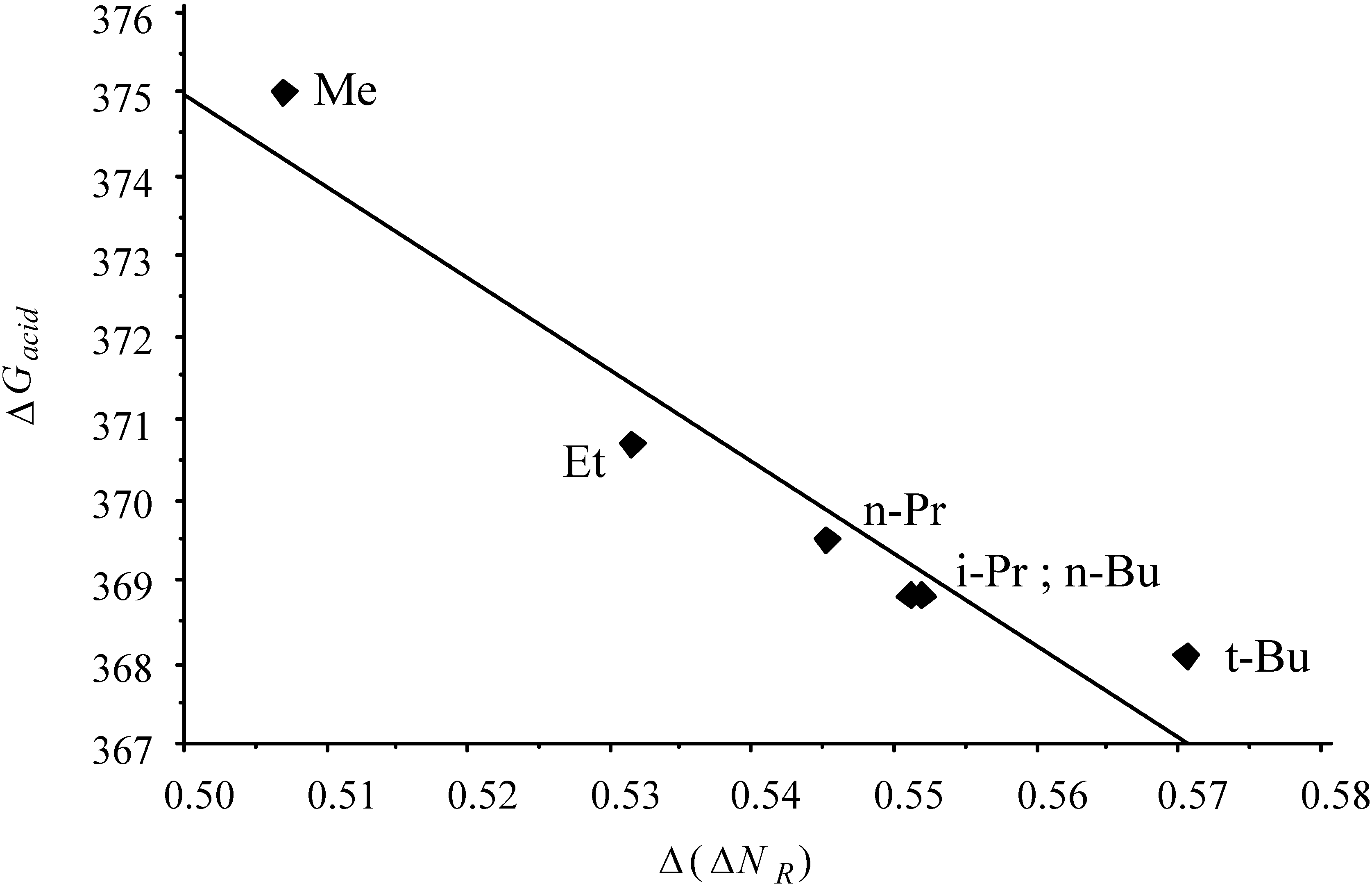
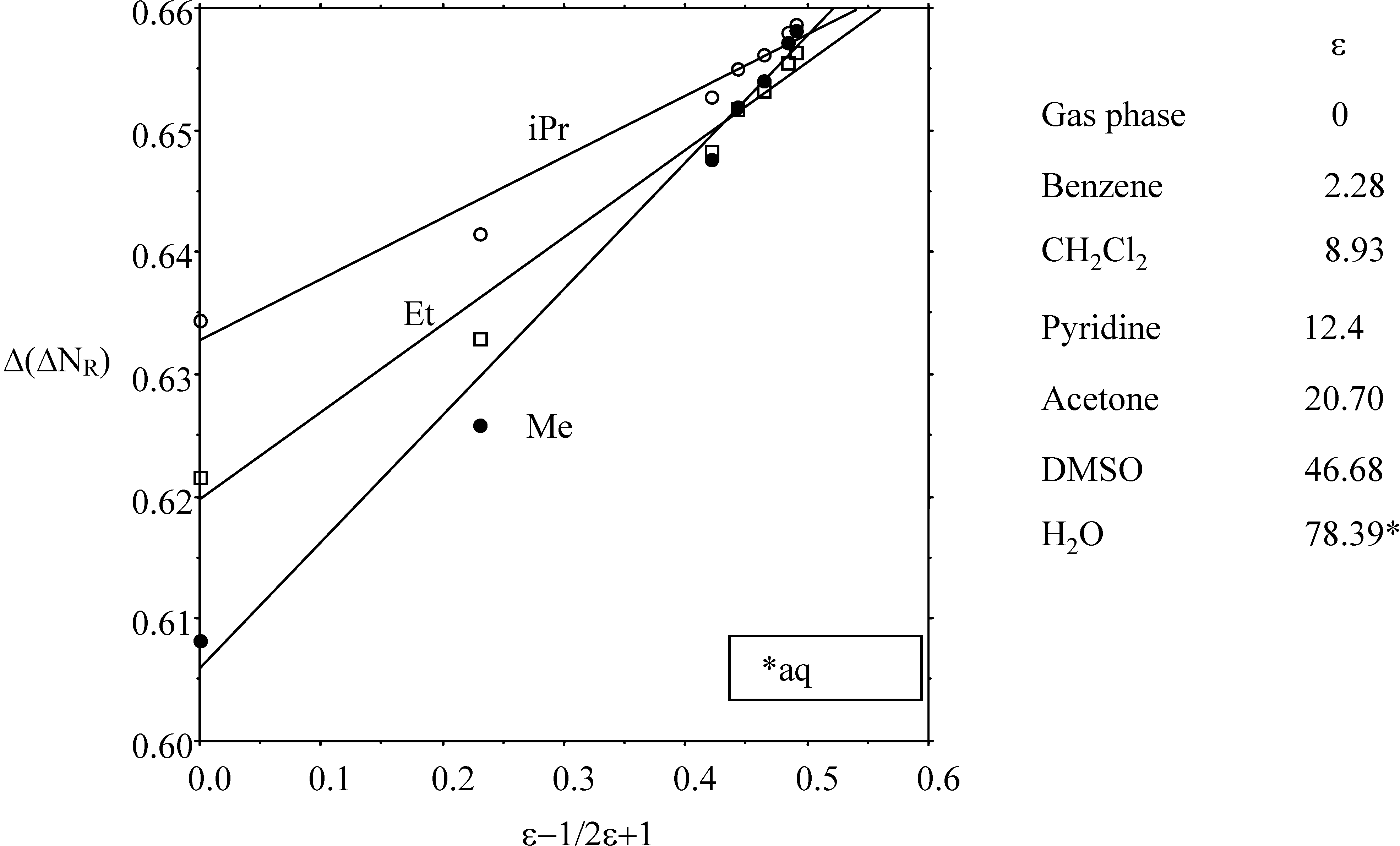
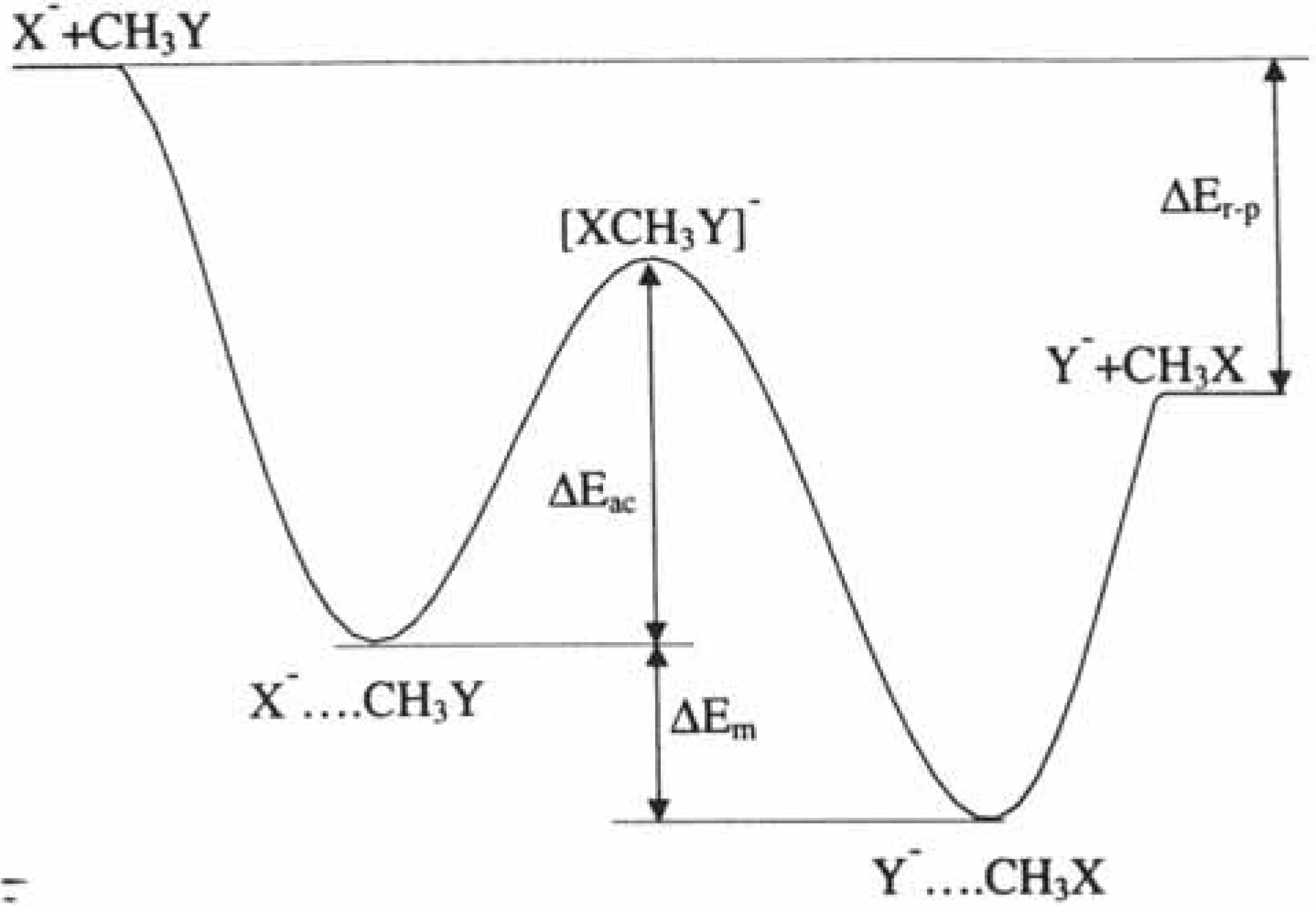
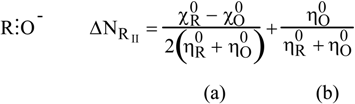2.3. Inorganic Chemistry: Zeolites
Zeolites are alumino-silicates consisting of SiO
4 and AlO
4 tetrahedra linked to each other by their corner oxygens. The alumino silicate structure is negatively charged due to the isomorphic subsitution of silicon by aluminum [
87]. This negative charge is balanced by exchangeable cations.
When protons are introduced as counterions, the zeolite becomes a Brϕnsted acid, the protons being positioned on an oxygen atom connecting an aluminum and a silicon atom. These bridging hydroxyls [
88] are at the origin of the acid catalysis application of zeolites [
89]. Another aspect of the structure of zeolites is the occurrence of large vacant interconnected spaces forming long, wide channels of varying size depending on the type considered allowing the crystal to act as a molecular sieve [
90].
For many years [
91] the concepts of electronegativity, softness, hardness …. were exploited by us in the study of the acidity of bridging hydroxyl in zeolites, of utmost importance in their catalytic properties (for a review see [
92]).
Due to space limitations and because some fundamental aspects of this kind of study are already present in § 2.2.1. on the acidity of alcohols, we concentrate in this paragraph on a recently aborded topic especially highlighting the possibilities of Computational Chemistry, switching at the end however again to Conceptual DFT : adsorption in zeolites.
The problem we recently addressed [
93,
94,
95] was the selectivity of adsorption of gases [
96] for which up to now no parameter free ab initio quantum chemical studies were performed yet [
90]. Below we summarize the results for the interaction of small molecules (such as N
2, O
2, CO), with a NaY faujasite type zeolite, more specifically with the α cage.
At sufficiently low pressure adsorption is governed by Henry's law [
96]
where q is the amount of substance per unit volume in the adsorbed phase and p the pressure. Henry's constant K can be written as [
97]
where B is the number of cavities in which adsorption can take place per unit mass of zeolite. The quantity “a” equals 1 for a monoatomic gas, 4π for a linear molecule and 8π
2 for a non linear molecule. The configuration integral I is defined as
where E represents the interaction energy of the adsorbing molecule with the zeolite cage at position
r and with orientation
. The integration is performed over the volume V of the supercage.
