.W.; Kumar, K.R. An Update on the Neurological Short Tandem Repeat Expansion Disorders and the Emergence of Long-Read Sequencing Diagnostics. Acta Neuropathol. Commun. 2021, 9, 1–20. [Google Scholar] [CrossRef]
- Maestri, S.; Maturo, M.G.; Cosentino, E.; Marcolungo, L.; Iadarola, B.; Fortunati, E.; Rossato, M.; Delledonne, M. A Long-read Sequencing Approach for Direct Haplotype Phasing in Clinical Settings. Int. J. Mol. Sci. 2020, 21, 9177. [Google Scholar] [CrossRef]
- Leitão, E.; Schröder, C.; Depienne, C. Identification and Characterization of Repeat Expansions in Neurological Disorders: Methodologies, Tools, and Strategies. Rev. Neurol. 2024, 180, 383–392. [Google Scholar] [CrossRef]
- Vollger, M.R.; Korlach, J.; Eldred, K.C.; Swanson, E.; Underwood, J.G.; Bohaczuk, S.C.; Mao, Y.; Cheng, Y.-H.H.; Ranchalis, J.; Blue, E.E.; et al. Synchronized Long-Read Genome, Methylome, Epigenome and Transcriptome Profiling Resolve a Mendelian Condition. Nat. Genet. 2025, 57, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Depienne, C.; Mandel, J.L. 30 Years of Repeat Expansion Disorders: What Have We Learned and What Are the Remaining Challenges? Am. J. Hum. Genet. 2021, 108, 764–785. [Google Scholar] [CrossRef]
- Ibañez, K.; Jadhav, B.; Zanovello, M.; Gagliardi, D.; Clarkson, C.; Facchini, S.; Garg, P.; Martin-Trujillo, A.; Gies, S.J.; Galassi Deforie, V.; et al. Increased Frequency of Repeat Expansion Mutations across Different Populations. Nat. Med. 2024, 30, 3357–3368. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, D.; Danzi, M.C.; Wilke, C.; Renaud, M.; Fazal, S.; Dicaire, M.-J.; Scriba, C.K.; Ashton, C.; Yanick, C.; Beijer, D.; et al. Deep Intronic FGF14 GAA Repeat Expansion in Late-Onset Cerebellar Ataxia. N. Engl. J. Med. 2023, 388, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, K.; Kawahara, R.; Asano, T.; Morishita, S. A Landscape of Complex Tandem Repeats within Individual Human Genomes. Nat. Commun. 2023, 14, 5530. [Google Scholar] [CrossRef]
- Gymrek, M. A Genomic View of Short Tandem Repeats. Curr. Opin. Genet. Dev. 2017, 44, 9–16. [Google Scholar] [CrossRef]
- Pellerin, D.; Iruzubieta, P.; Tekgül, Ş.; Danzi, M.C.; Ashton, C.; Dicaire, M.J.; Wandzel, M.; Roth, V.; Lamont, P.J.; Bonnet, C.; et al. Non-GAA Repeat Expansions in FGF14 Are Likely Not Pathogenic—Reply to: “Shaking Up Ataxia: FGF14 and RFC1 Repeat Expansions in Affected and Unaffected Members of a Chilean Family”. Mov. Disord. 2023, 38, 1575–1577. [Google Scholar] [CrossRef]
- Yoon, J.G.; Lee, S.; Cho, J.; Kim, N.; Kim, S.; Kim, M.J.; Kim, S.Y.; Moon, J.; Chae, J.H. Diagnostic Uplift through the Implementation of Short Tandem Repeat Analysis Using Exome Sequencing. Eur. J. Hum. Genet. 2024, 32, 584–587. [Google Scholar] [CrossRef] [PubMed]
- Austin-Tse, C.A.; Jobanputra, V.; Perry, D.L.; Bick, D.; Taft, R.J.; Venner, E.; Gibbs, R.A.; Young, T.; Barnett, S.; Belmont, J.W.; et al. Best Practices for the Interpretation and Reporting of Clinical Whole Genome Sequencing. NPJ Genom. Med. 2022, 7, 27. [Google Scholar] [CrossRef]
