Inhibition of Heme Peroxidase During Phenol Derivatives Oxidation. Possible Molecular Cloaking of the Active Center

1
Vytautas Magnus University, Vileikos 8, 44404, Kaunas, Lithuania
2
Institute of Biochemistry, Mokslininku 12, 08662 Vilnius, Lithuania
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2005, 6(9), 245-256; https://doi.org/10.3390/i6090245
Submission received: 1 March 2005
/
Accepted: 29 August 2005
/
Published: 20 October 2005
Abstract
:Ab initio quantum chemical calculations have been applied to the study of the molecular structure of phenol derivatives and oligomers produced during peroxidase-catalyzed oxidation. The interaction of substrates and oligomers with Arthromyces ramosus peroxidase was analyzed by docking methods. The most possible interaction site of oligomers is an active center of the peroxidase. The complexation energy increases with increasing oligomer length. However, the complexed oligomers do not form a precise (for the reaction) hydrogen bonding network in the active center of the enzyme. It seems likely that strong but non productive docking of the oligomers determines peroxidase inhibition during the reaction.
Introduction
Enzymatic oxidation of phenol substances has attracted considerable attention recently due to environmental effects and polymer synthesis [1,2]. The environmental effects are associated with large scale production and use of man-made phenol substances at concentrations that cause ecologically undesirable effects. Many of these compounds are highly resistant to biotic and abiotic degradation and, as a result, remain in the environment at toxic levels. Enzyme-catalyzed degradation of phenol substances has been considered as one of possibilities for detoxification of these congeners [1].
The synthesis of polyphenols is another area of investigation in the enzyme-catalyzed oxidation of phenol substances [2]. Several oxidoreductases, e.g. peroxidases, laccase and bilirubin oxidase, catalyze the oxidative polymerization of phenol derivatives [3]. Polyphenols with useful properties for electronics have been very recently synthesized using peroxidase [4].
The peroxidase-catalyzed phenol oxidation has several limitations, including permanent peroxidase inactivation by various undesirable side reactions [1]. In an attempt to decrease the inhibition rate, some additives were used [4,5]. It was shown that the yield of recombinant Coprinus cinereus peroxidase-catalyzed phenol derivative oxidation may increase significantly if albumins or non-ionic polymeric compounds were used [5].
In order to elucidate a possible inhibition mechanism in the process of phenol substance oxidation the interaction of intermediate products (oligomers) with peroxidase was explored by using ab initio quantum chemical and mechanical docking calculations. As peroxidase substrates three phenol derivatives, i.e. 1-naphthol, 2-naphthol and 4-hydroxybiphenyl were analyzed.
Computational Details
Structures and atomic charges of studied compounds were calculated with ab initio method on 3-21G basis set and Hartree-Fock theory with Gaussian 98W [6]. The spin densities of radicals were calculated with B3LYP DFT theory. The structure of Arthromyces ramosus peroxidase (ARP) that has in addition one terminal amino group compared to Coprinus cinereus peroxidase has been described [7]. The crystal data of ARP (PDB-ID: 1ARP) [7] with resolution of 1.9 Å was downloaded from the Protein Data Bank. All water molecules were removed, except the oxygen atom of one structural water molecule, which was left in the active center of ARP. In order to model a catalytically active state of ARP, i.e. compound I/II, the distance of the Fe=O bond was set to 1.77 Å, i.e. the average Fe=O distance of compounds I and II of horseradish peroxidase [8].
The simulations of substrate docking in the active center of ARP were performed with AutoDock 3.0.5 [9]. Atomic interaction energy grid maps were calculated with 0.6 Å grid spacing and 126 grid points forming a cubic box of about 75 Å centered on the geometric center of the peroxidase. The space of the cubic box covered the whole peroxidase and the space beyond. The electrostatic interaction energy grid used a distance-dependent dielectric function of Mehler-Solmajer [10]. The docking was accomplished using Lamarckian Genetic Algorithm. The number of individuals in populations was set to 50. Maximum number of energy evaluations that genetic algorithm should make was set to 1000000. Each docking was assigned to make 200 runs.
Results and Discussion
Structure of oligomers
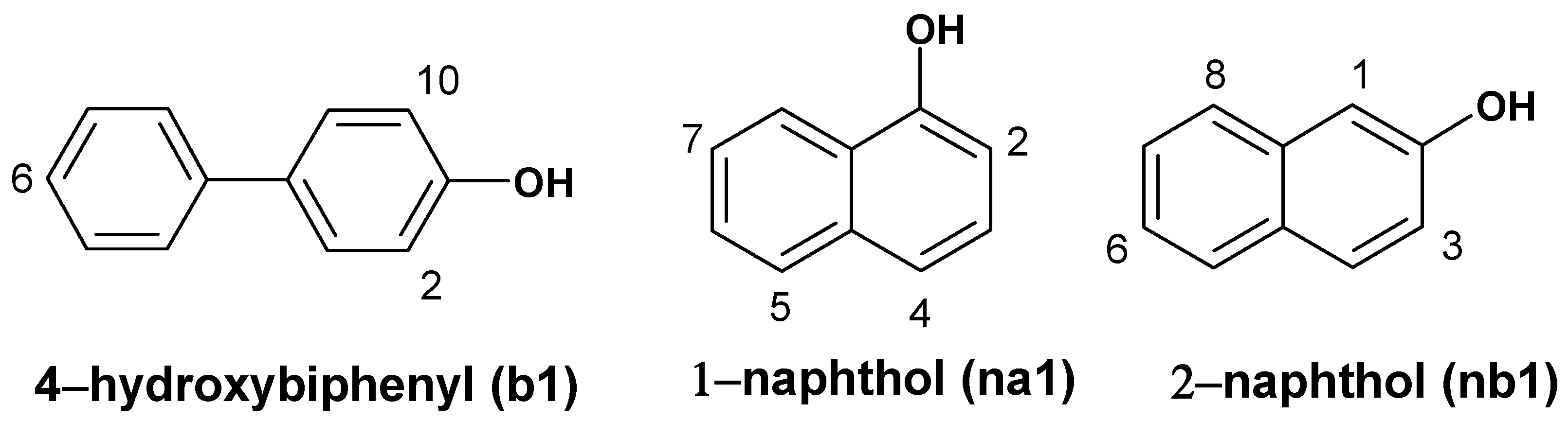
The structures of the studied substrates are depicted in Figure 1. The oxidation of these phenol derivatives is accomplished through radical formation. The recombination of radicals produces oligomer structures. To build possible structures of oligomers, the recombination of radicals through the position with the highest spin densities was performed. According to calculations the highest spin density was found in four positions of naphthols and three of 4-hydroxybiphenyl. The recombination of a radical through oxygen atom was also considered in 1-naphthol. A large number of oligomers can be produced during the recombination reactions and there is no possibility of examining them all, therefore our investigation was limited to mono-, di-, tri-, tetra- and pentaoligomers (Table 1). The calculations showed that 4-hydroxybiphenyl and its dimer b2a had totally planar structure, therefore they were set to be rigid in docking through C-C bonds connecting phenyl rings. The remaining derivatives of 4-hydroxybiphenyl have fully rotatable C-C bonds connecting phenyl rings.
Figure 1.
Structures of the substrates. (Numeration of 4-hydroxybiphenyl atoms does not match IUPAC nomenclature and it is used for convenience).
Figure 1.