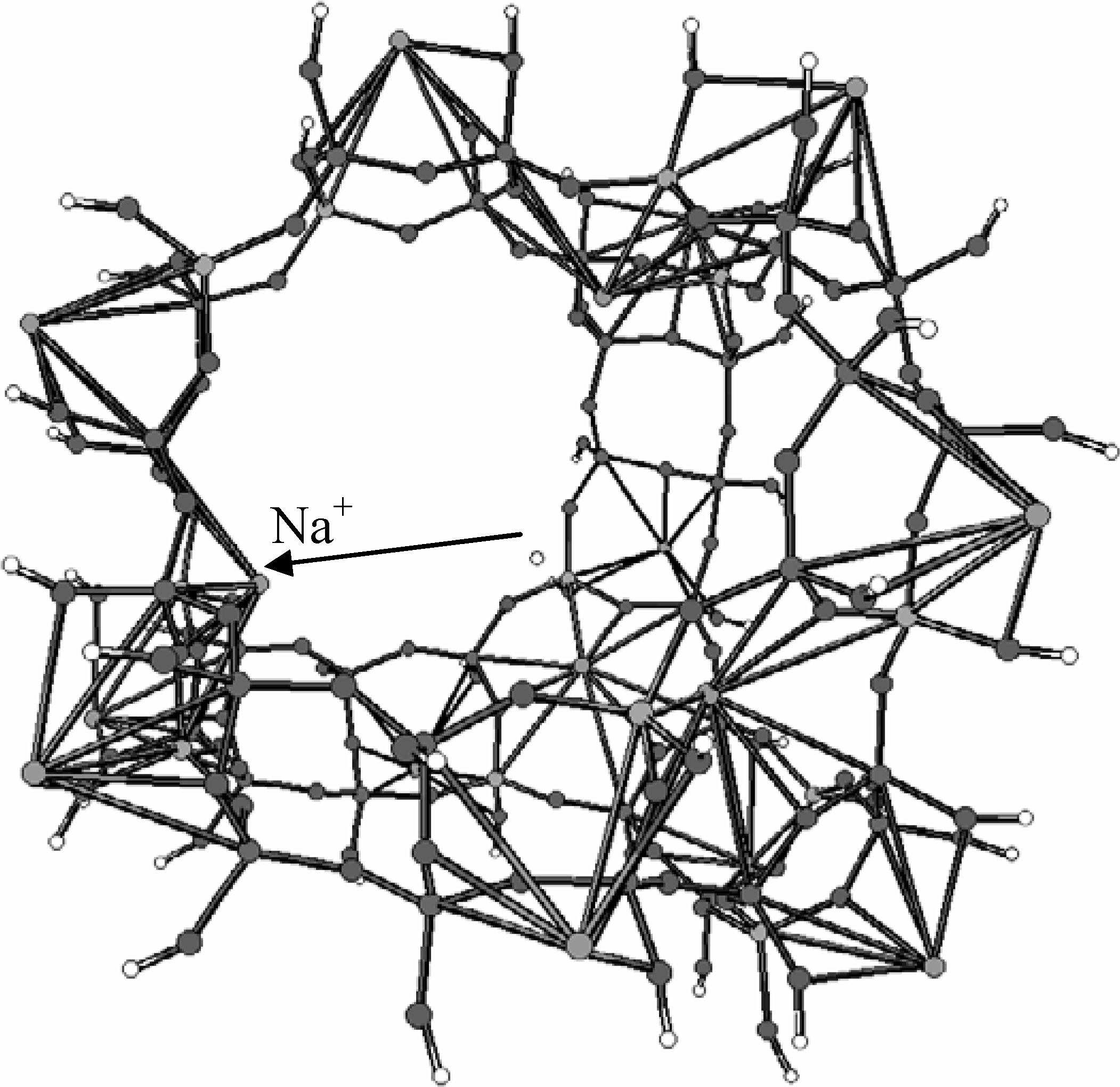
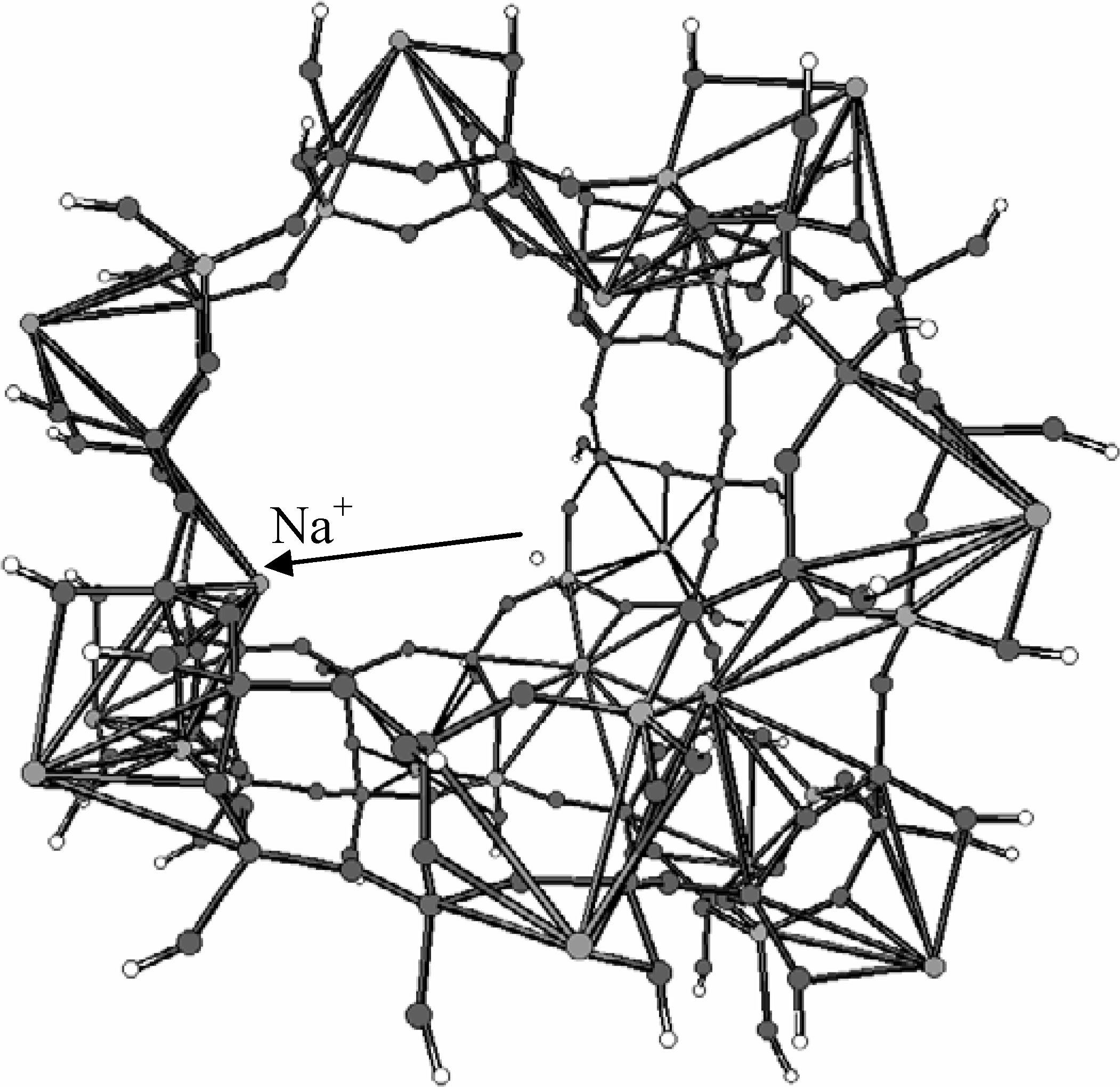
The cluster representing the supercage and its nearest environment was chosen to be "as large as possible" (within computational limits) and with an Si/Al ratio of 3. The 232 atoms cluster, involving OH groups as terminators is shown in
Figure 15, where also four cationic adsorption sites of type II, the ones "active" in the conditions that we consider are present, one of them being indicated.
Figure 15.
Cluster used to model the large cage of faujasite Y (Reprinted with permission by J. Wiley - Reference [
94])
Figure 15.
Cluster used to model the large cage of faujasite Y (Reprinted with permission by J. Wiley - Reference [
94])
An embedded cluster method was then adopted, considering the molecule in the field generated by the cage atom charges, the latter being obtained in a single run at Hartree Fock STO-3G or 3-21G level. The points at which the interaction energy is evaluated are selected by constructing a cubic grid with a grid distance of 0.50 Å in each direction and considering the center of each cube generated in this way. The expression (25) is thereby approximated as
where the three terms account for three mutually perpendicular orientations of the molecule, each one given an equal weight to simplify the orientational integration
.
equals 0.125 Å
3.
A supplementary condition was introduced imposing a minimal distance between an atom of the adsorbing molecule and a cage atom (cf. the treatment of the repulsive term in the potential, for details see [
94]). At these small distances E becomes positive and its contribution to I, via the exponential function, very small. This procedure leads to the consideration of not less than ± 3000 cage-points necessitating an in-depth search for an optimal quality/cost ratio in the procedure for the interaction energy evaluation. The optimal procedure, from a quality/cost ratio point of view, turned out the following one [
94,
95,
98]. The starting point was the energy expansion of a molecule in a non-uniform electric field [
99].
where F
α, F
αβ, ...represent the field, field gradient, ... components whereas μ
α, Θ
αβ, Ω
αβγ ,… stand for the dipole, quadrupole, octadecapole, … components and α
α, β
αβ, … are the dipole polarizability and the first hyperpolarizability. In the case of a neutral molecule of the C
∞,v type (CO) the expression simplifies to (the molecular axis is taken to be the z-axis)
Fi represents the electric field at the origin of the molecule due to the surrounding point charges. In D
∞,h cases (N
2, O
2, CO
2, …) terms in µ and Ω vanish.