- Chen, Z.; Morris, H.R.; Polke, J.; Wood, N.W.; Gandhi, S.; Ryten, M.; Houlden, H.; Tucci, A. Repeat Expansion Disorders. Pract. Neurol. 2024, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Genovese, L.M.; Geraci, F.; Corrado, L.; Mangano, E.; D’Aurizio, R.; Bordoni, R.; Severgnini, M.; Manzini, G.; De Bellis, G.; D’Alfonso, S.; et al. A Census of Tandemly Repeated Polymorphic Loci in Genic Regions Through the Comparative Integration of Human Genome Assemblies. Front. Genet. 2018, 9, 155. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Chu, J.Y. A Brief Review of Short Tandem Repeat Mutation. Genom. Proteom. Bioinform. 2007, 5, 7. [Google Scholar] [CrossRef]
- Ziaei Jam, H.; Li, Y.; DeVito, R.; Mousavi, N.; Ma, N.; Lujumba, I.; Adam, Y.; Maksimov, M.; Huang, B.; Dolzhenko, E.; et al. A Deep Population Reference Panel of Tandem Repeat Variation. Nat. Commun. 2023, 14, 6711. [Google Scholar] [CrossRef]
- Cui, Y.; Ye, W.; Li, J.S.; Li, J.J.; Vilain, E.; Sallam, T.; Li, W. A Genome-Wide Spectrum of Tandem Repeat Expansions in 338,963 Humans. Cell 2024, 187, 2336–2341.e5. [Google Scholar] [CrossRef]
- Laß, J.; Thomsen, M.; Borsche, M.; Lüth, T.; Prietzsche, J.C.; Schaake, S.; Milovanović, A.; Macpherson, H.; Gustavsson, E.K.; Awad, P.S.; et al. FGF14 Repeat Length and Mosaic Interruptions: Modifiers of SCA27b? medRxiv 2024. [Google Scholar] [CrossRef]
- Sehgal, A.; Jam, H.Z.; Shen, A.; Gymrek, M. Genome-Wide Detection of Somatic Mosaicism at Short Tandem Repeats. Bioinformatics 2024, 40, btae485. [Google Scholar] [CrossRef]
- Khristich, A.N.; Mirkin, S.M. On the Wrong DNA Track: Molecular Mechanisms of Repeat-Mediated Genome Instability. J. Biol. Chem. 2020, 295, 4134–4170. [Google Scholar] [CrossRef]
- Handsaker, R.E.; Kashin, S.; Reed, N.M.; Tan, S.; Lee, W.-S.; McDonald, T.M.; Morris, K.; Kamitaki, N.; Mullally, C.D.; Morakabati, N.R.; et al. Long Somatic DNA-Repeat Expansion Drives Neurodegeneration in Huntington’s Disease. Cell 2025, 188, 623–639.e19. [Google Scholar] [CrossRef]
- Rasmussen, A.; Hildonen, M.; Vissing, J.; Duno, M.; Tümer, Z.; Birkedal, U. High Resolution Analysis of DMPK Hypermethylation and Repeat Interruptions in Myotonic Dystrophy Type 1. Genes 2022, 13, 970. [Google Scholar] [CrossRef] [PubMed]
- Pešović, J.; Perić, S.; Brkušanin, M.; Brajušković, G.; Rakoč Ević -Stojanović, V.; Savić-Pavić Ević, D. Repeat Interruptions Modify Age at Onset in Myotonic Dystrophy Type 1 by Stabilizing DMPK Expansions in Somatic Cells. Front. Genet. 2018, 9, 601. [Google Scholar] [CrossRef] [PubMed]
- Morato Torres, C.A.; Zafar, F.; Tsai, Y.C.; Vazquez, J.P.; Gallagher, M.D.; McLaughlin, I.; Hong, K.; Lai, J.; Lee, J.; Chirino-Perez, A.; et al. ATTCT and ATTCC Repeat Expansions in the ATXN10 Gene Affect Disease Penetrance of Spinocerebellar Ataxia Type 10. Hum. Genet. Genom. Adv. 2022, 3, 100137. [Google Scholar] [CrossRef]