Structures of the substrates. (Numeration of 4-hydroxybiphenyl atoms does not match IUPAC nomenclature and it is used for convenience).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1-naphthol derivative | Bond | 2-naphthol derivative | Bond | 4-hydroxy-biphenyl derivative | Bond |
|---|---|---|---|---|---|
| Monomers | |||||
| na1 | - | nb2 | - | b1 | - |
| Dimers | |||||
| na2a | 5-5’ | nb2a | 6-6’ | b2a | 6-6’ |
| na2b | 5-4’ | nb2b | 3-3’ | b2b | 2-6’ |
| na2c | 4-4’ | nb2c | 6-1’ | ||
| na2d | 7-5’ | nb2d | 1-1’ | ||
| na2e | 7-7’ | nb2e | 3-1’ | ||
| na2f | 5-2’ | nb2f | 1-8’ | ||
| na2g | 7-2’ | nb2g | 8-6’ | ||
| na2h | 7-4’ | nb2h | 6-3’ | ||
| na2i | 4-2’ | nb2i | 8-8’ | ||
| na2j | 2-2’ | nb2j | 8-3’ | ||
| na2k | O-2’ | ||||
| na2l | O-4’ | ||||
| na2m | O-7’ | ||||
| Trimers | |||||
| na3a | 4-7’,5’-2” | nb3a | 6-6’,3’-6” | b3a | 6-6’,2-6” |
| na3b | 7-7’,5’-7” | nb3b | 6-6’,1’-6” | b3b | 2-6’,10-6” |
| na3c | 4-5’,2’-4” | nb3c | 3-3’,1’-3” | b3c | 2-6’,2’-6” |
| na3d | 7-7’,2’-7” | nb3d | 1-1’,3’-3” | ||
| na3e | O-4’,O’-7” | ||||
| na3f | O-4’,2’-O” | ||||
| Tetramers | |||||
| b4a | 2-6’,6-6”,2”-6”’ | ||||
| b4b | 6-6’,2-6”,10-6”’ | ||||
| Pentamers | |||||
| na5a | 5-7’,5’-7”,4”-4”’,2”’-7”” | nb5a | 6-3’,1’-6”,6’-6”’,3”’-6”” | ||
| na5b | 4-2’,5’-2”,4”-4”’,2”’-4”” | nb5b | 1-1’,6’-1”,6”-1”’,6”’-6”” | ||
| na5c | 7-5’,7’-7”,5”-7”’,5”’-7”” | nb5c | 3-1’,3’-3”,1”-1”’,3”’-3”” | ||
| na5d | 7-7’,5’-7”,2’-7”’,5”’-7”” | nb5d | 6-6’,1’-3”,3’-3”’,8”’-3”” | ||
| na5e | O-4',O'-4",2"-O"',7"'-O"" |
Docking of monomers
The most common reaction mechanism of phenol substrates oxidation with heme peroxidases that appears to be universally accepted today is depicted in the following scheme:
where E is a resting (native) form of a peroxidase, S stands for the substrate and S• for the corresponding radical. The resting ferric enzyme reacts rapidly with peroxide to form compound I, i.e. an oxy-ferryl (Fe=O) derivative, in which one electron has been withdrawn from the heme group to form a porphyrin π-cation radical. This intermediate is reduced in two sequential one-electron oxidation steps through compound II formation.
Compound I and II have bound an oxygen atom to a heme of iron forming Fe=O. The intermolecular orientation of substrate and (porphyrin)Fe=O complex must be favorable for an electron and a proton transfer. The experimental and modeling studies of ARP associated reactions showed that a proton transfer is determined by a proper hydrogen bonding network inside the active center [11]. The absence of hydrogen bonding between oxygen of ferryl group and hydroxyl group of phenol derivative produces “unproductive” complex that determines the decrease of the reaction rate almost thousand times [11].
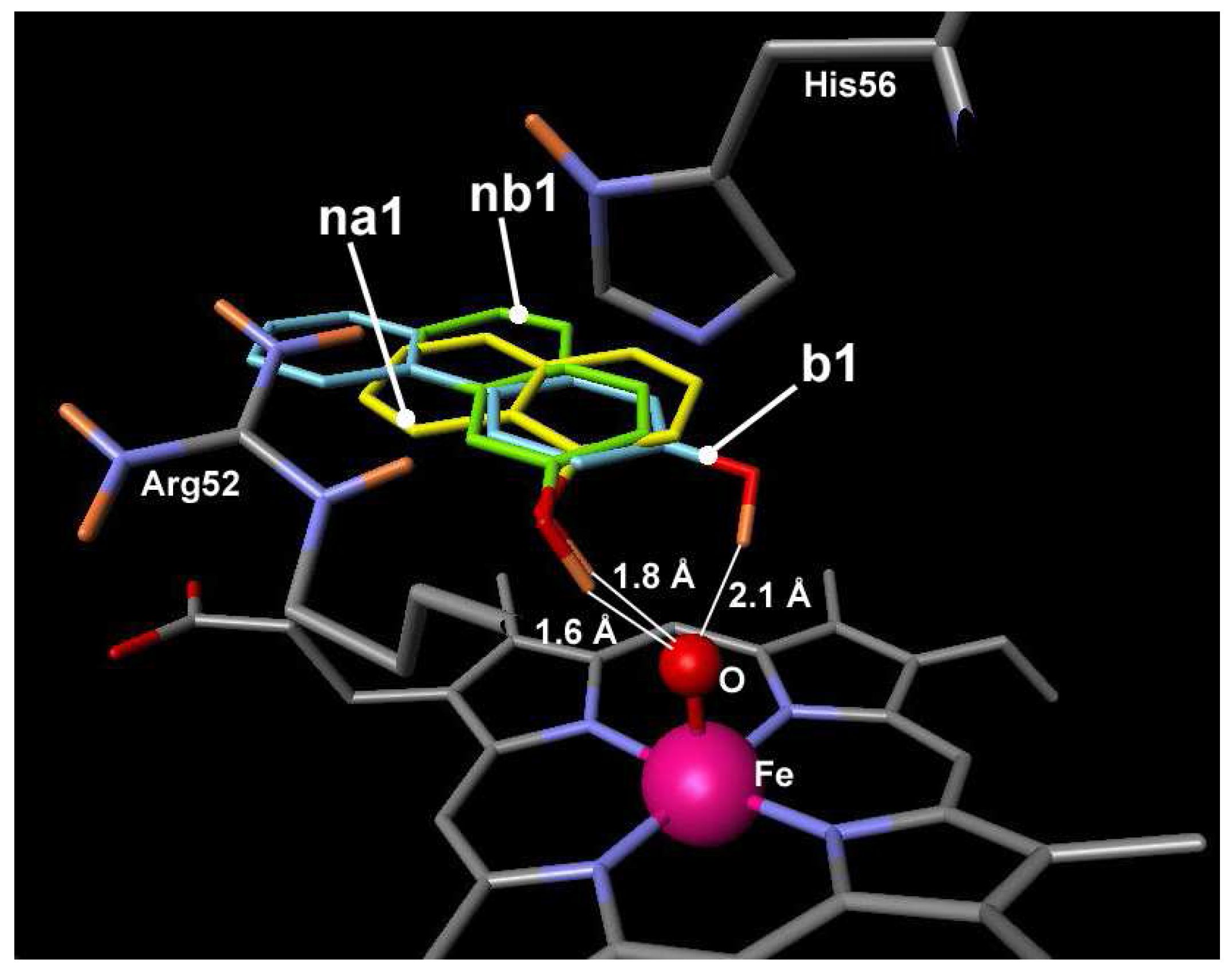
The dockings showed that phenol derivatives b1, na1 and nb1 docked in the active center with highest affinity, or the lowest docking energy, in the whole ARP structure. All monomers placed OH groups inside the active center near Fe=O and formed strong hydrogen bonds with the oxygen atom of Fe=O fragment (Figure 2). The lengths of hydrogen bonds are 2.1 Å, 1.8 Å and 1.6 Å respectively of 4-hydroxybiphenyl, 1-naphthol and 2-naphthol.
Figure 2.
The dockings of 1-naphthol (yellow), 2-naphthol (green) and 4-hydroxy-biphenyl (cyan) in the active center of ARP. Hydrogen bonds (length depicted in Å) are formed between OH and Fe=O groups.
Figure 2.
The dockings of 1-naphthol (yellow), 2-naphthol (green) and 4-hydroxy-biphenyl (cyan) in the active center of ARP. Hydrogen bonds (length depicted in Å) are formed between OH and Fe=O groups.
The docking energies of monomers were -7.0 kcal/mol, -6.4 kcal/mol and -6.4 kcal/mol (Table 2). The docking results show that phenol substances form a “productive” complex. For that reason they should be considered as active substrates. Experimental measurements confirm the high activity of these substances. The constants of 1-naphthol and 2-naphthol oxidation were 1.4·107 M-1·s-1 and 5.4·107 M-1·s-1 and approached the rate of diffusion limited for enzyme catalysis [5] .
Docking of oligomers
In this work the hypothesis was made that unproductive bonding of oligomers may determinate peroxidase inhibition during phenol derivatives oxidation. To confirm or reject this hypothesis the docking of oligomer structures in the active center of ARP has been performed. The calculations show that all oligomer structures dock in the active center of ARP with the highest affinity. The complexes of oligomers are even stronger since the docking energy is less in comparison to monomer substances (Table 2).