The quality of this approach in dependent on the number of terms retained in the expressions (27) and (28) and the quality of the multi-pole moments, polarizabilities … These were calculated with DFT methodology (B3LYP functional [
19]) combined with Dunning’s extremely large basis sets (the augmented -correlation consistent and polarized - valence - quadruple or quintuple basis sets (AVQZ or AV5Z) [
100,
101]).
This methodology was proved by us to yield multipole moments which are in excellent agreement with experiment [
98,
102].
In
Table 3 the main results are summarized for N
2, O
2, CO, CO
2, C
2H
2. Besides Henry constants, also separation constants α (ratio of two Henry constants) and the isosteric heats of adsorption are tabulated. The latter values were obtained via the Van't Hoff equation
for which K was calculated in the temperature interval between 260 and 340K with an increment of 10K and using a linear regression.
Table 3.
Henry constants, separation constants and isosteric heats of adsorption on NaY : comparison between theory and experiment [
94,
98].
| | N2 | O2 | CO | CO2 | C2H2 |
| K | 3.320 | 1.830 | 9.951 | 63.33 | 196.8 |
| Kexp | 31.4 | 15.4 | 85 | | |
| | | | | | |
| ΔH° | -12.9 | -7.9 | -18 | -27 | -29 |
| ΔHexp | -14 | -9.4 | -20 | -37 | |
| | N2/O2 | CO/N2 | CO2/CO |
| α | 1.81 | 2.99 | 6.36 |
| αexp | 2.04 | 2.70 | |
As a whole these results yield K values which are systematically one order of magnitude too small but showing correct sequences. The order of magnitude should be considered within the correct context : a uniform underestimation of the interaction energy of only 1.4 kcal mol-1 already yields an order of magnitude underestimation of K. This extremely high sensitivity is of course due to the sensitivity of the potential in the repulsion part, plugged into an exponential in the configuration integral. The ratio Kexp/K is almost constant with values of 9.45, 8.41, 8.54 respectively. As a consequence the separation constants are in excellent agreement with experiment. The isosteric heats of adsorption also show very good agreement in absolute value and reproduce the experimental sequence. In our opinion these investigations pave the way for future studies involving more complex molecules where the lines drawn here, together with increasing computer hard and software and DFT based methodology, may finally yield to calculations which will be of great use in the design of zeolites for well defined purposes (e.g. gas separation).