- Dolzhenko, E.; van Vugt, J.J.F.A.; Shaw, R.J.; Bekritsky, M.A.; Van Blitterswijk, M.; Narzisi, G.; Ajay, S.S.; Rajan, V.; Lajoie, B.R.; Johnson, N.H.; et al. Detection of Long Repeat Expansions from PCR-Free Whole-Genome Sequence Data. Genome Res. 2017, 27, 1895–1903. [Google Scholar] [CrossRef] [PubMed]
- Rajan-Babu, I.-S.; Dolzhenko, E.; Eberle, M.A.; Friedman, J.M. Sequence Composition Changes in Short Tandem Repeats: Heterogeneity, Detection, Mechanisms and Clinical Implications. Nat. Rev. Genet. 2024, 25, 476–499. [Google Scholar] [CrossRef]
- Mangin, A.; de Pontual, L.; Tsai, Y.C.; Monteil, L.; Nizon, M.; Boisseau, P.; Mercier, S.; Ziegle, J.; Harting, J.; Heiner, C.; et al. Robust Detection of Somatic Mosaicism and Repeat Interruptions by Long-Read Targeted Sequencing in Myotonic Dystrophy Type 1. Int. J. Mol. Sci. 2021, 22, 2616. [Google Scholar] [CrossRef]
- Sullivan, R.; Chen, S.; Saunders, C.T.; Yau, W.Y.; Goh, Y.Y.; O’Connor, E.; Dominik, N.; Deforie, V.G.; Morsy, H.; Cortese, A.; et al. RFC1 Repeat Expansion Analysis from Whole Genome Sequencing Data Simplifies Screening and Increases Diagnostic Rates. medRxiv 2024. [Google Scholar] [CrossRef]
- Nethisinghe, S.; Kesavan, M.; Ging, H.; Labrum, R.; Polke, J.M.; Islam, S.; Garcia-moreno, H.; Callaghan, M.F.; Cavalcanti, F.; Pook, M.A.; et al. Interruptions of the Fxn Gaa Repeat Tract Delay the Age at Onset of Friedreich’s Ataxia in a Location Dependent Manner. Int. J. Mol. Sci. 2021, 22, 7507. [Google Scholar] [CrossRef]
- Kaplun, L.; Krautz-Peterson, G.; Neerman, N.; Stanley, C.; Hussey, S.; Folwick, M.; McGarry, A.; Weiss, S.; Kaplun, A. ONT Long-Read WGS for Variant Discovery and Orthogonal Confirmation of Short Read WGS Derived Genetic Variants in Clinical Genetic Testing. Front. Genet. 2023, 14, 1145285. [Google Scholar] [CrossRef]
- Vegezzi, E.; Facchini, S.; Bragg, D.C.; Sharma, N.; Cortese, A.; Vegezzi, E.; Ishiura, H.; Cristopher Bragg, D.; Pellerin, D.; Magrinelli, F.; et al. Neurological Disorders Caused by Novel Non-Coding Repeat Expansions: Clinical Features and Differential Diagnosis. Lancet Neurol. 2024, 23, 725–764. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, D.; Wilke, C.; Traschütz, A.; Nagy, S.; Currò, R.; Dicaire, M.J.; Garcia-Moreno, H.; Anheim, M.; Wirth, T.; Faber, J.; et al. Intronic FGF14 GAA Repeat Expansions Are a Common Cause of Ataxia Syndromes with Neuropathy and Bilateral Vestibulopathy. J. Neurol. Neurosurg. Psychiatry 2023, 95, 175. [Google Scholar] [CrossRef]
- Ibañez, K.; Polke, J.; Hagelstrom, R.T.; Dolzhenko, E.; Pasko, D.; Thomas, E.R.A.; Daugherty, L.C.; Kasperaviciute, D.; Smith, K.R.; Deans, Z.C.; et al. Whole Genome Sequencing for the Diagnosis of Neurological Repeat Expansion Disorders in the UK: A Retrospective Diagnostic Accuracy and Prospective Clinical Validation Study. Lancet Neurol. 2022, 21, 234–245. [Google Scholar] [CrossRef]