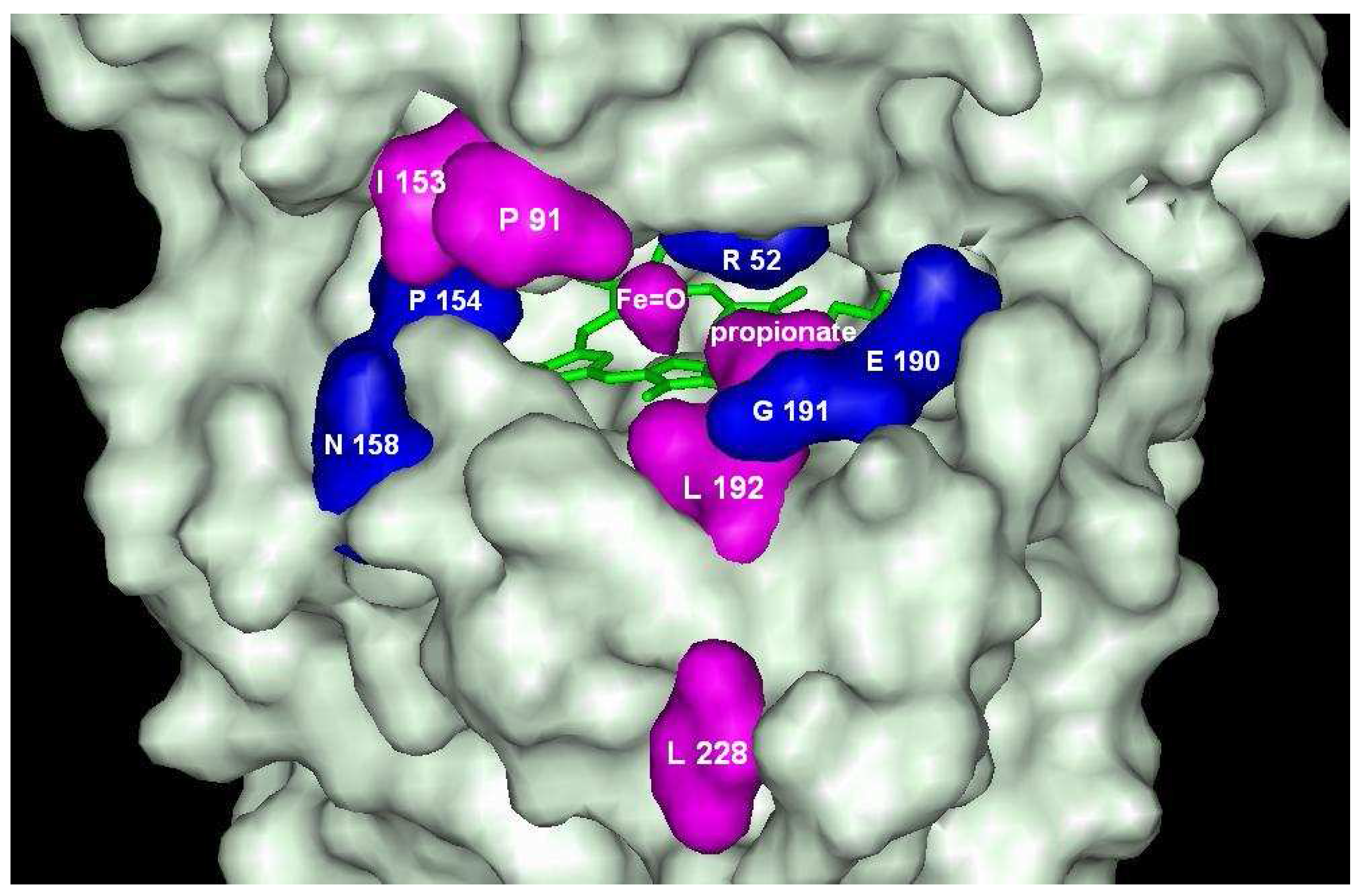
The dockings revealed some amino acids that are capable to make hydrogen bonds with hydroxyl group of the oligomers. All oligomers are capable to make hydrogen bonds with the amino acids that are common for 4-hydroxybiphenyl, 1-naphthols and 2-naphthols compounds: Arg52, Pro154, Asn158, Glu190, Gly191 (blue in Figure 3). All interacting amino acids make the heme pocket environment or are the part of the active site itself. Among these amino acids only Arg52 is hydrophilic, while other amino acids are hydrophobic. Hydrogen bonding is made through oxygen atom of C=O group in peptide fragment of the amino acids. Naphthols have more amino acids capable to participate in H-bonding and almost all of these amino acids are hydrophobic too: Ile153, Pro91, Leu192, Leu228. Naphthols make hydrogen bonding with oxygen atom from Fe=O and propionate residue of heme, too.
Figure 3.
Amino acids of ARP forming hydrogen bonding with oligomers. Amino acids in blue are common for 1- naphthol, 2-naphthol and 4-hydroxybiphenyl oligomers. Amino acids in magenta are characteristic to naphthols oligomers.
Figure 3.
Amino acids of ARP forming hydrogen bonding with oligomers. Amino acids in blue are common for 1- naphthol, 2-naphthol and 4-hydroxybiphenyl oligomers. Amino acids in magenta are characteristic to naphthols oligomers.
| 1-naphthol derivative | Docking energy, kcal/mol | 2-naphthol derivative | Docking energy, kcal/mol | 4-hydroxy-biphenyl derivative | Docking energy, kcal/mol |
|---|---|---|---|---|---|
| na1 | -6.4 | nb2 | -6.4 | b1 | -7.0 |
| na2a | -8.4 | nb2a | -7.6 | b2a | -9.0 |
| na2b | -5.6 | nb2b | -6.8 | b2b | -10.5 |
| na2c | -5.4 | nb2c | -9.1 | ||
| na2d | -9.3 | nb2d | -8.5 | ||
| na2e | -9.9 | nb2e | -7.1 | ||
| na2f | -8.6 | nb2f | -8.8 | ||
| na2g | -8.9 | nb2g | -10.0 | ||
| na2h | -9.2 | nb2h | -9.1 | ||
| na2i | -8.7 | nb2i | -9.1 | ||
| na2j | -8.6 | nb2j | -7.1 | ||
| na2k | -8.9 | ||||
| na2l | -8.3 | ||||
| na2m | -8.4 | ||||
| na3a | -5.0 | nb3a | -11.6 | b3a | -11.8 |
| na3b | -10.4 | nb3b | -8.6 | b3b | -11.6 |
| na3c | -8.3 | nb3c | -9.3 | b3c | -12.4 |
| na3d | -11.5 | nb3d | -8.8 | ||
| na3e | -10.3 | ||||
| na3f | -9.5 | ||||
| b4a | -10.5 | ||||
| b4b | -12.2 | ||||
| na5a | -7.6 | nb5a | -11.4 | ||
| na5b | -13.3 | nb5b | -13.9 | ||
| na5c | -8.0 | nb5c | -8.1 | ||
| na5d | -7.3 | nb5d | -11.4 | ||
| na5e | -9.8 |
The oligomers of 1-naphthol are docked at the entrance of the active center, but are not able to make hydrogen bond with Fe=O group of ARP. The complex formation energies vary between –5.5 kcal/mol and –9.9 kcal/mol for ten C-dimers and three O-dimers of naphthol derivatives. The lowest energies are found for dimers having bonds through position 7 (Figure 1), i.e. for na2d, na2e, na2g and na2h. The highest energy is calculated for compounds na2b and na2c having bonding in position 5. Despite distinct differences in affinities, these compounds occupy almost the same position at the entrance of the heme pocket (Figure 4). Dockings show that the difference in docking energies are determined by docking peculiarities of the naphthol fragment outside the heme pocket. All dimers have no ability to make hydrogen bonds with Fe=O fragment with exception of derivative na2e and na2i. These derivatives may form a complex in which OH groups is located at larger than 2.5 Å distance from Fe=O. In this case hydrogen bond should be very weak. The lowest docking energies for trimers were found for na3b (-10.4 kcal/mol), na3d (-11.5 kcal/mol) and na3e (-10.3 kcal/mol) compounds, which have bonding at the position 7. The remaining trimers made bonding mostly at position 4. The lowest docking energy –13.3 kcal/mol was indicated for pentamer na5b.
The studied oligomeric O-derivatives of 1-naphthols have only one OH group. Therefore, these compound have limited abilities to form hydrogen bond inside the active center of ARP. However oligomeric O-derivatives have similar and lower docking energies than other oligomeric C-derivatives, which have two and more OH groups. The current finding reveals that hydrogen bonding is not the main factor responsible for binding of oligomers and the hydrophobic forces play much more important role.
