Introduction
Electron capture is the dominant inelastic process during the interaction of multiply charged ions and neutral atoms and molecules at energies less than 25 keV/amu. There is currently strong interest in these interactions due their importance in many astrophysical and terrestrial environments. For example, electron capture by solar wind ions interacting with cometary gases [
1], by auroral ions interacting with planetary atmospheres [
2], and stellar wind ions with the interstellar medium [
3], results in the formation of excited states of either or both collision partners with the resultant visible, UV and X-ray emissions. The thermal and ionisation balance of astrophysical plasmas are influenced by one-electron capture processes [
4] involving a wide variety of partially ionised ions. Modeling of ion transport and radiative cooling processes in the divertor region of current large Tokamak fusion plasma devices requires detailed information on the relevant electron capture processes [
5].
There have been a large number of experimental measurements of total one-electron capture cross sections by multiply charged ions in atomic and molecular targets (see for example [
6], [
7]). Experimental studies of state selective electron capture have been carried out primarily via photon emission spectroscopy (PES) or translational energy spectroscopy (TES), see for example [
8,
9,
10]. Theoretical calculations of electron capture cross sections have been carried out using both semi-classical and quantal approaches. See for example [
11], [
12].
In many of the collision systems investigated, a comparison of theoretical and experimental data was complicated by the presence of unknown fractions of low-lying metastables in the primary ion beams used in the experiments. These metastable ions can have a significantly different capture probability than ground state ions [
13]. Therefore, to obtain information on collision processes of ground state ions in atomic and molecular targets, it is necessary to either determine the metastable fraction of the ion beam or produce an ion beam free from metastable contamination. We have developed an experimental system that makes it possible to produce pure primary ion beams in either ground or metastable states using double translational energy spectroscopy (DTES) [
14]. This technique has been used to identify the significant capture channels and determine their relative importance, of a wide variety of multiply charged ions in both ground and metastable states in atomic and molecular targets (see [
15], [
16]). A particular feature of the DTES technique is its ability, in the case of molecular targets, to determine the relative importance of capture involving both dissociative and non-dissociative processes where the projectile is left in either a ground or metastable state. Information can also be obtained on the distribution of the final vibrational state population of the target product ion [
17].
Experimental method
A schematic diagram of the DTES system is shown in
figure 1.
Figure 1.
Schematic Diagram of DTES apparatus.
Figure 1.
Schematic Diagram of DTES apparatus.
In order to illustrate the technique, the production of a ground state beam of N
2+(2s
22p)
2P
o ions is described. A beam of ions is produced by an all-permanent magnet 9-10.5GHz ECR ion source [
18]. A characteristic of such sources is the existence of a positive plasma potential that must be accounted for in defining the final energy of the beam. In our case, the source plasma tube is held at a small negative potential approximately equal and opposite to the positive plasma potential. The plasma potential is measured using a retarding potential analyzer (RPA) at the rear of energy analyzer 1. The ion beam is then extracted from the ECR source via a ‘floating beam-line accelerator’ in which the beam-line is held at a potential of –4kV. N
3+ ions are selected using a 90° double focusing analysing magnet. A switching magnet is used to deflect the beam into the DTES apparatus. After entering the DTES beamline, the N
3+ ions are decelerated by a cylindrical lens system L1 into the first hemispherical energy analyser EA1. The beam passes through EA1 and then is accelerated by lens system L2 into the first target T1, containing helium. The N
3+ ions undergo one-electron capture collisions in this first gas target and the resulting beam, now containing N
2+ and N
3+ ions in both ground and metastable states, is accelerated by lens system L3 into a second energy analyser, EA2. Applying a suitable retarding voltage on L3, L4, and EA2 ensures only the N
2+ ions pass through. By scanning this voltage, a translational energy spectrum is obtained of the N
2+ ions as shown in
figure 2.
Figure 2.
Energy change spectrum for one-electron capture by N
3+ in He at 3 keV [
19].
Figure 2.
Energy change spectrum for one-electron capture by N
3+ in He at 3 keV [
19].
It is possible to select product N
2+ (
2P) ions from the main observed channel:
corresponding to peak C. The excited N
2+ (
2P) ions decay rapidly (1.4ns) to the ground state within the minimum flight time to the second target (10μs), and so produce a beam of ground state N
2+ ions. This is the first stage of double translational energy spectroscopy.
The ion beam, now consisting of only ground state N
2+ ions, is accelerated by lens system L4 into the second target region (T2). T2 consists of an aluminium cell coupled to a microwave-driven atomic hydrogen source that can be used to provide beams of highly dissociated hydrogen [
20] or other target species as required. A schematic diagram of this T2 region is shown in
figure 3. It should be noted that at this stage the beam energy is uniquely defined and independent of any plasma potential changes. The N
2+ ions undergo charge transfer collisions in the hydrogen target and the product N
+ ions, forward scattered within an angular range of ±3º, pass through a set of horizontal and vertical deflection plates into L5, where they are decelerated into the final energy analyser EA3. Again, by applying a suitable retarding voltage on L5 and EA3, only the passage of the product N
+ ions is allowed. The N
+ ions pass through EA3 and are detected by a position sensitive detector (PSD).
Figure 3.
Schematic diagram of the microwave driven atomic hydrogen source.
Figure 3.
Schematic diagram of the microwave driven atomic hydrogen source.
Again, by scanning the retarding voltage on L5 and EA3, a translational energy spectrum of the product N+ ions can be obtained whilst maintaining a constant energy resolution of 0.5q eV (q=primary ion charge). The translational energy change is equal to the energy defect of the particular capture channel with a small correction for non-zero degree scattering and target recoil.