- Roy, S.; Coldren, C.; Karunamurthy, A.; Kip, N.S.; Klee, E.W.; Lincoln, S.E.; Leon, A.; Pullambhatla, M.; Temple-Smolkin, R.L.; Voelkerding, K.V.; et al. Standards and Guidelines for Validating Next-Generation Sequencing Bioinformatics Pipelines: A Joint Recommendation of the Association for Molecular Pathology and the College of American Pathologists. J. Mol. Diagn. 2018, 20, 4–27. [Google Scholar] [CrossRef] [PubMed]
- Scriba, C.K.; Stevanovski, I.; Chintalaphani, S.R.; Gamaarachchi, H.; Ghaoui, R.; Ghia, D.; Henderson, R.D.; Jordan, N.; Winkel, A.; Lamont, P.J.; et al. RFC1 in an Australasian Neurological Disease Cohort: Extending the Genetic Heterogeneity and Implications for Diagnostics. Brain Commun. 2023, 5, fcad208. [Google Scholar] [CrossRef] [PubMed]
- Dominik, N.; Magri, S.; Currò, R.; Abati, E.; Facchini, S.; Corbetta, M.; MacPherson, H.; Di Bella, D.; Sarto, E.; Stevanovski, I.; et al. Normal and Pathogenic Variation of RFC1 Repeat Expansions: Implications for Clinical Diagnosis. Brain 2023, 146, 5060–5069. [Google Scholar] [CrossRef]
- Nobile, V.; Pucci, C.; Chiurazzi, P.; Neri, G.; Tabolacci, E. DNA Methylation, Mechanisms of FMR1 Inactivation and Therapeutic Perspectives for Fragile X Syndrome. Biomolecules 2021, 11, 296. [Google Scholar] [CrossRef]
- Hunter, J.E.; Berry-Kravis, E.; Hipp, H.; Todd, P.K. FMR1 Disorders. In GeneReviews®; University of Washington: Seattle, WA, USA, 2024. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1384/ (accessed on 10 February 2025).
- Wallenius, J.; Kafantari, E.; Jhaveri, E.; Gorcenco, S.; Ameur, A.; Karremo, C.; Dobloug, S.; Karrman, K.; de Koning, T.; Ilinca, A.; et al. Exonic Trinucleotide Repeat Expansions in ZFHX3 Cause Spinocerebellar Ataxia Type 4: A Poly-Glycine Disease. Am. J. Hum. Genet. 2023, 111, 82. [Google Scholar] [CrossRef]
- Neerman, N.; Faust, G.; Meeks, N.; Modai, S.; Kalfon, L.; Falik-Zaccai, T.; Kaplun, A. A Clinically Validated Whole Genome Pipeline for Structural Variant Detection and Analysis. BMC Genom. 2019, 20, 1–8. [Google Scholar] [CrossRef]
- Variantyx Genomic Unity® Test A Whole Genome Clinical Validation Study. 2019. Available online: https://www.variantyx.com/wp-content/uploads/2022/04/Genomic-Unity-Clinical-Validation-Study.pdf (accessed on 2 February 2025).
- De Coster, W.; Höijer, I.; Bruggeman, I.; D’Hert, S.; Melin, M.; Ameur, A.; Rademakers, R. Medically Relevant Tandem Repeats in Nanopore Sequencing of Control Cohorts. medRxiv 2024. [Google Scholar] [CrossRef]
- Read, J.L.; Davies, K.C.; Thompson, G.C.; Delatycki, M.B.; Lockhart, P.J. Challenges Facing Repeat Expansion Identification, Characterisation, and the Pathway to Discovery. Emerg. Top. Life Sci. 2023, 7, 339. [Google Scholar] [CrossRef] [PubMed]
- Friedreich Ataxia (FRDA) via the FXN GAA Repeat Expansion Test—PreventionGenetics. Available online: https://www.preventiongenetics.com/testInfo?val=Friedreich-Ataxia-%28FRDA%29-via-the-FXN-GAA-Repeat-Expansion (accessed on 6 March 2025).