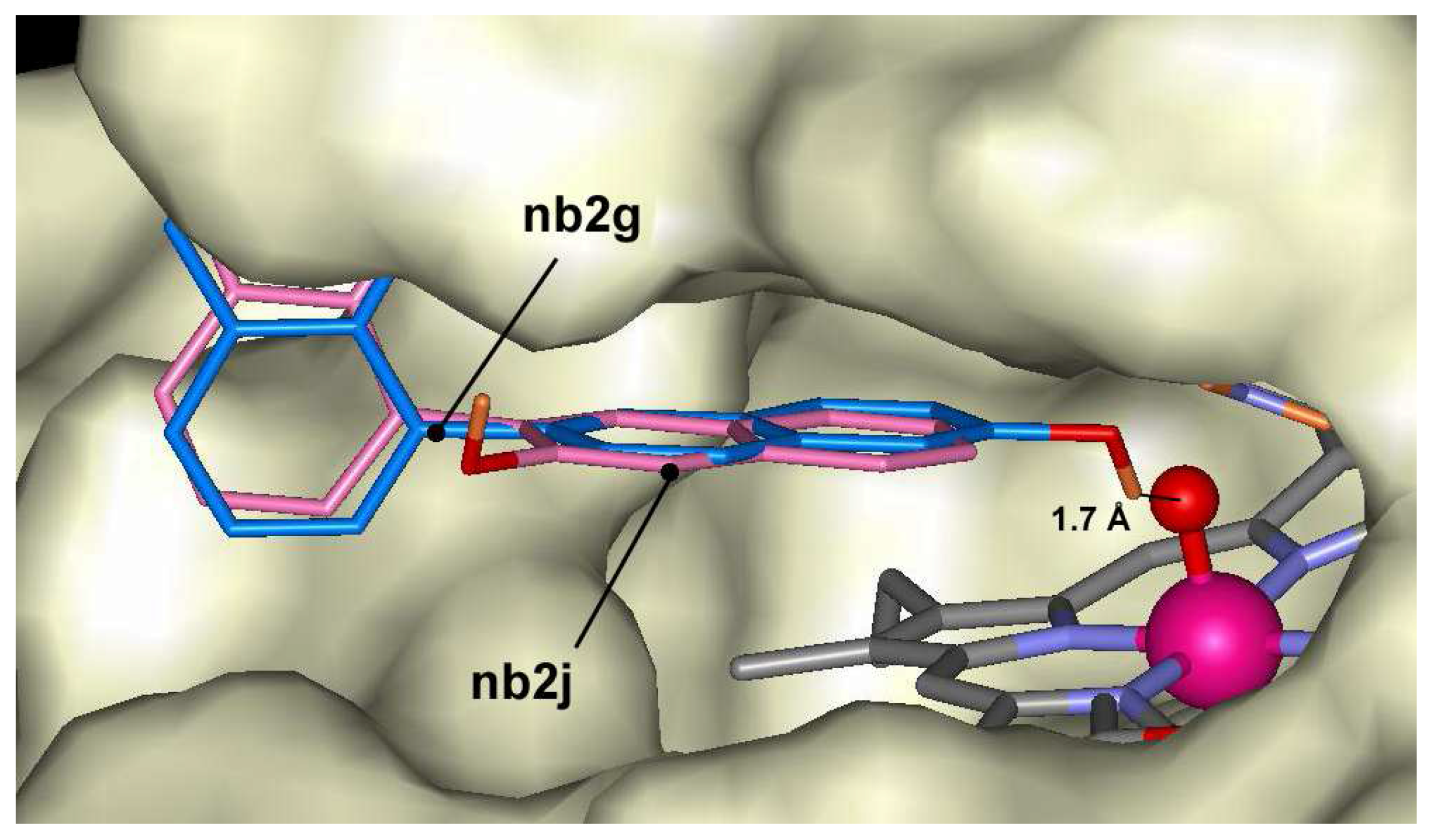
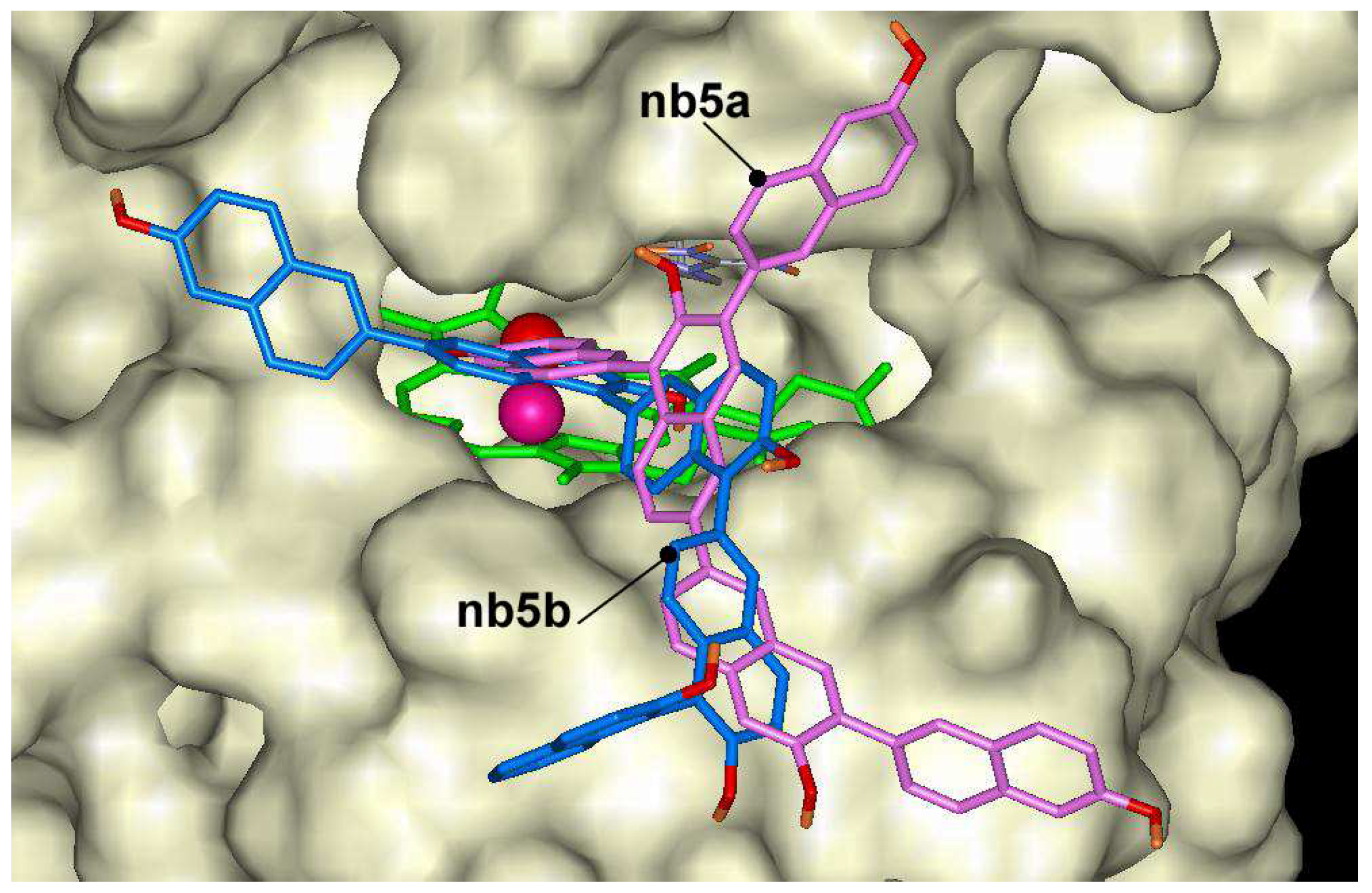
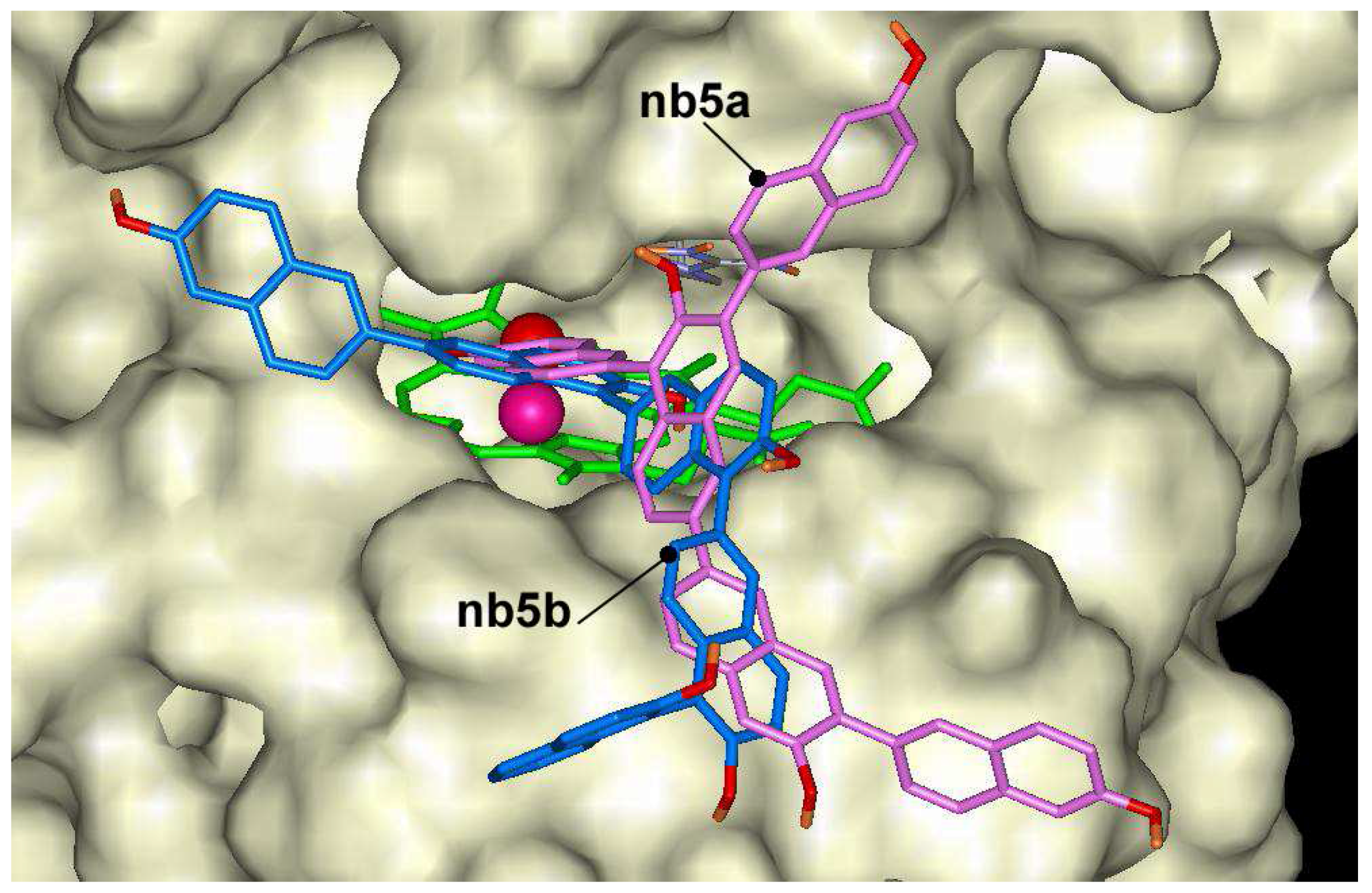
2-naphthol dimers may form 10 dimers through C-C bonding, and the docking of these dimers was calculated in ARP structure. The lowest docking energy was found for compound nb2g (-10.0 kcal/mol) and some other dimers having bonding at positions 6 and 8 like compounds nb2i, nb2h or nb2c. The highest energies were calculated for compounds nb2b, nb2e and nb2j having bonding at position 3. All mentioned dimers occupy the same position in the heme pocket (Figure 5). The possible explanation of energy differences might be related to the fact that nb2b, nb2e and nb2j compounds do not form hydrogen bond with Fe=O group like as nb2g, nb2h or nb2c. The last three compounds are exception among