An alternative for the interaction energy evaluation including electrostatic and polarization effects was proposed in a conceptual DFT context. Using a DFT perturbational approach we obtained the following expression for the interaction energy
The first term corresponds to the electrostatic interaction V(
Ri) as given by the molecular electrostatic potential at position
Ri where charge q
i is located, multiplied by this charge. The second term is the polarization term [
103] in which both the total softness, S, and the fukui function, f(
r), appear. Further work to evaluate in an efficient way the integral in the second term is in progress, the basic requirements for the evaluation of the interaction energy being the molecular electrostatic potential and fukui function (V(
r) and f(
r)) and the total softness nowadays ready available at a high precision level.
Conceptual DFT can thus be exploited in the study of adsorption behaviour of zeolites, as also especially witnessed in the work by Chatterjee and coworkers using the condensed Fukui function and local softness to estimate and rationalize the interaction energy of several small molecules with a zeolitic framework [
104,
105,
106]. An attempt was made to explain selective permeation of these molecules [
105] and the choice of the best template for a particular zeolite synthesis by estimating the reactivity of the templating molecule [
106].
2.4. Biochemistry : Influence of point mutations on the catalytic activity of subtilisin
As an example of our work in biochemistry we summarize very briefly recent studies in which the embedded cluster approach presented in § 2.3 was used to study the active sites in enzymes, more precisely the catalytic triad in subtilisin [
107] and Ribonuclease T
1 [
108].
The methodology followed in these studies shows similarities with part of the zeolite adsorption studies (high level quantum chemical calculation on that part of the system at which the active site is situated and a point charge environment for the residues farther away). The results of the subtilisin study are given as an example.
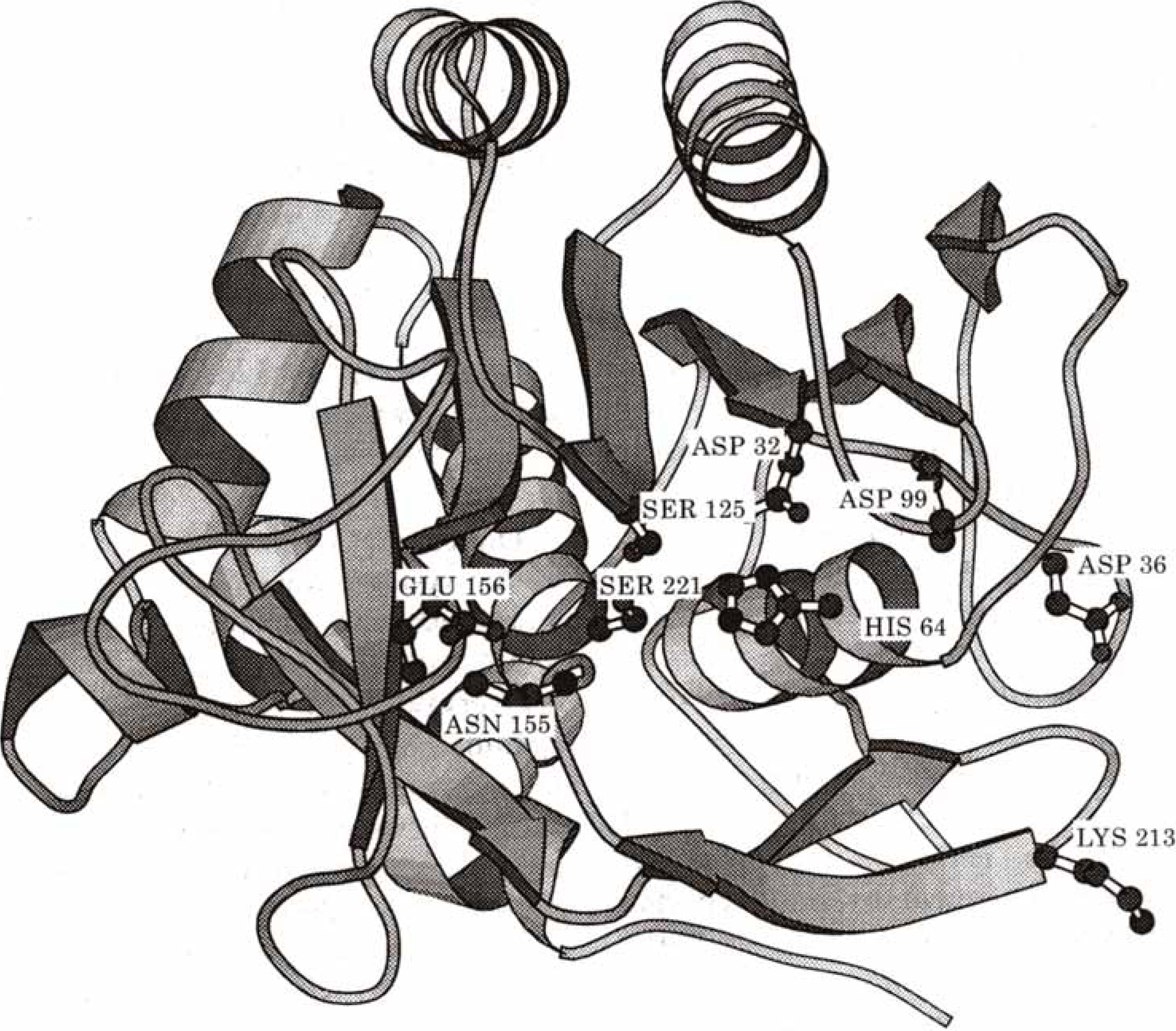
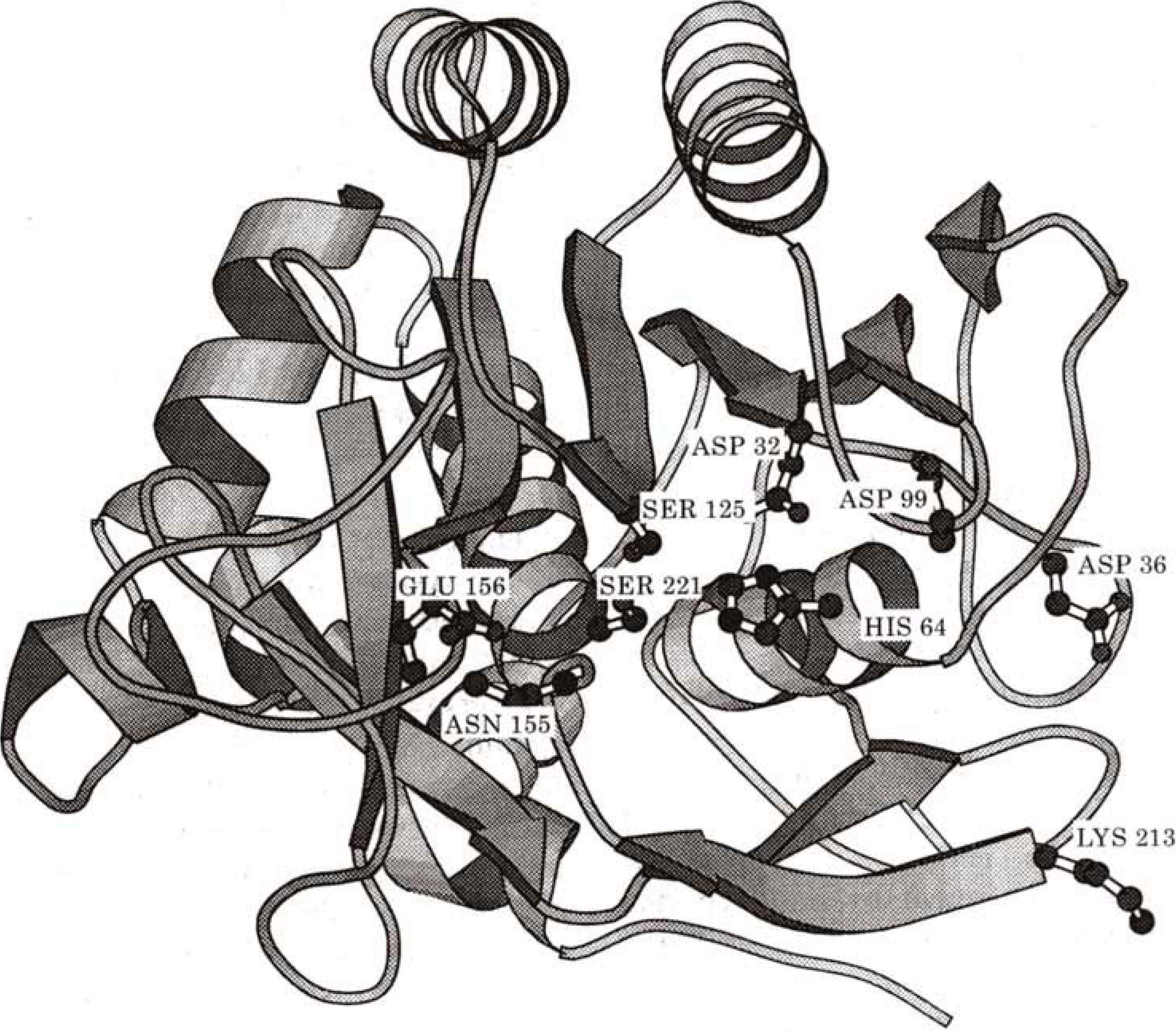
Subtilisin is a bacterial enzyme belonging to the class of the serine proteases characterized by a catalytical apparatus consisting of three amino acid residues, serine, histidine and aspartate : the catalytic triad (Asp32 - His64 - Ser221 for subtilisin) (
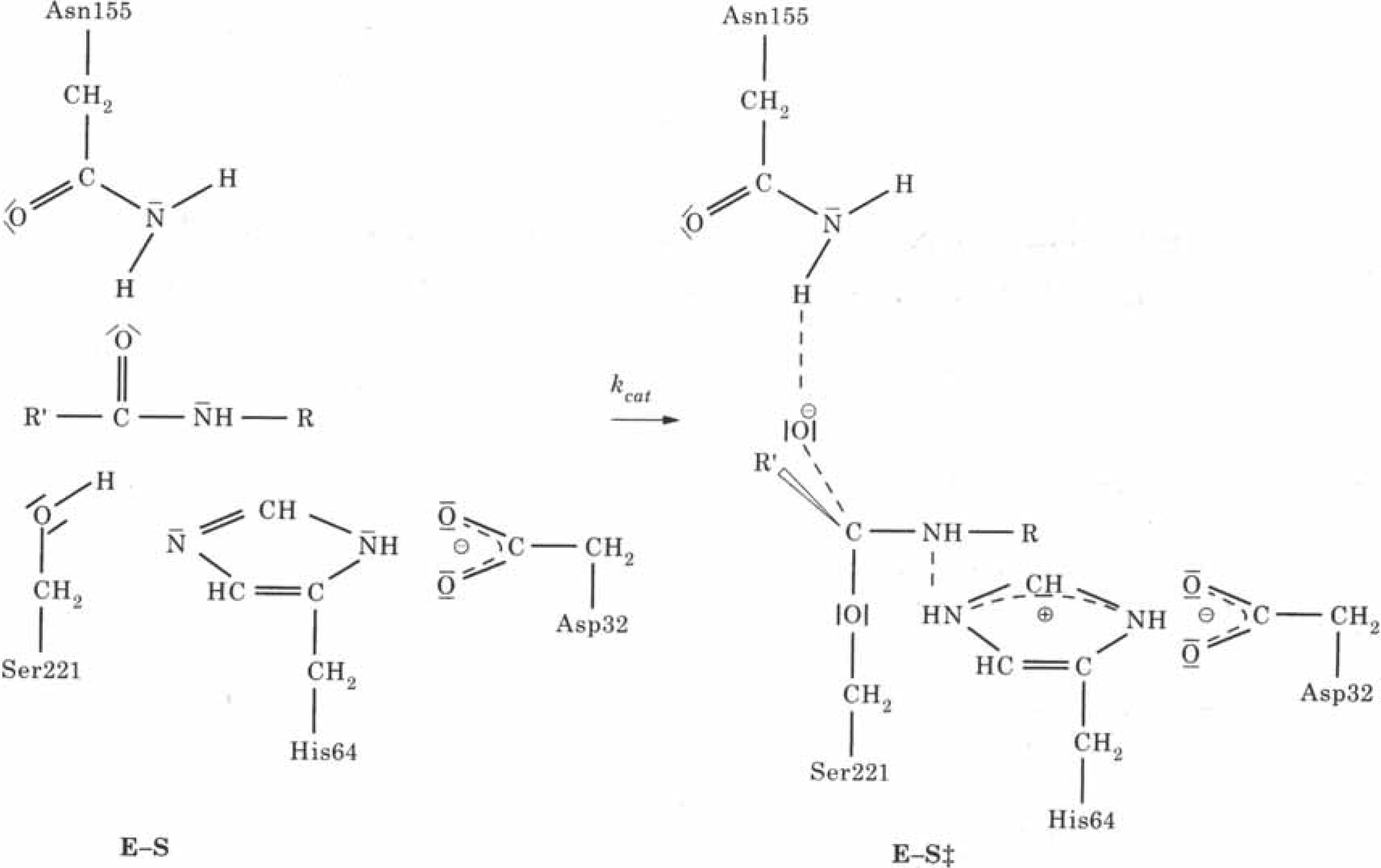
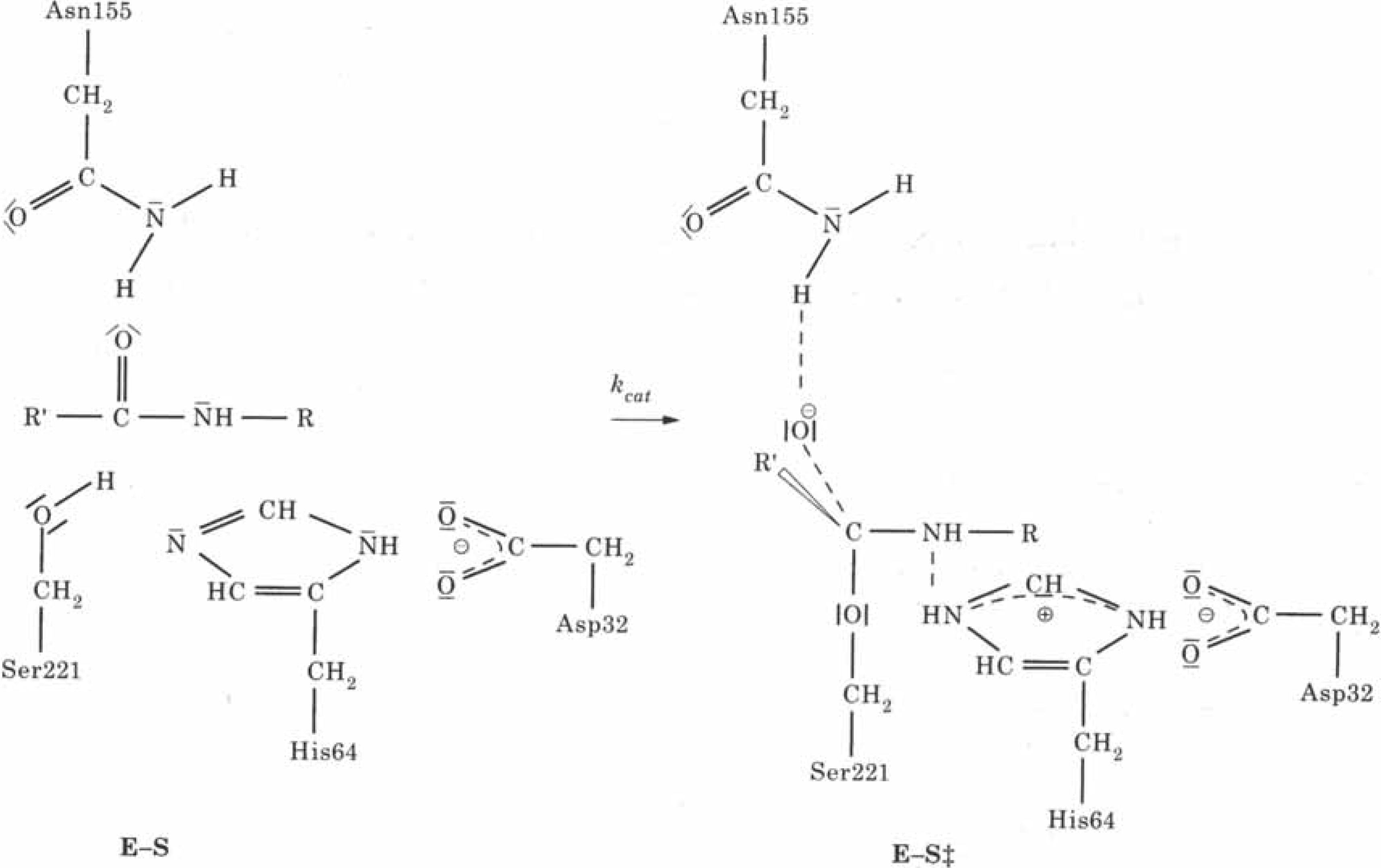