- Seifert, B.A.; Reddi, H.V.; Kang, B.E.; Bean, L.J.H.; Shealy, A.; Rose, N.C. Myotonic Dystrophy Type 1 Testing, 2024 Revision: A Technical Standard of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2024, 26, 101145. [Google Scholar] [CrossRef] [PubMed]
- 620114: SCA1 (ATXN1) Genetic Testing (Repeat Expansion)|Labcorp Labcorp. Available online: https://www.labcorp.com/tests/620114/sca1-atxn1-genetic-testing-repeat-expansion (accessed on 17 March 2025).
- Rudaks, L.I.; Yeow, D.; Ng, K.; Deveson, I.W.; Kennerson, M.L.; Kumar, K.R. An Update on the Adult-Onset Hereditary Cerebellar Ataxias: Novel Genetic Causes and New Diagnostic Approaches. Cerebellum 2024, 23, 2152–2168. [Google Scholar] [CrossRef] [PubMed]
- PacBio—Sequence with Confidence. Available online: https://www.pacb.com/ (accessed on 3 March 2025).
- Welcome to Oxford Nanopore Technologies. Available online: https://nanoporetech.com/ (accessed on 3 March 2025).
- Oehler, J.B.; Wright, H.; Stark, Z.; Mallett, A.J.; Schmitz, U. The Application of Long-Read Sequencing in Clinical Settings. Hum. Genom. 2023, 17, 73. [Google Scholar] [CrossRef]
- Illumina Complete Long Reads Portfolio. Available online: https://www.illumina.com/products/by-brand/complete-long-reads-portfolio.html (accessed on 3 March 2025).
- Genome Reference Consortium Human Build 38—NCBI. Available online: https://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.26/ (accessed on 5 January 2023).
- GnomAD. Available online: https://gnomad.broadinstitute.org/ (accessed on 3 February 2025).
- Halman, A.; Dolzhenko, E.; Oshlack, A. STRipy: A Graphical Application for Enhanced Genotyping of Pathogenic Short Tandem Repeats in Sequencing Data. Hum. Mutat. 2022, 43, 859–868. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No Long-Read Sequencing Required | Long-Read Sequencing Required | Total | |
|---|---|---|---|
| Cases with STR expansion | 184 (9.0%) | 154 (7.6%) | 338 (16.6%) |
| Cases with Other variant types (No STR) | 1527 (74.9%) | 173 (8.5%) | 1700 (83.4%) |
| Total | 1711 (84.0%) | 327 (16.0%) | 2038 (100.00%) |
| Ranges | Reported Variants | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| STR Locus | Normal Range | Mutable Normal | Intermediate/Uncertain Range | Reduced Penetrance Range | Pathogenic Threshold | Repeat Unit Length | No Long-Read Sequencing | Long-Read Sequencing | Total |
| AR | <34 | 35 | 36–37 | 38 | 3 | 1 | 0 | 1 | |
| ATN1 | 6–35 | 36–47 | 48 | 3 | 1 | 1 | 2 | ||
| ATXN1 | 6–35 (36–44 **) | 36–38 | 39 (46 *) | 3 | 11 | 5 | 16 | ||
| ATXN10 | 10–32 | 33–280 | 280–800 | 800 | 5 | 3 | 2 | 5 | |
| ATXN2 | <30 | 30–32 *** | 33–34 | 35 | 3 | 23 | 5 | 28 | |