oligomers and may form hydrogen bonds with Fe=O group at distances 1.7 Å, 2.0 Å and 1.9 Å, respectively. Trimers of 2-naphthol have the lowest docking energies between –8.6 kcal/mol and –11.6 kcal/mol. Like as dimers the lowest docking energy was calculated for nb3a producing oligomers at 6 th position. The pentamers showed lower docking energies. The lowest docking energy was calculated for pentamer nb5b (–13.9 kcal/mol) that is formed with bonds at position 6 and 1 (Figure 6). In general, the lowest affinities of 2-naphthol oligomers were found for ones having bonding through position 3.
Figure 4.
Dockings of na2b (magenta) and na2d (dark orange) in the active center of ARP.
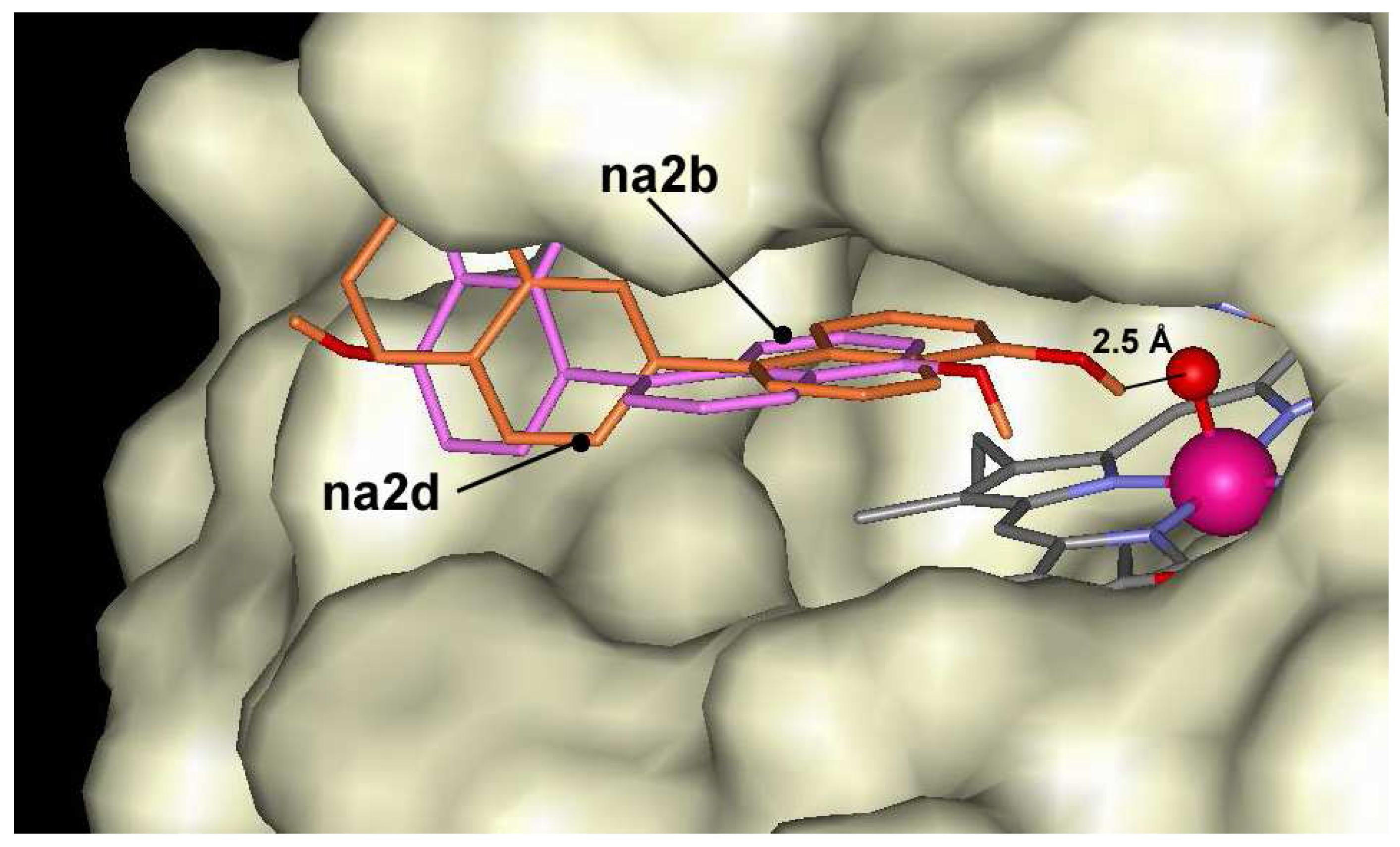
Figure 5.
Dockings of nb2g (blue) and nb2j (magenta) in the active center of ARP.