Fig. 16). In the rate determining step of the hydrolysis reaction (see
Fig. 17) the hydroxyl proton of Ser221 is transferred to the N
ε2 atom of His64. Simultaneously a nucleophilic attack by the hydroxyl oxygen of Ser221 at the carbon atom of the scissile peptide bond occurs. The role of the aspartate residue is to enhance the nucleophilicity of Ser221 due to the electric field of the charged aspartate side chain and to provide electrostatic stabilization of the tetrahedral intermediate.
The role of the catalytic triad amino acids was studied by Carter and Wells : both single and double alanine substitutions of these residues led to a lowering of the catalytic rate constant k
cat [
109]. Russell and Fersht studied the effect on k
cat for mutations occurring outside the catalytic triad [
110].
These effects were studied by placing the catalytic triad into an environment of ChelpG point charges representing all atoms of the amino acids within a 15 Å sphere around His64, and obtained from ab initio calculations on isolated amino acids; the structural data on the wild type enzyme were taken from X-ray diffraction studies [
111], mutations were carried out
in computero. The nucleophilicity of the Ser221 oxygen was investigated using local softness and (models for) the local hardness : the charge on the oxygen atom and the MEP.
Local softness turned out to be not successful when correlating
with k
cat values in line with Fersht's statement that the nucleophilic attack of the serine on the substrate can be considered as an attack on a hard nucleophilic center [
112]. We now further concentrate on local hardness descriptors. When comparing the wild type enzyme and the His64 Ala mutant with, respectively, the Asp32 Ala and Asp32 Ala : His64 Ala mutants,
Table 4 shows a less negative charge and, much more pronounced, a less negative MEP value, indicating that the interaction of the catalytic serine with an electrophile is less advantageous in the aspartate mutants. These results are in agreement with experiments pointing out an enhancement of the serine nucleophlicity by the aspartate residue [
113].
Table 4.
Serine oxygen charge (qO) (au) and MEP minimum around this oxygen atom (V(R)min) (in kcalmol-1) calculated for the wild-type and mutant enzymes (3-21G Hartree Fock with ChelpG charges).
Table 4.
Serine oxygen charge (qO) (au) and MEP minimum around this oxygen atom (V(R)min) (in kcalmol-1) calculated for the wild-type and mutant enzymes (3-21G Hartree Fock with ChelpG charges).