Results and discussion
In the present study we have used both single (TES) and double (DTES) translational energy spectroscopy to study state selective electron capture processes in collisions of C
2+, N
2+, He
2+, N
5+, and O
6+ in H
2, and He
2+ ions in CO at energies < 1keV/amu. In the case of C
2+ and N
2+ where metastable contamination is evident in beams derived directly from ion sources [
21], the experimental system was operated in the DTES mode. The TES mode was used for measurements with the other ions where metastables either do not exist or are too high-lying to contribute to the one electron capture spectra.
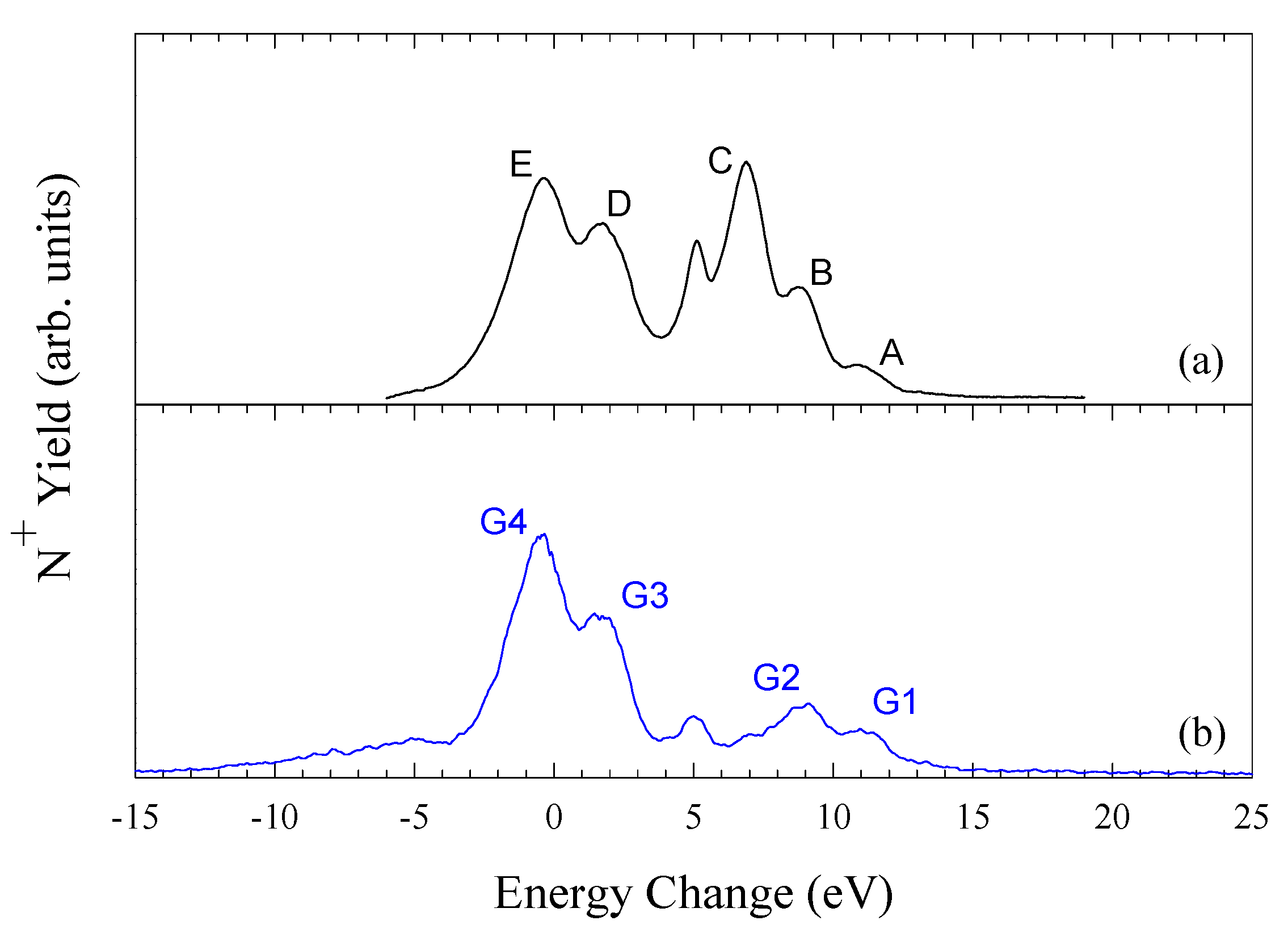
N2+ + H2
Figure 4 shows the translational energy spectra [
22] obtained for (a) a mixed beam containing both ground state and metastable N
2+ ions and (b) a pure ground state beam of N
2+ ions in H2 at 428eV/amu.
Table 1 lists the energy defects for each peak shown in
figure 4. It can be seen in
figure 5 that the unknown metastable content of the ion beam greatly influences the total measured N
+ signal. DTES is therefore an effective way of ensuring that the primary ion beams are free from metastable contamination. Full details are given in [
19]. Previous measurements with beams containing both ground state and metastable N
2+ ions must be treated with caution.
Figure 4.
Energy change spectra for electron capture by (a) a mixed beam of ground state and metastable N2+ ions and (b) a pure ground state beam of N2+ ions in H2 at 428 eV/amu.
Figure 4.