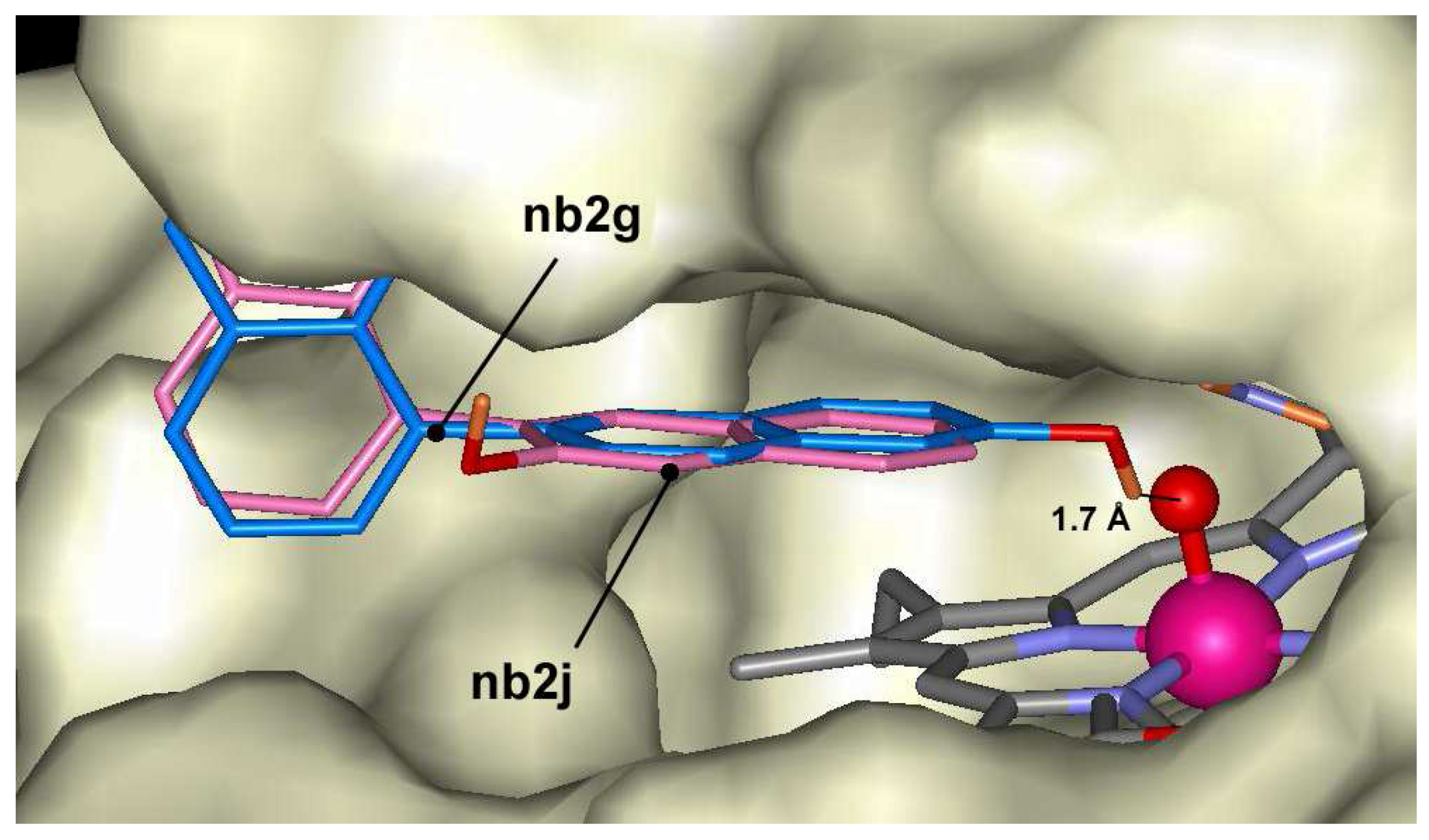
Figure 6.
Dockings of nb5a (magenta) and nb5b (blue) in the active center of ARP.
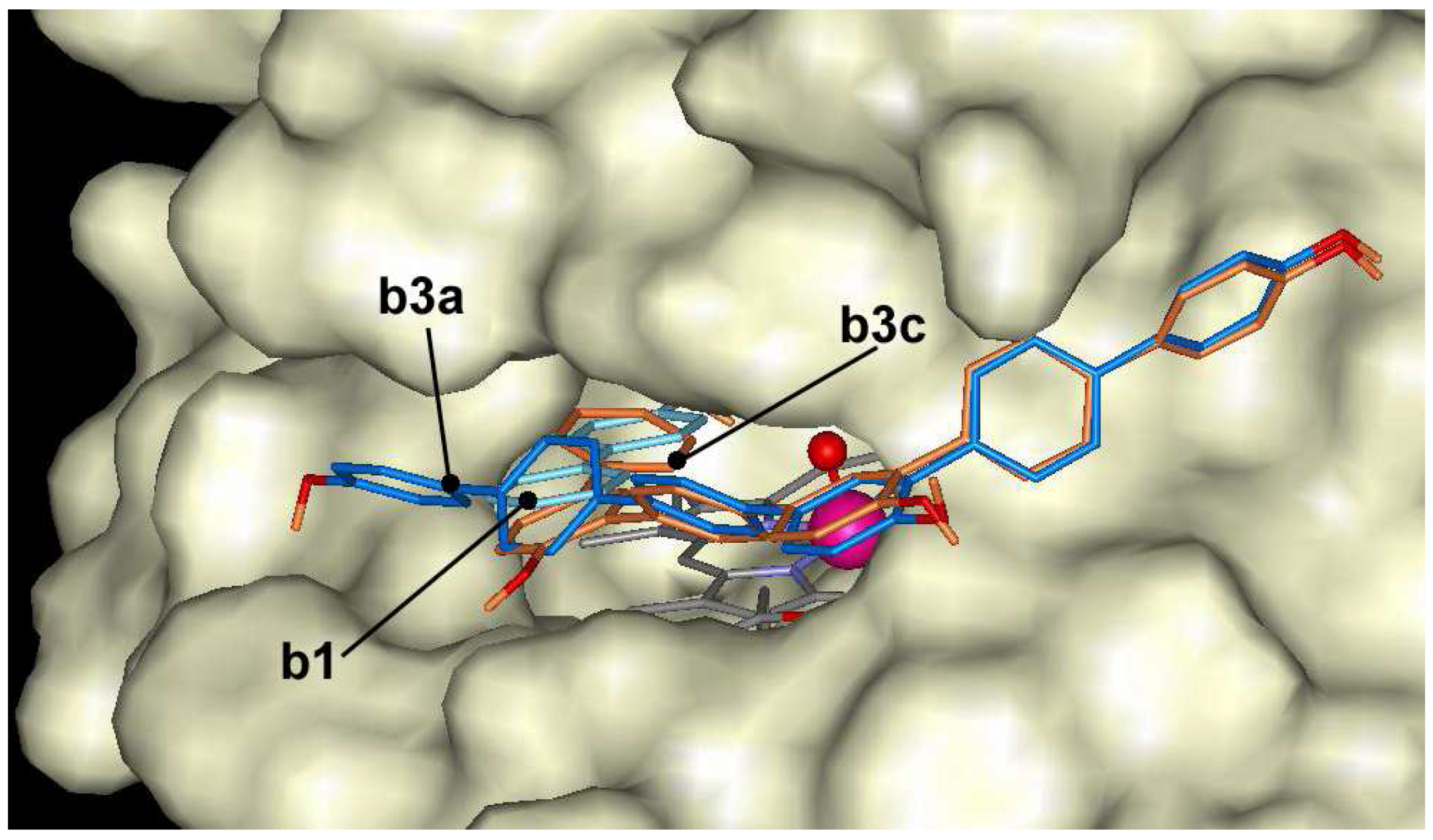
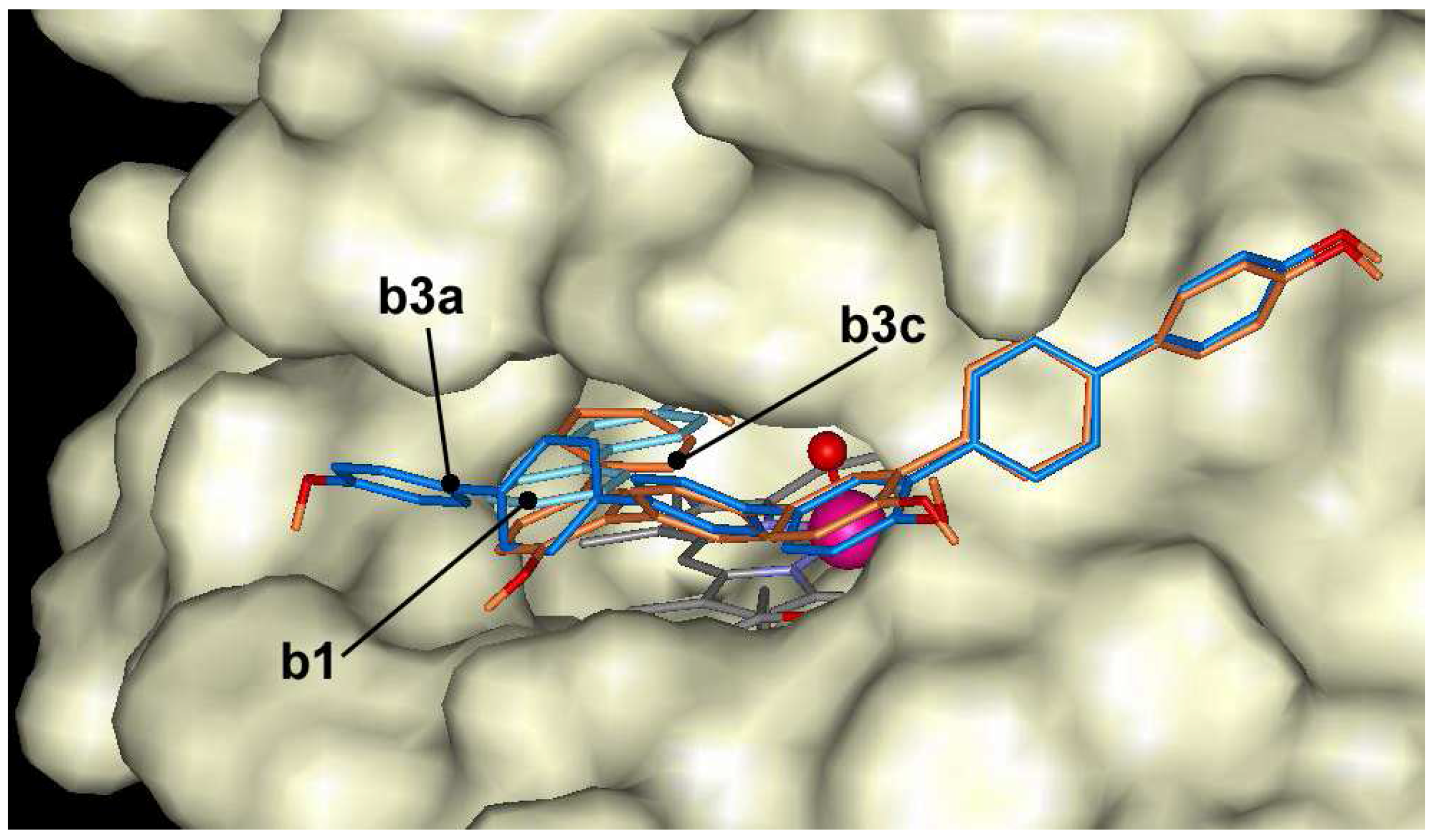
The oligomers of 4-hydroxybiphenyl dock at the entrance of the active center, but are not able to make hydrogen bond with Fe=O group of ARP like as naphthol derivatives. The docking energies vary in the range of –7.0 and –12.4 kcal/mol. 4-hydroxybiphenyl derivatives with linear-shape like b2a, b3a and b4a dock across the entrance to the heme pocket blocking the entrance and can not form hydrogen bond with Fe=O group (Figure 7). Compounds with crooked shape like b2b, b3b and b4b dock across the entrance to the heme pocket with some hydroxyls in the heme pocket. However, these hydroxyls are located too far from Fe=O group to form hydrogen bond. The compounds with crooked shape have about 1 kcal/mol higher affinity to the active center of ARP than linear compounds.
Figure 7.
The docking of 4-hydroxybiphenyl (b1 - cyan) and trimer (b3a - blue, b3c – dark orange) at the entrance of the active center of ARP.
Figure 7.
The docking of 4-hydroxybiphenyl (b1 - cyan) and trimer (b3a - blue, b3c – dark orange) at the entrance of the active center of ARP.
