Theoretical Study of the Diastereofacial Isomers of Aldrin and Dieldrin

Abstract
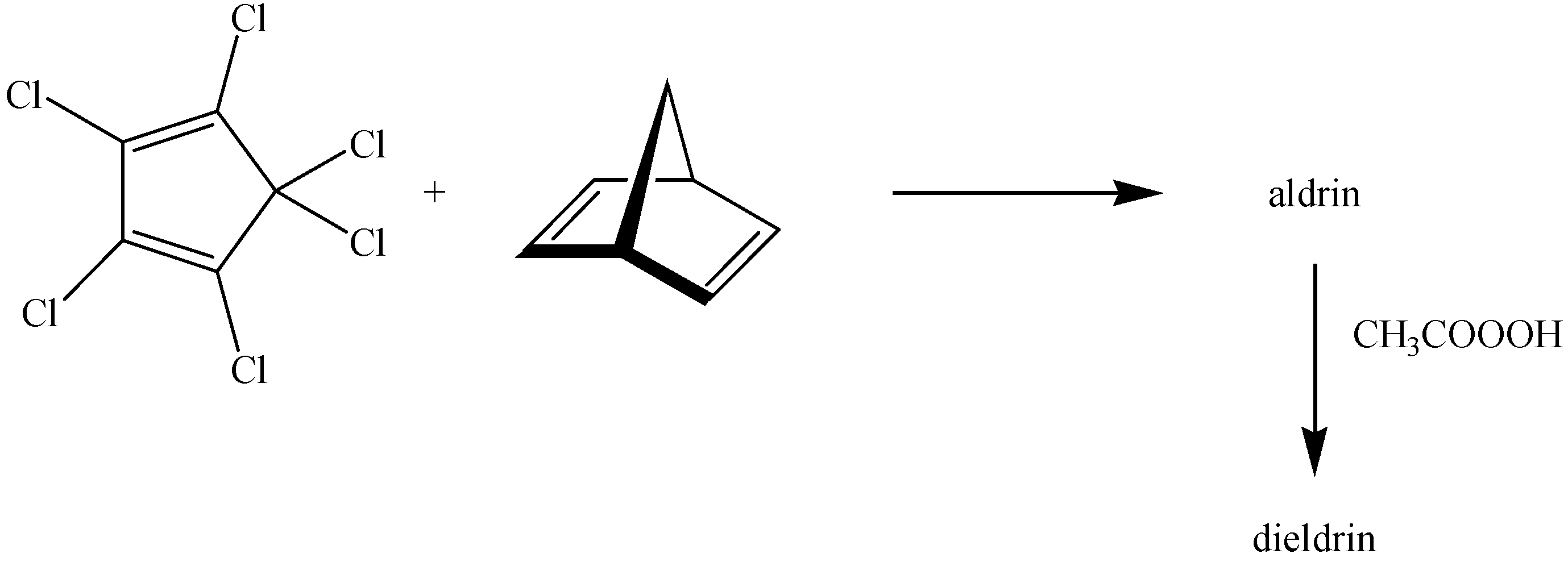
:1. Introduction
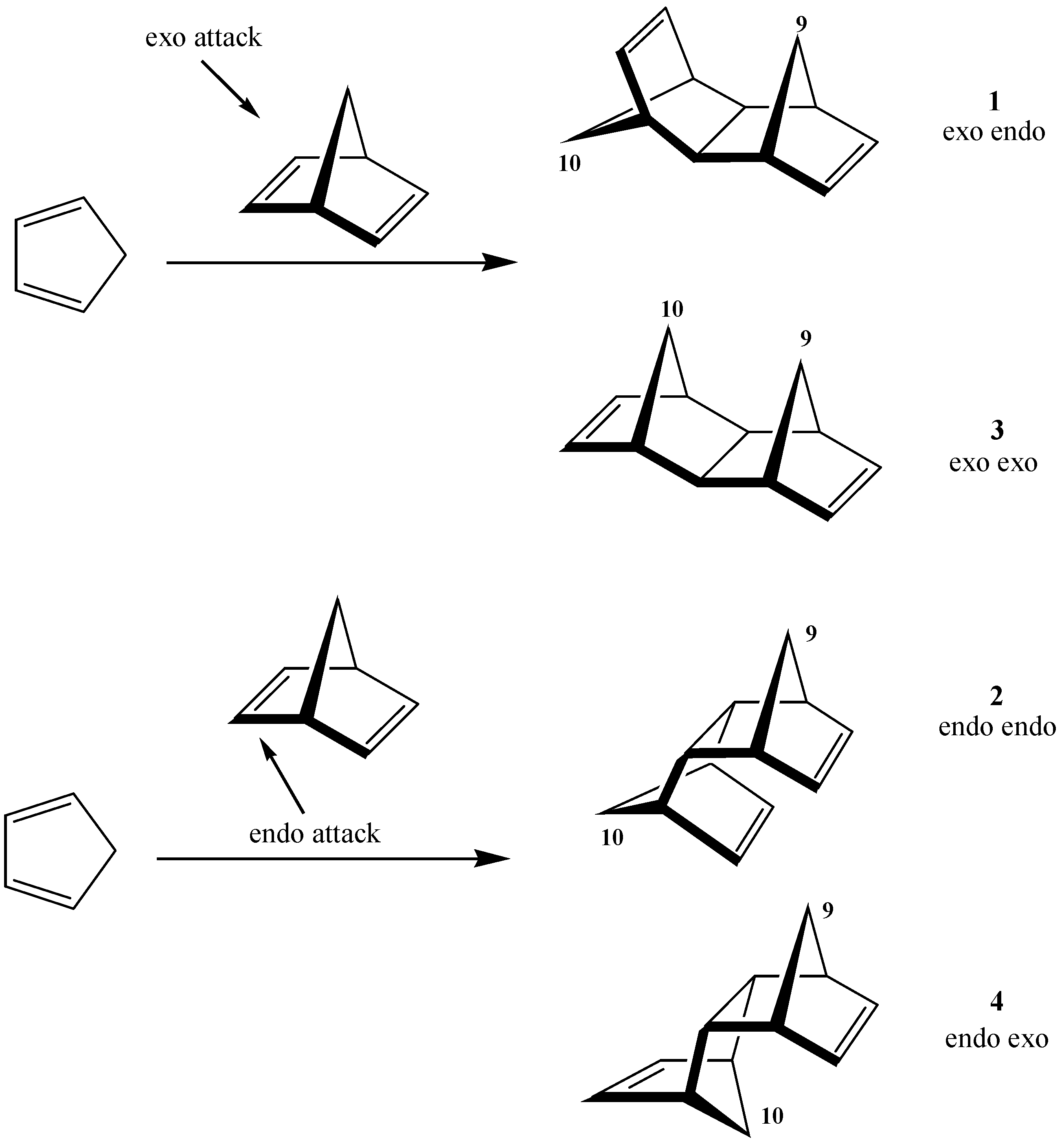