Energy change spectra for electron capture by (a) a mixed beam of ground state and metastable N2+ ions and (b) a pure ground state beam of N2+ ions in H2 at 428 eV/amu.
Table 1.
Main reaction channels for electron capture by N2+ ions in H21Σg+
Table 1.
Main reaction channels for electron capture by N2+ ions in H21Σg+
| Peak | Reaction channels | Energy defects / eV |
|---|
| | N2+ (1s22s22p) 2Po + H2 X 1Σg+→ | |
| | | N+ (1s22s22p2) 3P + H2+2Σg+ | 14.18 (v=0) – 11.53 (Dissociation limit DL) |
| A, G1 | | N+ (1s22s22p2) 1D + H2+ " | 12.28 (v=0) – 9.63 (DL) |
| B, G2 | | N+ (1s22s22p2) 1S + H2+ " | 10.12 (v=0) – 7.47 (DL) |
| D, G3 | | N+ (1s22s2p3) 3Do + H2+ " | 2.74 (v=0) – 0.09 (DL) |
| E, G4 | | N+ (1s22s2p3) 3Po + H2+ " | 0.63 (v=0) – -2.02 (DL) |
| | | N+ (1s22s2p3) 1Do + H2+ " | -3.70 (v=0) – -6.35 (DL) |
| | N2+ (1s22s2p2) 4P + H2 X 1Σg+ 1sσ→ | |
| B | | N+ (1s22s2p3) 3Do + H2+2Σg+ | 9.83 (v=0) – 7.18 (DL) |
| C | | N+ (1s22s2p3) 3Po + H2+ " | 7.72 (v=0) – 5.07 (DL) |
| D | | N+ (1s22s22p3s) 3Po + H2+ " | 2.80 (v=0) – 0.15 (DL) |
| D, E | | N+ (1s22s2p3) 3So + H2+ " | 2.03 (v=0) – -0.62 (DL) |
| E | | N+ (1s22s22p3p) 3D + H2+ " | 0.62 (v=0) – -2.03 (DL) |
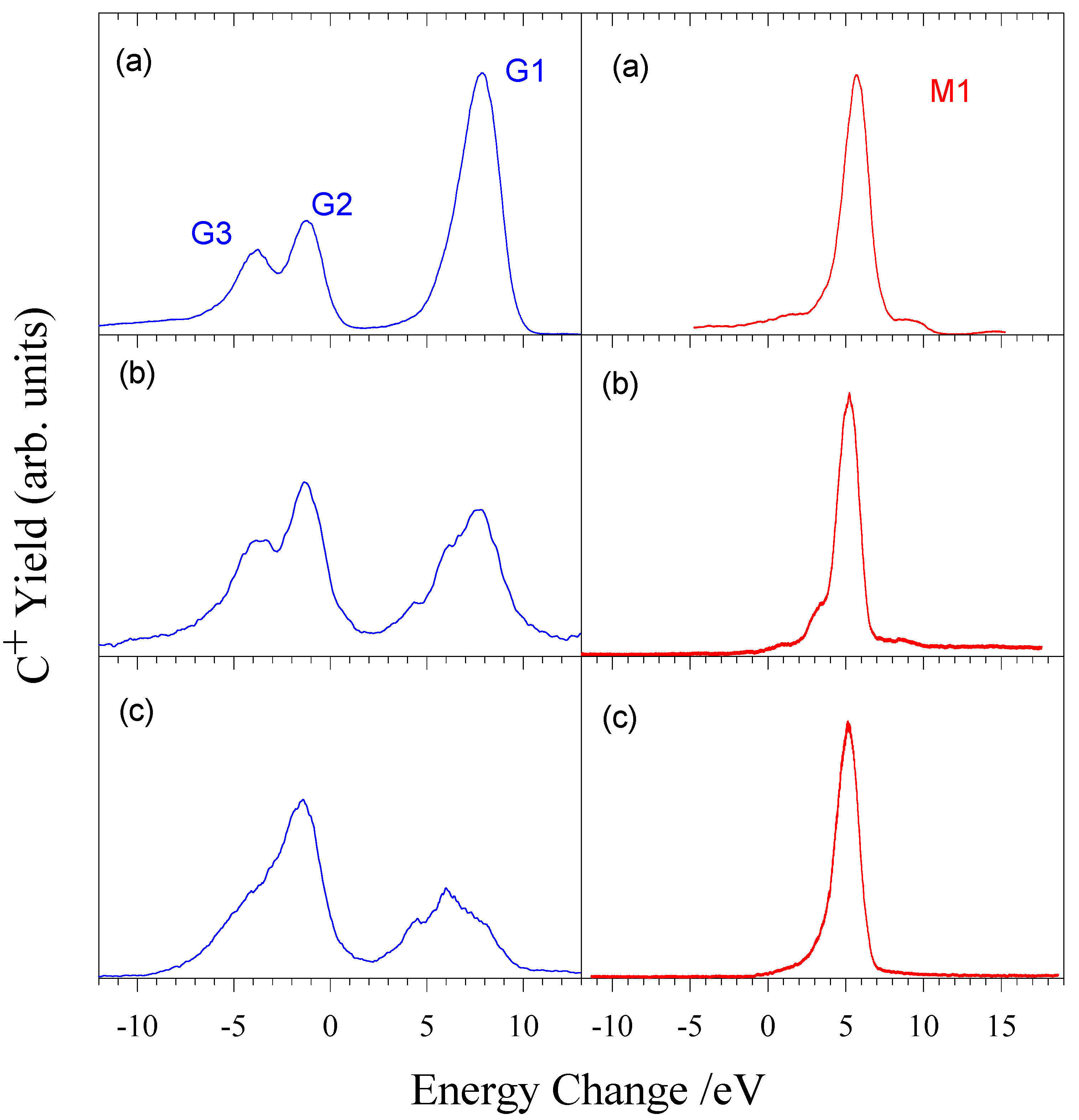
C2+ + H2
Figure 5 shows the measured energy change spectrum obtained for C
2+ ions in H
2 at 500 eV/amu. The main reaction channels are listed in table 2.