1-naphthol and 2-naphthol derivatives are more compact compounds and have lower flexibility degree than 4-hydroxybiphenyl and its derivatives, because naphthol derivatives do not have one extra flexible bond. However, 4-hydroxybiphenyl tetramers have too many freely rotating bonds and the docking results is distorted due to high flexibility. The decreasing affinity of pentamers and tetramers can be related to increasing multi-center binding of oligomers. Multi-center binding limits the possibility to exploit all favorable docking places in ARP, especially to compounds with a linear shape.
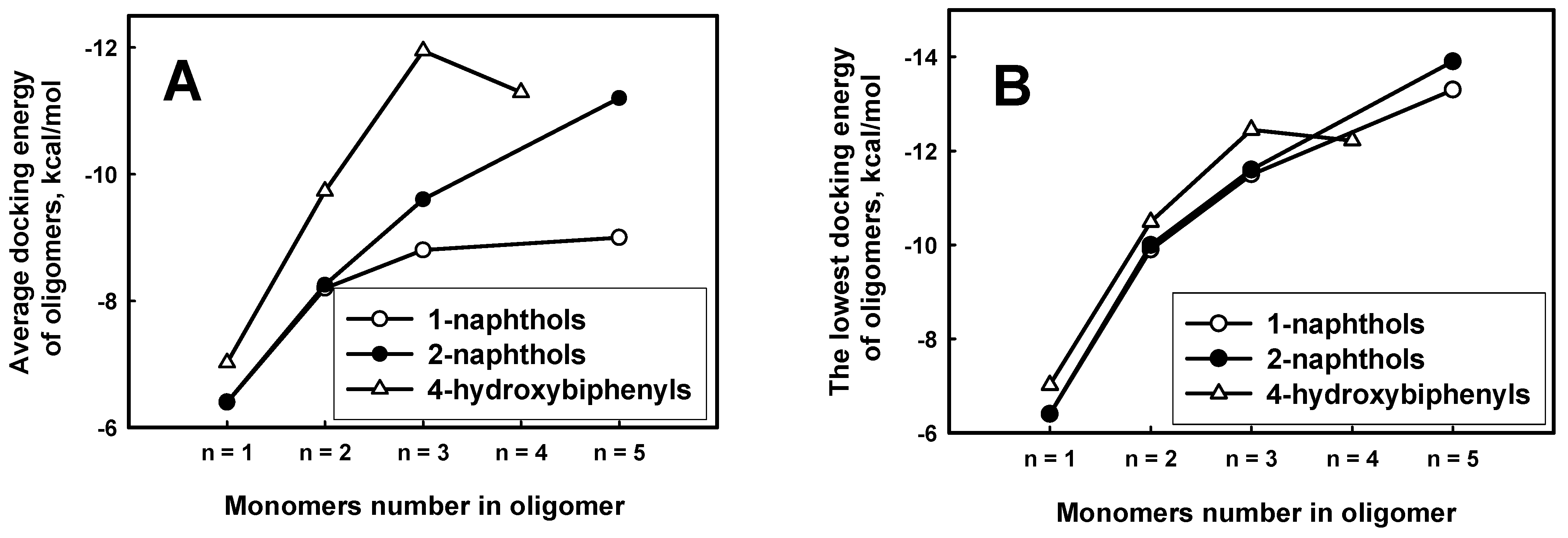
The calculated docking energies of phenol derivatives revealed the obvious tendency that the larger molecular weight of oligomer had the large affinity to ARP (Figure 8 A,B). This dependence was demonstrated for all derivatives of naphthols. There is also tendency that the average docking energy of explored 4-hydroxybiphenyl oligomers drops faster along with growing oligomer length. It means that 4-hydroxybiphenyl oligomers binds stronger than 1-naphthols and 2-naphthols. According to the average docking energies the oligomers can be placed in decreasing order of binding to ARP: 4-hydroxybiphenyl, 1-naphthols and 2-naphthols. That order is comparable to the decreasing monomer length: the longer monomer length in oligomers chain, the higher affinity to ARP. However, the dependence of lowest docking energies on oligomer lengths is similar to 4-hydroxybiphenyl, 1-naphthols and 2-naphthols (Figure 8 B) compounds than it is expressed in Figure 8A.
Figure 8.