| | qO | V(R)Min |
| Wild Type | -0.7719 | -121.72 |
| Asp32 Ala | -0.7440 | -75.64 |
| His64 Ala | -0.7665 | -113.13 |
| Asp32 Ala : His64 Ala | -0.7487 | -67.92 |
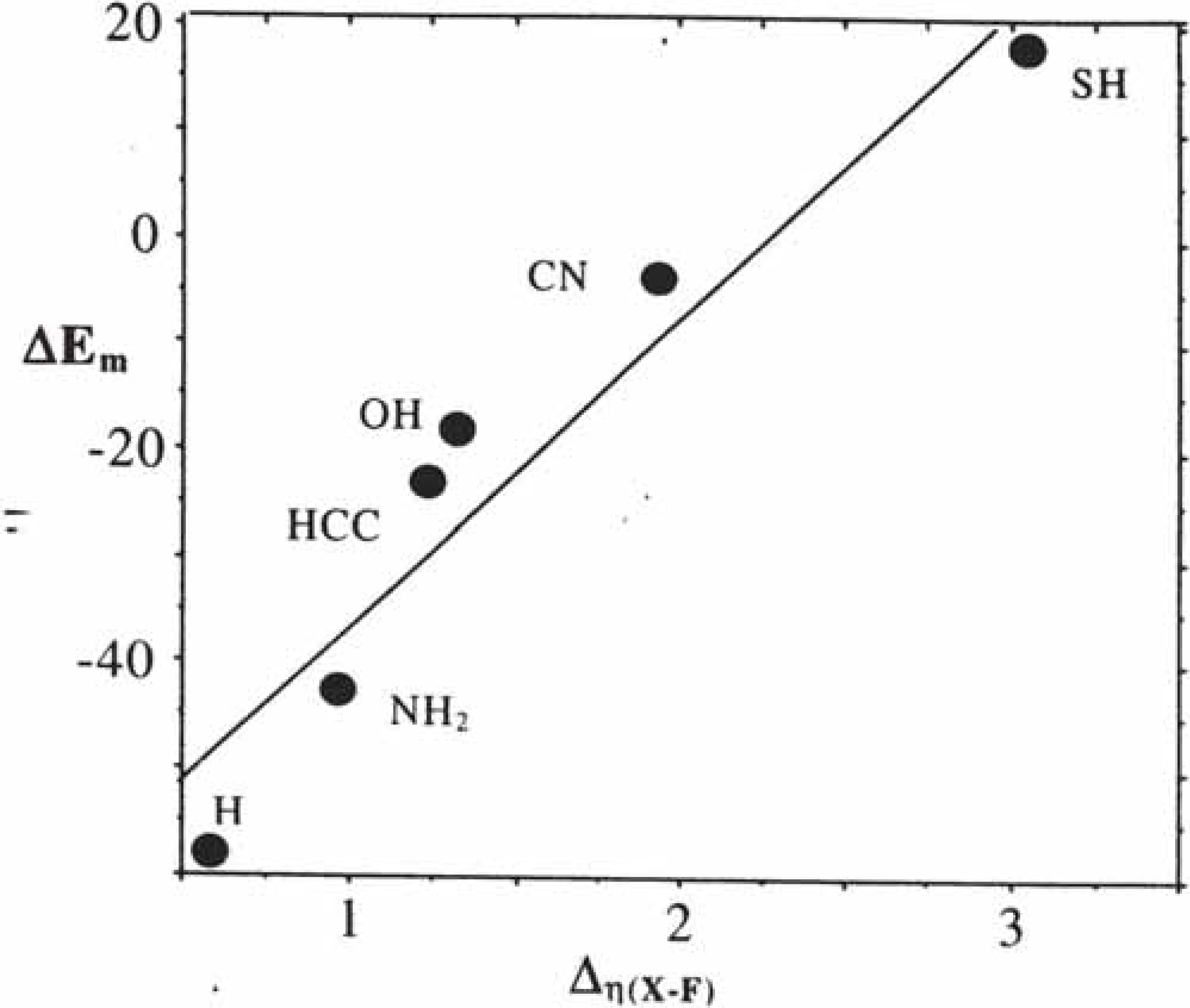
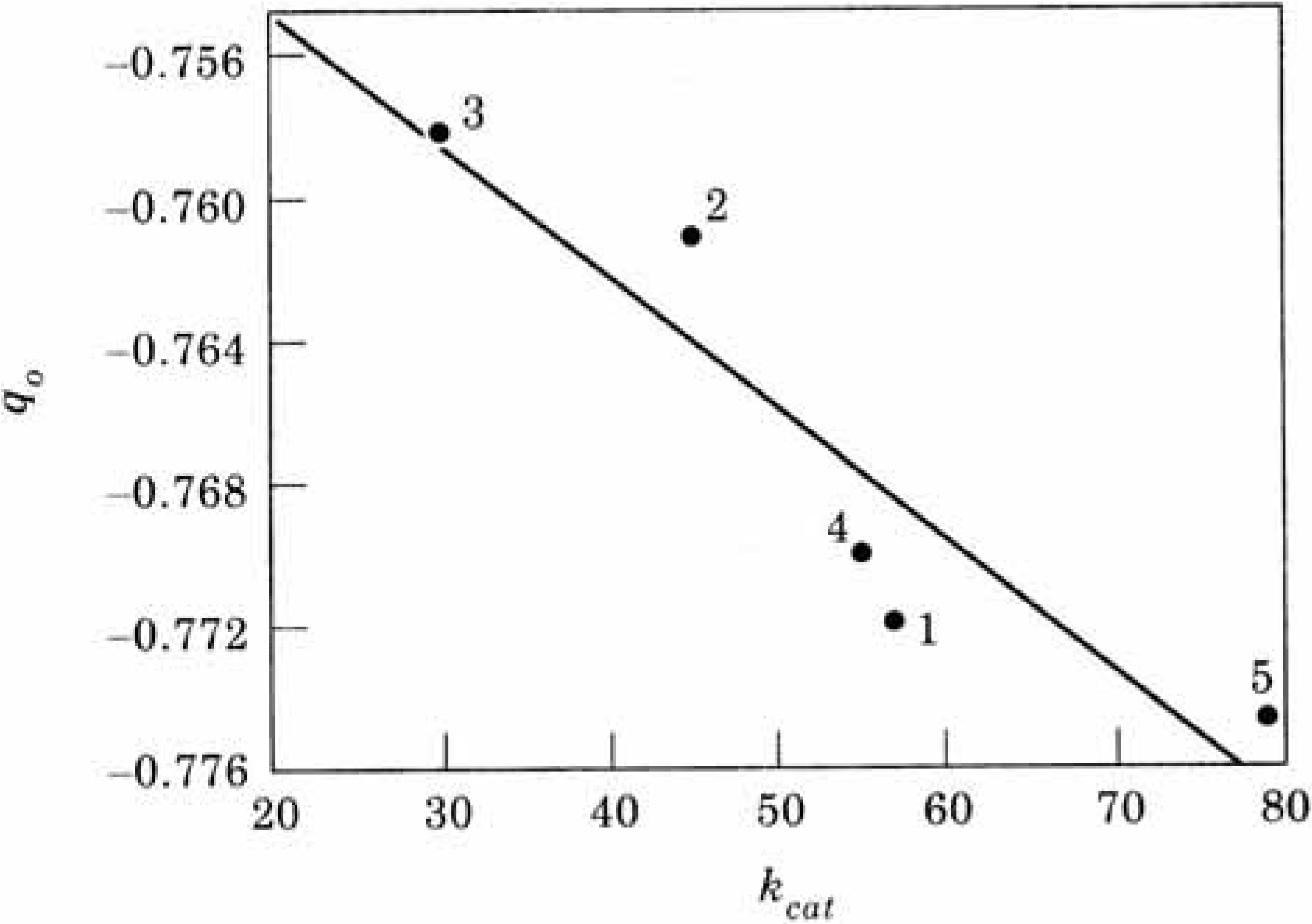
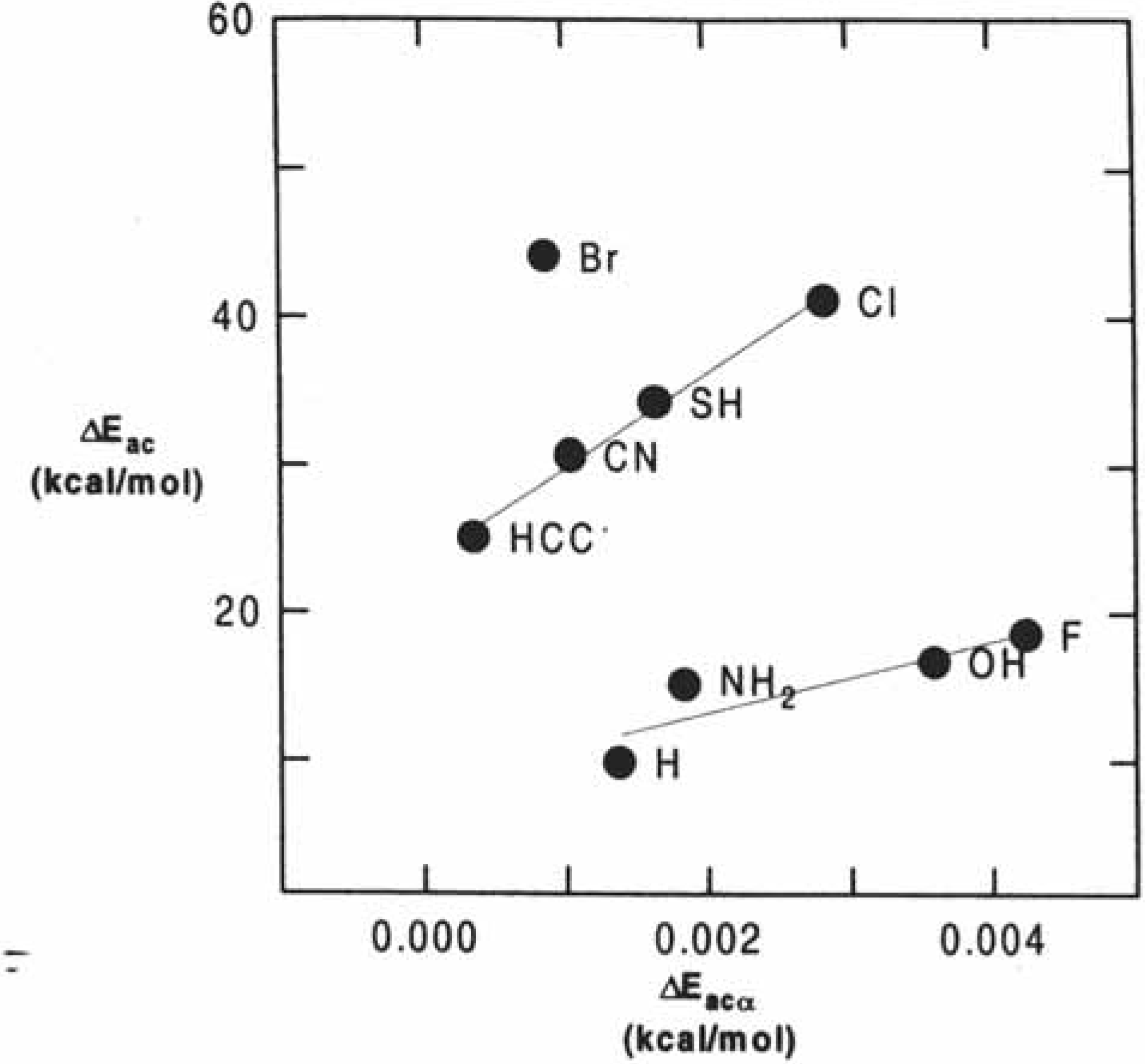
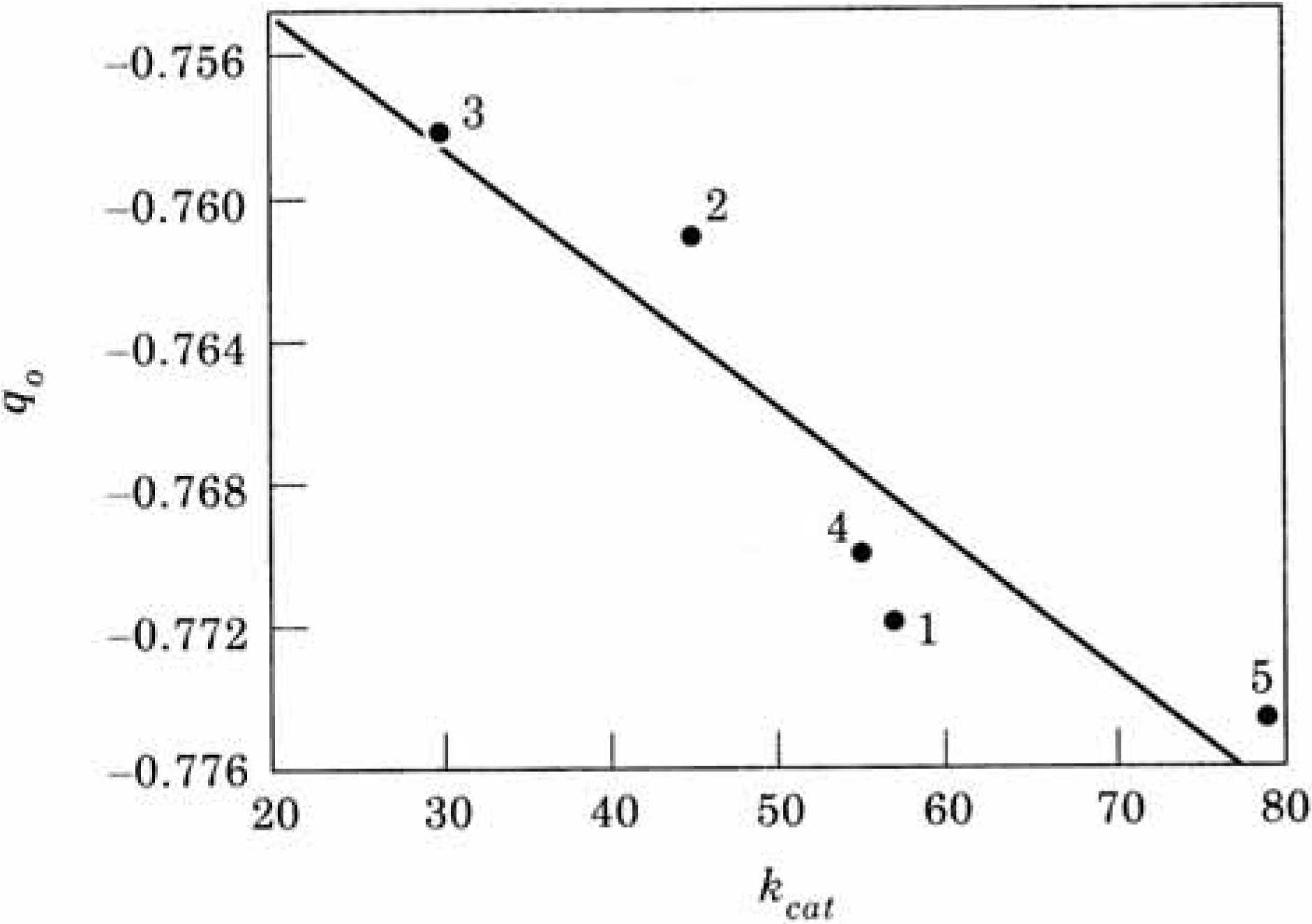
A more quantitative picture is obtained when studying the atomic charges and MEP vs. the experimental k
cat values [
109] for the enzymes when the mutations were performed in the environment of the catalytic triad (
Table 5). A correlation coefficient of 0.927 was obtained in the case of the charges (
Figure 18).
Table 5.
Experimental kcat values (s-1) and calculated charges qO (au) and MEP minimum V(R)min (kcal mol-1) for the serine oxygen for wild-type, aspartate 99 and glutamate 156 mutant enzymes.
Table 5.
Experimental kcat values (s-1) and calculated charges qO (au) and MEP minimum V(R)min (kcal mol-1) for the serine oxygen for wild-type, aspartate 99 and glutamate 156 mutant enzymes.
| | kcat | qO | V(R)min |
| 1. Wild Type | 57 | -0.7719 | -121.72 |
| 2. Asp99 Ser | 45 | -0.7610 | -121.33 |
| 3. Asp99 Lys | 30 | -0.7581 | -121.30 |
| 4. Glu156 Ser | 55 | -0.7700 | -122.96 |
| 5. Glu156 Lys | 79 | -0.7748 | -124.57 |
Figure 16.
Schematic drawing of subtilisin. Side chains of the residues of importance in the discussion are shown explicitely (Reprinted with permission by Academic Press, Reference [
107]).
Figure 16.
Schematic drawing of subtilisin. Side chains of the residues of importance in the discussion are shown explicitely (Reprinted with permission by Academic Press, Reference [
107]).
Figure 17.
Schematic representation of the enzymatic reaction of serine proteases; E-S is the enzyme substrate complex, E-S
≠ the tetrahedral reaction intermediate (Reprinted with permission by Academic Press - Reference [
107])
Figure 17.
Schematic representation of the enzymatic reaction of serine proteases; E-S is the enzyme substrate complex, E-S
≠ the tetrahedral reaction intermediate (Reprinted with permission by Academic Press - Reference [
107])
Figure 18.
Atomic charge on the serine oxygen atom q
0 vs. experimental k
cat (see
Table 5).
Figure 18.
Atomic charge on the serine oxygen atom q
0 vs. experimental k
cat (see
Table 5).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}