Figure 5.
Energy change spectra for electron capture by (a) a mixed beam of ground state and metastable C
2+ ions, (b) a pure ground state beam of C
2+ ions, and (c) a pure metastable C
2+ beam [
22].
Figure 5.
Energy change spectra for electron capture by (a) a mixed beam of ground state and metastable C
2+ ions, (b) a pure ground state beam of C
2+ ions, and (c) a pure metastable C
2+ beam [
22].
Table 2.
Main reaction channels for one electron capture by C
2+ ions in H
21Σ
g+ [
22]
Table 2.
Main reaction channels for one electron capture by C2+ ions in H21Σg+ [22]
| Peak | Product channels | Energy defects / eV |
|---|
| C2+ (1s22s2) + H21Σg+→ |
| G1 | C+ (1s22s22p) + H2+2Σg+ | 8.96 (v=0) – 6.31 (Dissociation limit DL) |
| G2 | C+ (1s22s2p2) + H2+ " | -0.33 (v=0) – -2.98 (DL) |
| G3 | C+ (1s22s2p2) + H2+ " | -3.01 (v=0) – -5.66 (DL |
| G3 | C+ (1s22s2p2) + H2+ " | -4.76 (v=0) – -7.41 (DL) |
| G3 | C+ (1s22s23s) + H2+ " | -5.49 (v=0) – -8.14 (DL) |
| C2+ (1s22s2p) + H21Σg+→ |
| | C+ (1s22s22p) + H2+ " | 15.45 (v=0) – 12.80 (DL) |
| | C+ (1s22s2p2) + H2+ " | 10.12 (v=0) – 7.47 (DL) |
| M1 | C+ (1s22s2p2) + H2+ " | 6.16 (v=0) – 3.51 (DL) |
| | C+ (1s22s2p2) + H2+ " | 3.49 (v=0) – 0.84 (DL) |
| | C+ (1s22s2p2) + H2+ " | 1.73 (v=0) – -0.92(DL) |
It can clearly be seen that one electron capture is dominated by capture from metastable C
2+ ions.
Figure 6 shows the energy dependence of both ground state and metastable reaction channels. It can be seen in the ground state spectra that the channel corresponding to peak 1 is decreasing in importance with decreasing energy, while peak 2 is increasing. In contrast, the metastable spectra show very little energy dependence. Leputsch et al [
17] have carried out a TES study of this system with an ion kinetic energy resolution ≤ 200meV enabling the final vibrational state distribution of the H
2+ target product ion to be identified.
Figure 6.
Energy change spectra obtained for C
2+ ground and metastable ions in H
2 at (a) 500 eV/amu, (b) 250 eV/amu, and 125 eV/amu [
22].
Figure 6.
Energy change spectra obtained for C
2+ ground and metastable ions in H
2 at (a) 500 eV/amu, (b) 250 eV/amu, and 125 eV/amu [
22].
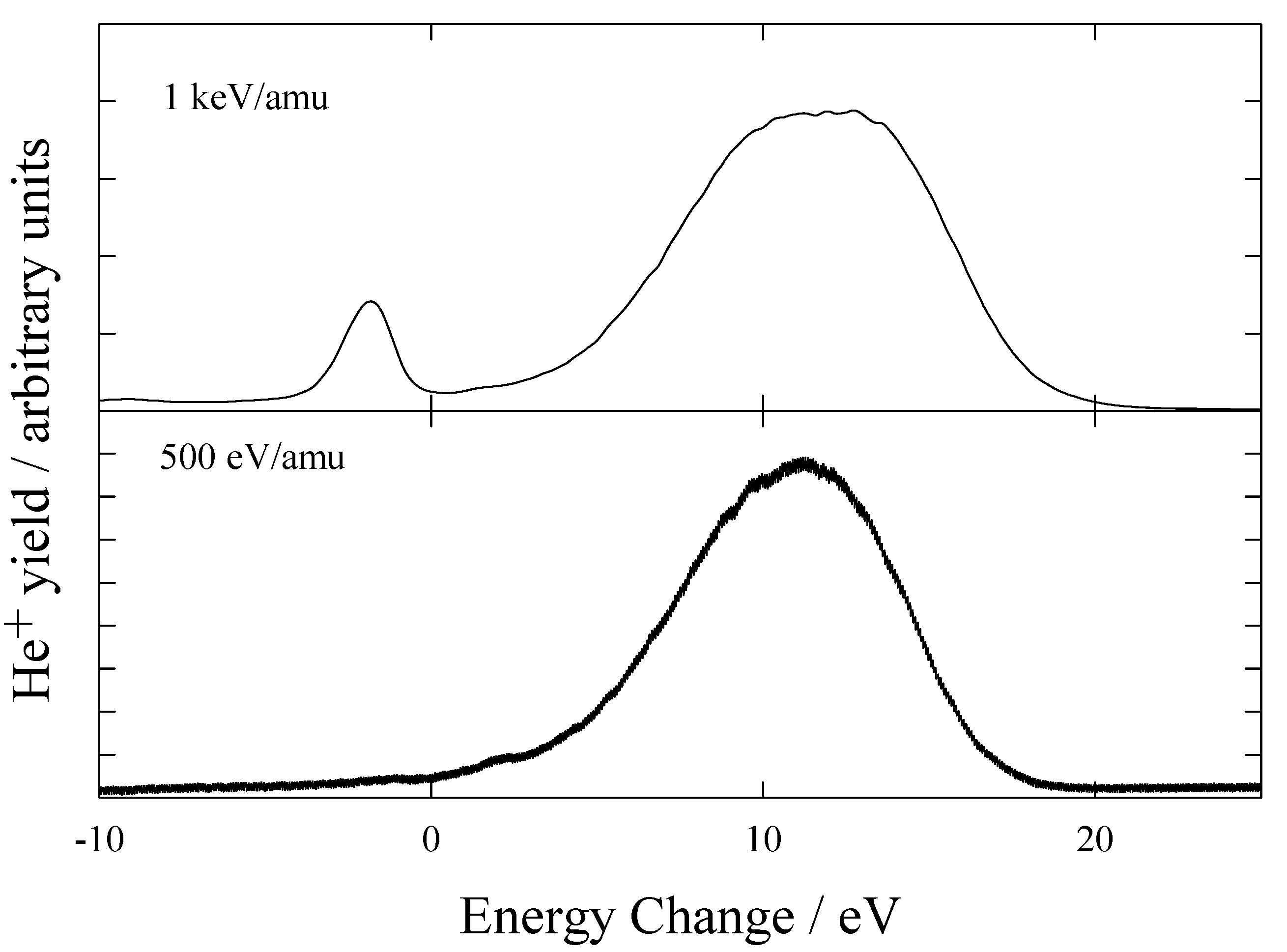
He2+ H2
Figure 7 shows the measured energy change spectra obtained for He
2+ ions in H
2 at energies of 1 keV/amu and 500 eV/amu.
Table 3.
Main product channels for one electron capture by He2+ ions in H21Σg+
Table 3.
Main product channels for one electron capture by He2+ ions in H21Σg+
| Product channels | Energy defects / eV |
|---|
| He+(n=2) + H2+ 2Σg | -1.79 - -4.44 |
| He+(1s) + H+ + H(2s) | 4.30 - 18.36 |
| He+(1s) + H+ + H(2p) | 4.40 - 12.73 |
Figure 7.