The average (A) and the lowest (B) docking energies of 1-hydroxybiphenyl, 1-naphthol and 2-naphthol oligomers through carbon for each length of oligomer.
Figure 8.
The average (A) and the lowest (B) docking energies of 1-hydroxybiphenyl, 1-naphthol and 2-naphthol oligomers through carbon for each length of oligomer.
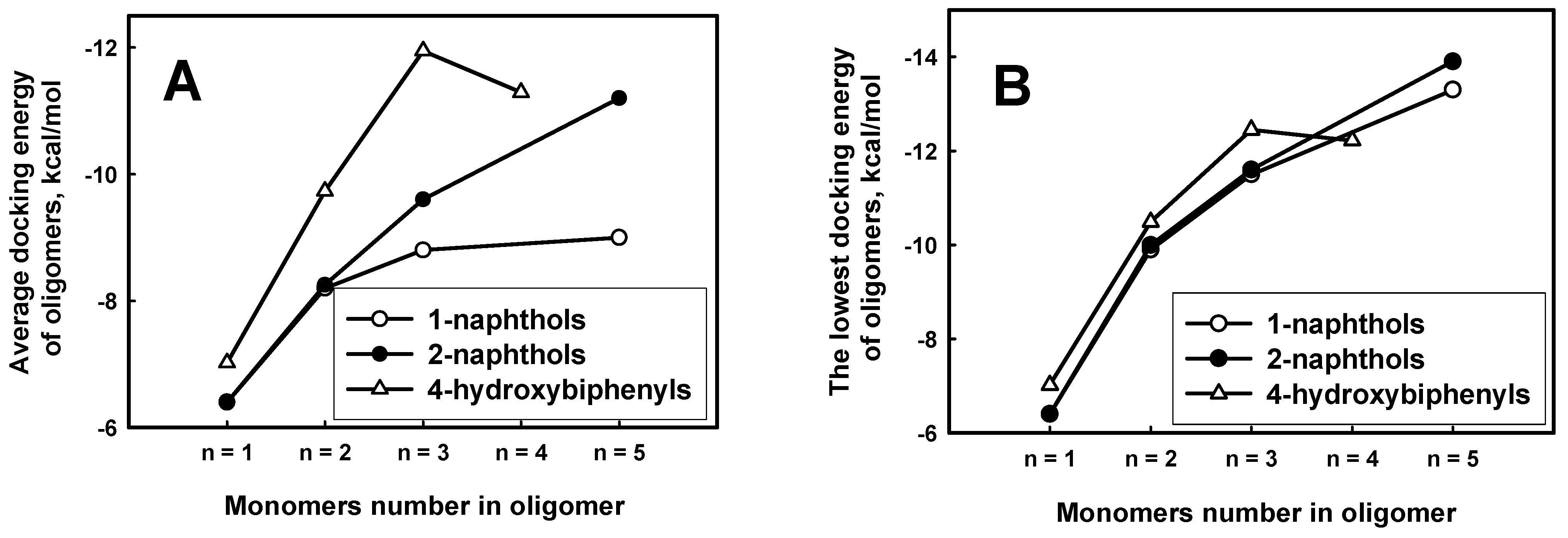
The docking results reveal that all explored oligomers have the highest binding affinity in the active center of ARP. It means that oligomer might act as substrates of ARP. Even more, the calculations show that they form a stronger complexes in comparison to monomers. The calculations, however, revealed that the most of these compounds did not form hydrogen bonding network inside the active center of ARP. Three 2-naphthol dimers can make weak hydrogen bonds in the active center of ARP, but oligomers of this compound with elongated chain like as trimers, tetramers or pentamers do not form hydrogen bonds at all.
The docking results of oligomers reveal that oligomers do not fulfill the demands for productive binding, which is crucial for substrate oxidation by ARP [11]. Even the dimer stage for 1-naphthol and 4-hydroxybiphenyl and the trimer stage for 2-naphthol forms unproductive complex. The absence of the proper hydrogen bonding network does not make favorable structure for proton transfer in order an oxidation reaction could appear. Therefore, oligomers of 4-hydroxybiphenyl, 1-naphthol and 2-naphthol serve as concurrent inhibitors, which block the entrance to the active site of ARP and prevent the binding of monomer. The higher affinity of oligomer derivatives guarantees the stable but unproductive oligomer ARP complex formation. That could lead further to the formation of the aggregates of ARP with oligomers or the aggregates of pure oligomers.
Conclusions
The docking calculations reveal that phenol derivatives bind in the active site of ARP with various degree of affinity. The oligomers of 4-hydroxybiphenyl, 1-naphthol and 2-naphthol bind with higher affinity than monomers. The main binding force is a hydrophobic interaction. The calculations reveal that during complexation the oligomers do not form hydrogen bonds between hydroxyl and Fe=O group that is necessary for effective reaction. For this reason it is predicted that many oligomers would be unreactive as substrates but function well as reversible inhibitors of the peroxidase.
Acknowledgements
This research was supported by Lithuanian State Science and Studies Foundation, project C-03020.
References and Notes
- Ghioureliotis, M.; Nicell, J.A. Assessment of soluble products of peroxidase-catalyzed polymerization of aqueous phenol. Enzyme Microb. Technol. 1999, 25, 185–193, (and citations therein). [Google Scholar] [CrossRef]
- Kobayashi, S. Enzymatic polymerization: a new method of polymer synthesis. J. Pol. Sci. A. 1999, 37, 3041–3056. [Google Scholar] [CrossRef]
- Kauffmann, C.; Petersen, B.R.; Morten, J.B. Enzymatic removal of phenols from aqueous solutions by Coprinus cinereus peroxidase and hydrogen peroxide. J. Biotech. 1999, 73, 71–74. [Google Scholar] [CrossRef]
- Liu, W.; Kumar, J.; Tripathy, S.; Lee, S.-H.; Samuelson, L.A. Peroxidase-catalyzed polymerization of 1-hydroxypyrene. J. Macromol. Science 2003, A40, 1407–1413. [Google Scholar] [CrossRef]
- Bratkovskaja, I.; Vidziunaite, R.; Kulys, J. Oxidation of phenolic compounds by peroxidase in the presence of soluble polymers. Biochemistry (Moscow) 2004, 69(9), 985–992. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; et al. Gaussian 98, Revision A.9, Gaussian, Inc.: Pittsburgh PA, 1998.
- Kunishima, N.; Fukuyama, K.; Matsubara, H.; Hatanaka, H.; Shibano, Y.; Amachi, T. Crystal structure of the fungal peroxidase from Arthromyces ramosus at 1.9 Å resolution. Structural comparisons with the lignin and cytochrome c peroxidases. J. Mol. Biol. 1994, 235, 331–334, PDB ID: 1ARP. [Google Scholar] [CrossRef]
- Berglund, G.I.; Carlsson, G.H.; Smith, A.T.; Szoke, H.; Henriksen, A.; Hajdu, J. The catalytic pathway of horseradish peroxidase at high resolution. Nature 2002, 417, 6887, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Goodsell, D.S.; Olson, A.J. Automated docking of substrates to proteins by simulated annealing. Proteins:Str.Func.Gen. 1990, 8, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Mehler, E.L.; Solmajer, T. Electrostatic effects in proteins: comparison of dielectric and charge models. Protein Eng. 1991, 4, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Kulys, J.; Ziemys, A. A role of proton transfer in peroxidase-catalyzed process elucidated by substrates docking calculations. BMC Structural Biology 2001, 1, 3. [Google Scholar] [CrossRef] [PubMed]
© 2005 by MDPI (http://www.mdpi.org).
Share and Cite
MDPI and ACS Style
Ziemys, A.; Kulys, J. Inhibition of Heme Peroxidase During Phenol Derivatives Oxidation. Possible Molecular Cloaking of the Active Center. Int. J. Mol. Sci. 2005, 6, 245-256. https://doi.org/10.3390/i6090245
AMA Style
Ziemys A, Kulys J. Inhibition of Heme Peroxidase During Phenol Derivatives Oxidation. Possible Molecular Cloaking of the Active Center. International Journal of Molecular Sciences. 2005; 6(9):245-256. https://doi.org/10.3390/i6090245
Chicago/Turabian StyleZiemys, Arturas, and Juozas Kulys. 2005. "Inhibition of Heme Peroxidase During Phenol Derivatives Oxidation. Possible Molecular Cloaking of the Active Center" International Journal of Molecular Sciences 6, no. 9: 245-256. https://doi.org/10.3390/i6090245