| ATXN3 | 44 | 45–49 | 50–55 | 56 | 3 | 18 | 8 | 26 | |
| ATXN7 | 27 | 28–33 | 34–36 | 37 | 3 | 3 | 3 | 6 | |
| ATXN8OS | 15–50 | 51–53 | 54 ^ | 54 ^ | 3 | 23 | 11 | 34 | |
| BEAN1 ^^ | - | - | 110 | 5 | 1 | 0 | 1 | ||
| C9ORF72 | ≤24 | 25–60 | 24–60 | 61 | 6 | 6 | 3 | 9 | |
| CACNA1A | 18 | 19 | 19–19 | 20 | 3 | 25 | 3 | 28 | |
| CNBP | 26 | 27–74 | 75 | 4 | 1 | 3 | 4 | ||
| CSTB | 2–3 | 12–17 | 30 | 12 | 0 | 1 | 1 | ||
| DIP2B | 6–23 | 139–206 | 250 | 3 | 0 | 1 | 1 | ||
| DMPK | 5–34 | 35–49 | 50 | 3 | 1 | 0 | 1 | ||
| FGF14 | 180–319 | 320 | 3 | 22 | 54 | 76 | |||
| FMR1 | 5–44 | 45–54 | 55–200 ^^^ | 201 | 3 | 6 | 10 | 16 | |
| FXN | 5–33 | 34–65 | 66 | 3 | 29 | 13 | 42 | ||
| GLS | 5–38 | - | 680 | 3 | 0 | 1 | 1 | ||
| HTT | 26 | 27–35 | 36–39 | 40 | 3 | 2 | 7 | 9 | |
| JPH3 | 28 | 29–39 | 40 | 3 | 0 | 2 | 2 | ||
| NOP56 | 3–14 | 15–649 | 650 | 6 | 0 | 3 | 3 | ||
| NOTCH2NLC | ≤40 | 41–59 | 60 | 3 | 0 | 1 | 1 | ||
| PABPN1 | 10 | 11–11 homozygous | 11–18 | 3 | 2 | 1 | 3 | ||
| PPP2R2B | 7–32 | 40–50 | 51 | 3 | 2 | 0 | 2 | ||
| RFC1 | 11–200 | 400 | 5 | 5 | 15 | 20 | |||
| TBP | 25–40 | 41–48 | 49 | 3 | 12 | 3 | 15 | ||
| TCF4 | 40 | 40–50 | 51 | 3 | 1 | 0 | 1 | ||
| ZFHX3 | 31 | 31–41 | 42 | 3 | 1 | 2 | 3 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaplun, L.; Krautz-Peterson, G.; Neerman, N.; Schindler, Y.; Dehan, E.; Huettner, C.S.; Baumgartner, B.K.; Stanley, C.; Kaplun, A. ONT in Clinical Diagnostics of Repeat Expansion Disorders: Detection and Reporting Challenges. Int. J. Mol. Sci. 2025, 26, 2725. https://doi.org/10.3390/ijms26062725
Kaplun L, Krautz-Peterson G, Neerman N, Schindler Y, Dehan E, Huettner CS, Baumgartner BK, Stanley C, Kaplun A. ONT in Clinical Diagnostics of Repeat Expansion Disorders: Detection and Reporting Challenges. International Journal of Molecular Sciences. 2025; 26(6):2725. https://doi.org/10.3390/ijms26062725
Chicago/Turabian StyleKaplun, Ludmila, Greice Krautz-Peterson, Nir Neerman, Yocheved Schindler, Elinor Dehan, Claudia S. Huettner, Brett K. Baumgartner, Christine Stanley, and Alexander Kaplun. 2025. "ONT in Clinical Diagnostics of Repeat Expansion Disorders: Detection and Reporting Challenges" International Journal of Molecular Sciences 26, no. 6: 2725. https://doi.org/10.3390/ijms26062725
APA StyleKaplun, L., Krautz-Peterson, G., Neerman, N., Schindler, Y., Dehan, E., Huettner, C. S., Baumgartner, B. K., Stanley, C., & Kaplun, A. (2025). ONT in Clinical Diagnostics of Repeat Expansion Disorders: Detection and Reporting Challenges. International Journal of Molecular Sciences, 26(6), 2725. https://doi.org/10.3390/ijms26062725