Translational energy spectra obtained for He2+ ions in H2 at 1 keV/amu and 500 eV/amu.
Figure 7.
Translational energy spectra obtained for He2+ ions in H2 at 1 keV/amu and 500 eV/amu.
The peak positions were compared with calculated energy defects shown in
table 3. The energy spectra contain 2 features. Firstly, a sharp endothermic peak corresponding to He
+(n=2) formation with H
2+ in the
2Σ
g state with vibrational excitation. Secondly, a broad distribution corresponding to dissociative capture processes:
It can be seen that at low energies only dissociative capture processes are important. Hoekstra et al [
23] suggested that at energies >5keV/amu, capture into excited states of He
+ is dominant, while at lower energies capture into the ground state of He
+ with dissociation and excitation of the target dominates. These results confirm this suggestion.
He2+ + CO
Figure 8 shows the translational energy spectra obtained for the He
2+ + CO system at 1keV/amu and 200eV/amu.
Table 4.
Main endothermic product channels for the He2+ + CO 1Σ+ system.
Table 4.
Main endothermic product channels for the He2+ + CO 1Σ+ system.
| Product channels | Energy defects / eV |
|---|
| He+(n=2) + CO+[X2Σ+] | - 0.42 (v=0) |
| He+(n=2) + CO+[A2Πi] | - 2.94 (v = 0) − -3.13 (v =1) |
| He+(n=2) + CO+[B2Σ+] | - 6.07 (v = 0) |
| He+(n=2) + CO+[C2Σ+] | - 9.41 (v = 0) − - 9.60 (v =1) |
| He+(n=3) + CO+[X2Σ+] | - 7.98 (v = 0) − - 8.24 (v = 1) |
| He+(n=3) + CO+[A2Πi] | -10.69 (v =2) − - 10.87 (v = 3) |
| He+(n=3) + CO+[B2Σ+] | - 13.83 (v = 1) |
Figure 8.
Translational energy spectra obtained for He2+ ions in CO at energies of 200 eV/amu and 1 keV/amu.
Figure 8.
Translational energy spectra obtained for He2+ ions in CO at energies of 200 eV/amu and 1 keV/amu.
At 1keV the spectrum the spectrum exhibits two main features. Firstly, a group of peaks corresponding to energy defects below zero, resulting from capture into He
+(n=2) and He
+(n=3) states with the target CO
+ ions in a variety of excited states with low levels of vibrational excitation. These capture channels are listed in
table 4 where energy defects shown were calculated from potential energy curves [
24], [
25] and energy level tabulations [
26].
Secondly, a broad distribution of peaks in the energy change region from 0 – 20eV, corresponding to dissociative capture processes of the type:
These dissociative capture channels become more important as the energy decreases and the non-dissociative channels become insignificant. The measurements of Folkerts et al [
27] have confirmed the importance of these channels. At 4keV/amu, they found that fragmentation channels account for about 46% of the total products of the ionization of CO.
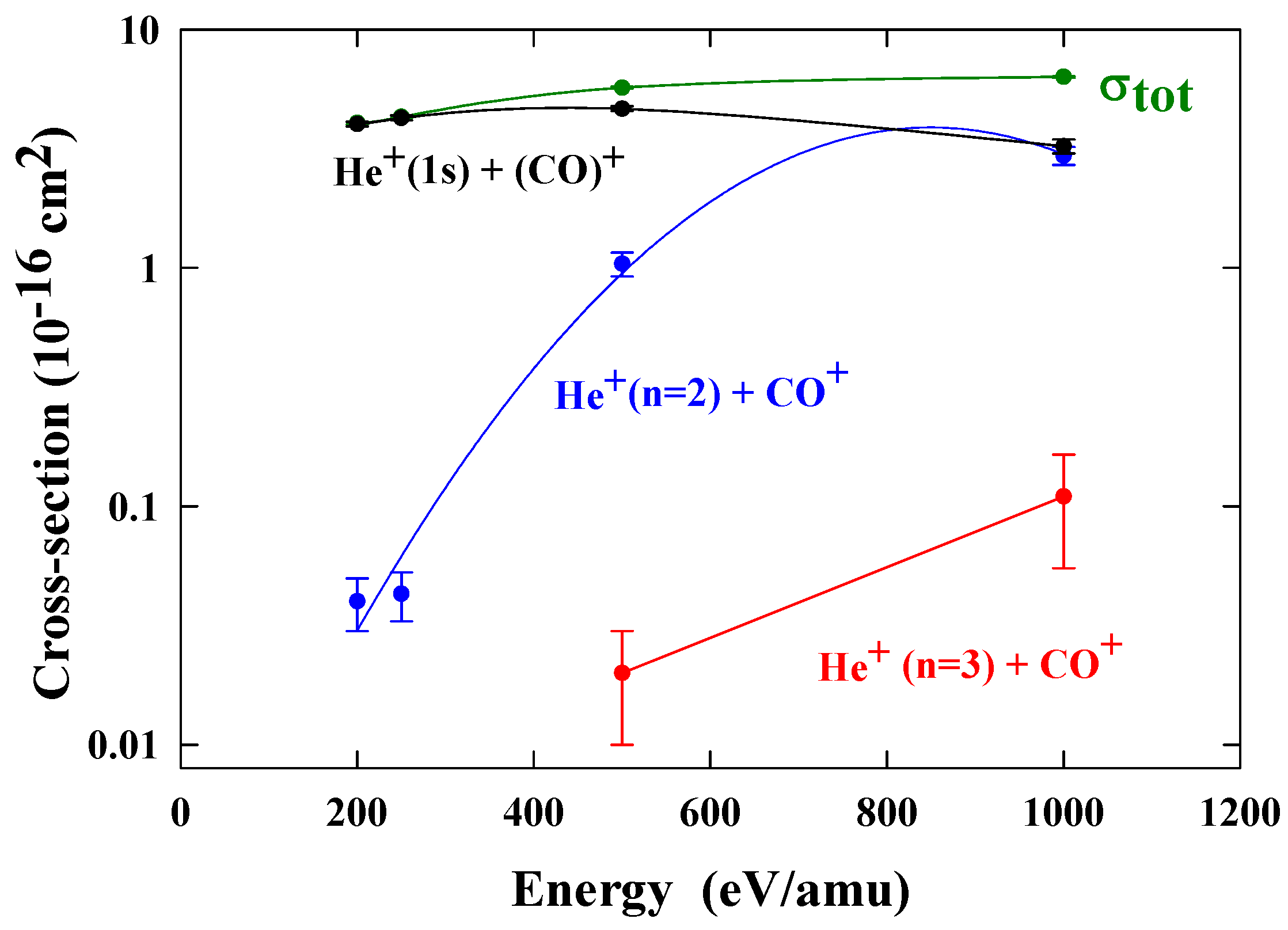
Relative contributions to electron capture from each of the channels have been obtained. By normalising these relative contributions to the total cross sections [
28], we obtained the partial cross sections shown in
figure 10.
These measurements are relevant to studies of the tail of Hale-Bopp. The characteristic blue emission from the tail of comet Hale-Bopp results from transitions from the
2Π
i to the B
2Σ
+ states of CO
+ [
29].
Figure 10.
Cross sections for one electron capture by He
2+ ions in CO. The present data are normalized to total cross sections σ
TOT [
28].
Figure 10.
Cross sections for one electron capture by He
2+ ions in CO. The present data are normalized to total cross sections σ
TOT [
28].
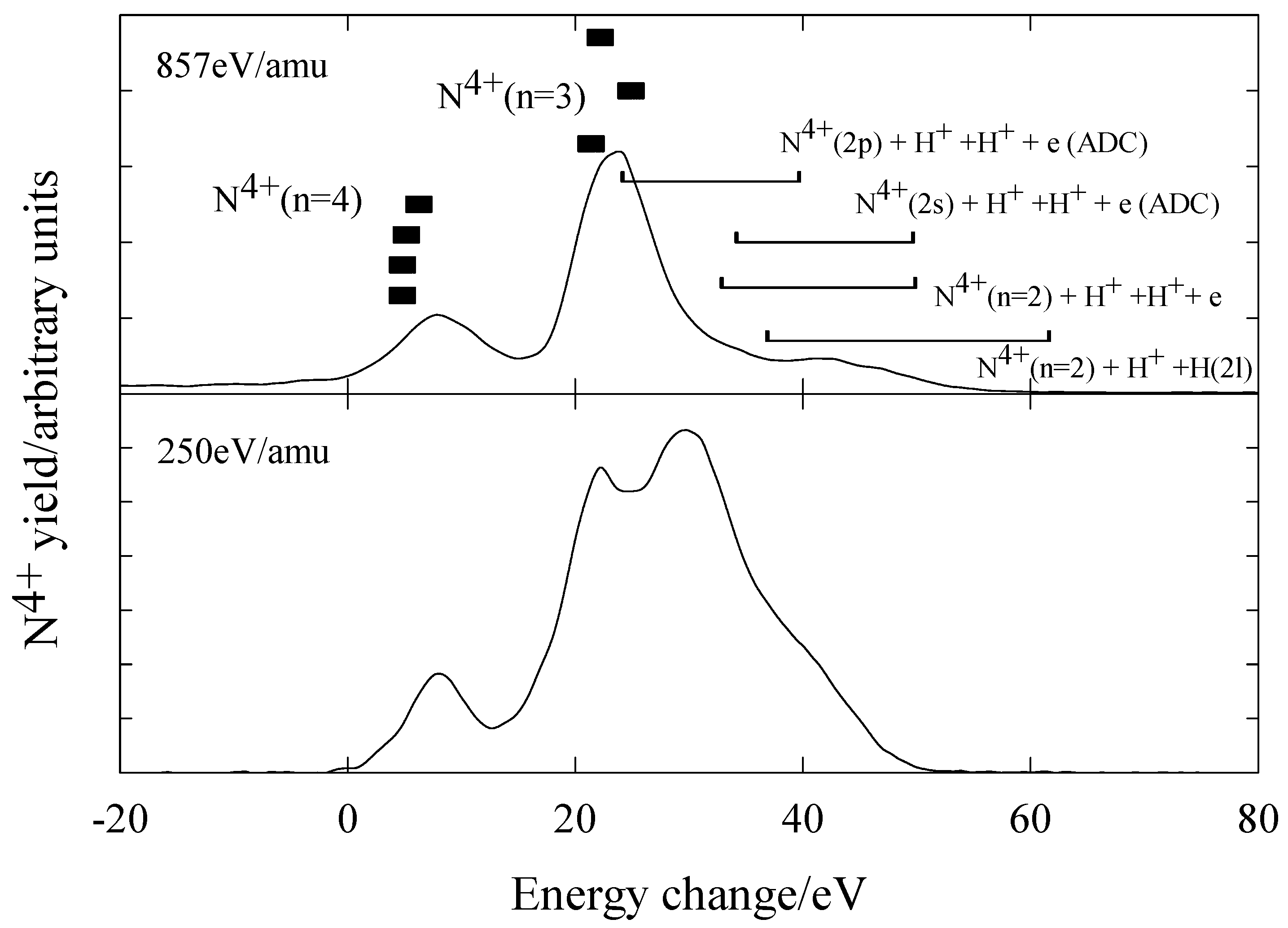
N5+ + H2
Figure 11 shows the measured energy change spectra for N
5+ ions in H
2 at energies of 857eV/amu and 250eV/amu. The main product channels are listed in
table 5 where energy defects shown were calculated from potential energy curves [
30] and energy level tabulations [
26]. The main features of this system are as follows. At 857eV/amu, capture occurs predominantly into N
4+(n=3) and N
4+(n=4) levels with the H
2+ product ion in the
2Σ
g state. This agrees with theoretical predictions [
31]. There is also some evidence of dissociative capture channels, either by direct capture or autoionising double capture (ADC), with N
4+(n=2) formation. At 250eV/amu the dissociative electron capture channels are a prominent feature.
Figure 11.
Translational energy spectra obtained for N5+ ions in H2 at energies of 857 eV/amu and 250 eV/amu.
Figure 11.
Translational energy spectra obtained for N5+ ions in H2 at energies of 857 eV/amu and 250 eV/amu.
Table 5.
Main product channels for the N5+(1s2) 1S + H21Σg+ system.
Table 5.
Main product channels for the N5+(1s2) 1S + H21Σg+ system.
| Product channels | Energy defects / eV |
|---|
| N4+(1s23s) + H2+ 2Σg | 25.91 (v=0) - 23.86 (v=10) |
| N4+(1s23p) + H2+ " | 23.22 (v=0) - 21.17 (v=10) |
| N4+(1s23d) + H2+ " | 22.40 (v=0) - 20.35 (v=10) |
| N4+(1s24s) + H2+ " | 7.29 (v=0) - 5.23 (v=10) |
| N4+(1s24p) + H2+ " | 4.14(v=0) − 6.19 (v=10) |
| N4+(1s24d) + H2+ " | 3.80(v=0) − 5.85 (v=10) |
| N4+(1s24f) + H2+ " | 3.78(v=0) − 5.83 (v=10) |
| Dissociative channels |
| N4+(n=2) + H+ + H(2l) | 36.87 − 61.61 |
| N4+(n=2) + H+ + H+ + e | 32.87 − 49.86 |
| Capture into autoionising states (ADC): |
| N3+(**) + H+ + H+ → |
| N4+(1s22s) + H+ + H+ + e | 34.152 − 49.664 |
| N4+(1s22p) + H+ + H+ + e | 24.152 − 39.664 |
O6+ + H2
Figure 12 shows the preliminary measurements for O
6+ ions in H
2 at 900eV/amu and 225 eV/amu with the energy defects shown listed in
table 6. The spectra are consistent with capture occurring predominantly into O
5+(n=4) with possible dissociative capture into n=3 states. In contrast to the N
5+ + H
2 system, there is no strong evidence of dissociative capture.
Figure 12.
Translational energy spectra obtained for O6+ ions in H2 at 900 eV/amu and 225 eV/amu.
Figure 12.
Translational energy spectra obtained for O6+ ions in H2 at 900 eV/amu and 225 eV/amu.
Table 6.
Main product channels for the O6+ (1s2) 1S + H21Σg+ system.
Table 6.
Main product channels for the O6+ (1s2) 1S + H21Σg+ system.
| Product channels | Energy defects / eV |
|---|
| O5+(1s23s) + H2+ 2Σg | 43.37 (v=0) - 41.31 (v=10) |
| O5+(1s23p) + H2+ " | 40.13 (v=0) - 38.07 (v=10) |
| O5+(1s23d) + H2+ " | 39.08 (v=0) - 37.02 (v=10) |
| O5+(1s24s) + H2+ " | 17.00 (v=0) - 14.94 (v=10) |
| O5+(1s24p) + H2+ " | 15.67(v=0) - 13.62 (v=10) |
| O5+(1s24d) + H2+ " | 15.24 (v=0) - 13.18 (v=10) |
| O5+(1s24f) + H2+ " | 15.21 (v=0) - 13.16 (v=10) |
| Dissociative channels |
| O5+(n=3) + H+ + H(1s) | 15.47 − 28.26 |
| Capture into autoionising states (ADC): |
| O4+(**) + H+ + H+ → |
| O5+(1s22p) + H+ + H+ + e | 66.03 − 76.03 |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
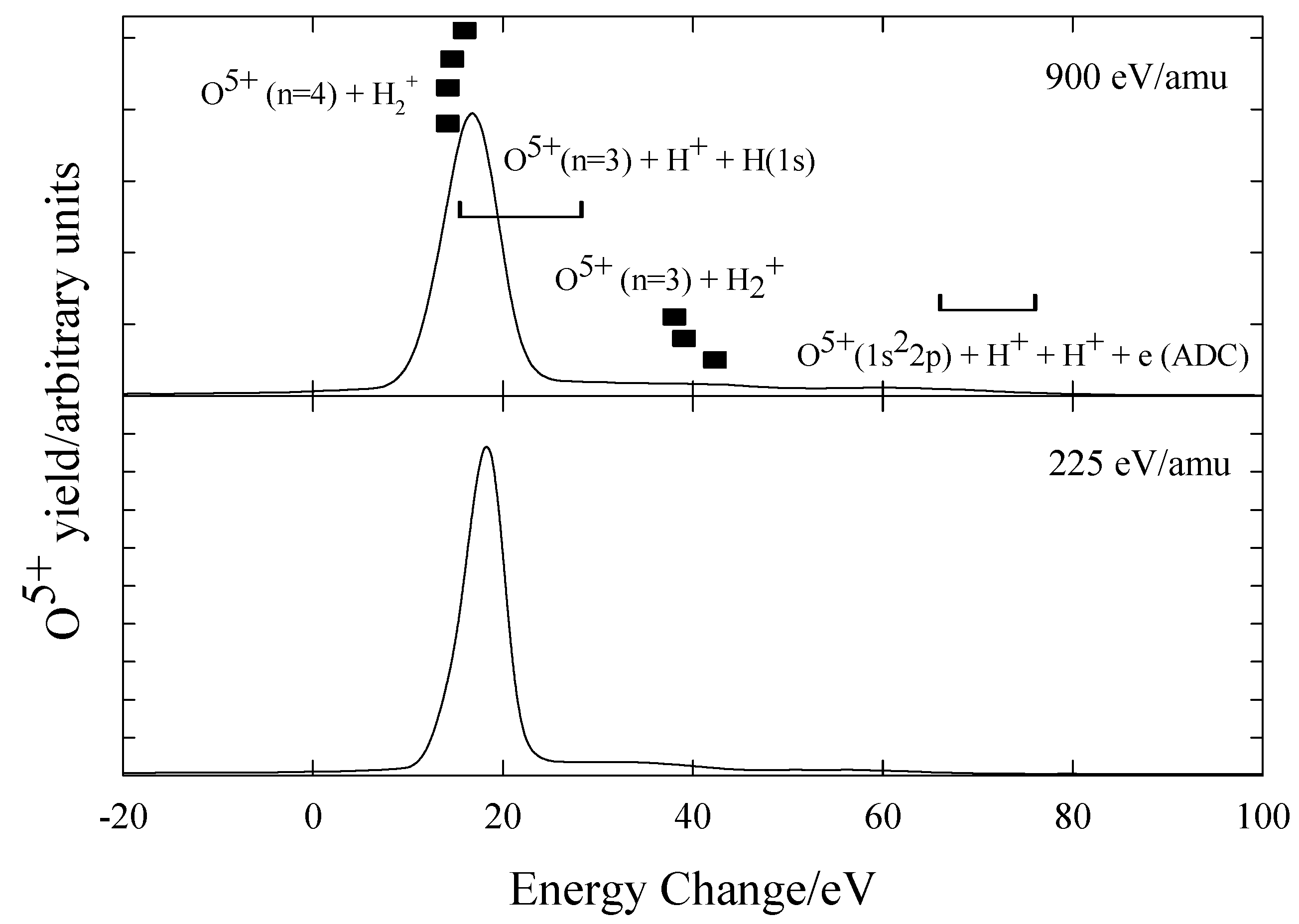{kind=link}