2. Results and Discussion




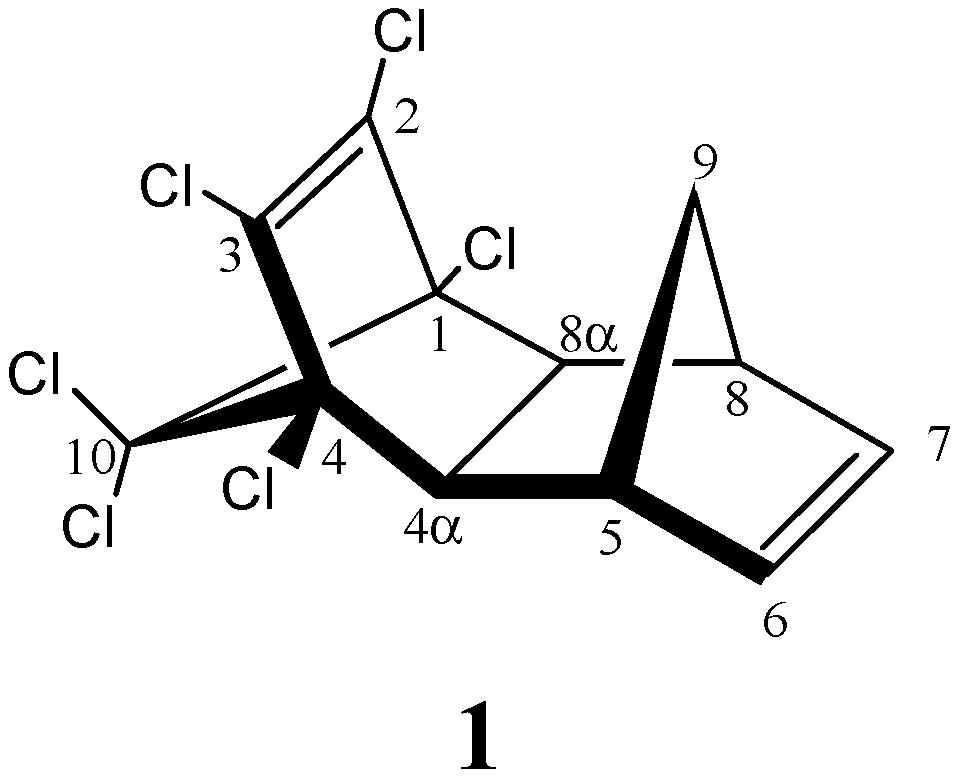
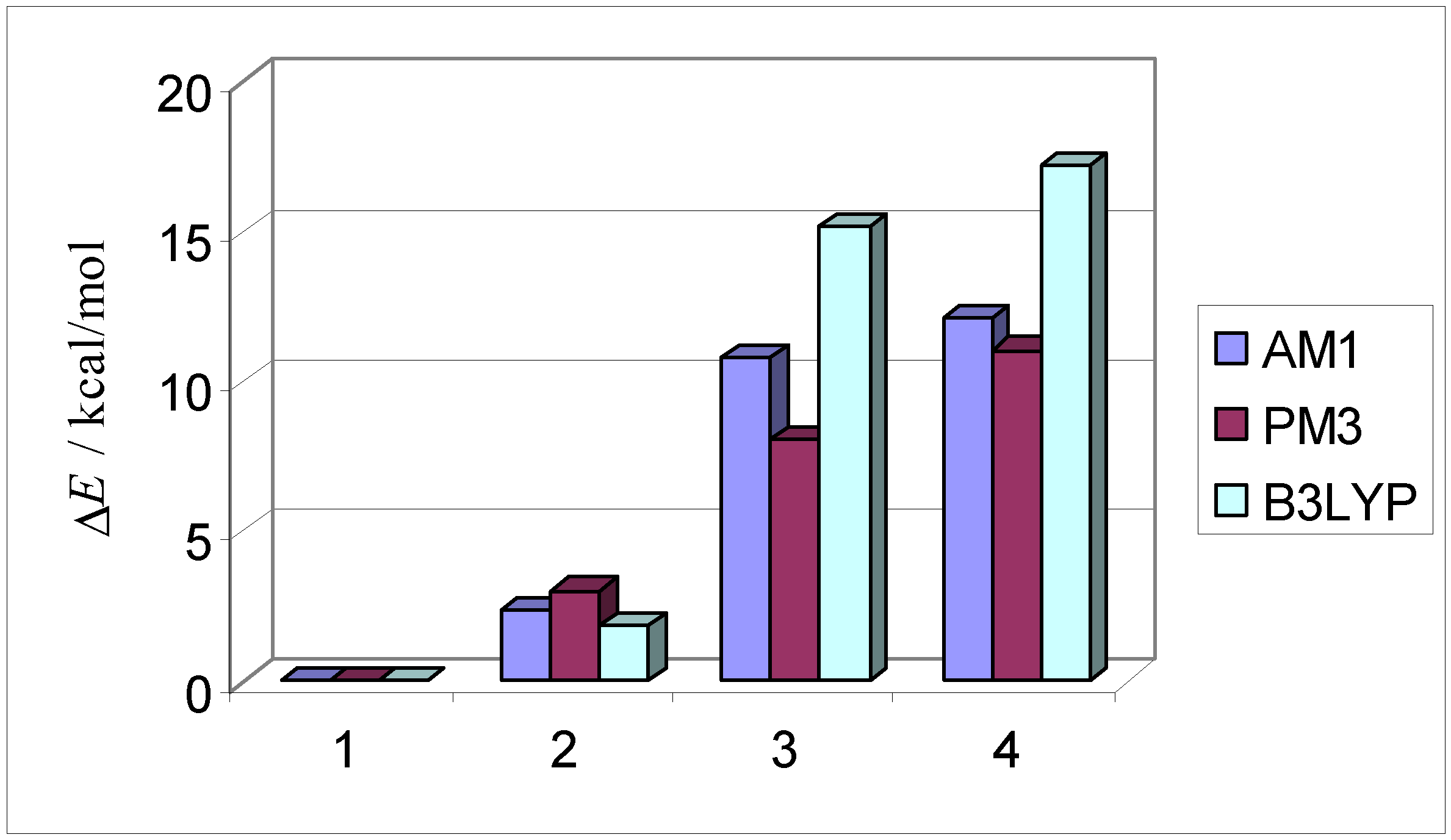
| AM1 structure | E(AM1)/ kcal/mol | ΔE(AM1)/ kcal/mol | E(PM3)/ kcal/mol | ΔE(PM3)/ kcal/mol | |
|---|---|---|---|---|---|
| 1 | –2587.2 | 0.0 | –2597.5 | 0.0 | |
| exo endo aldrin | |||||
| 2 | –2584.8 | 2.4 | –2594.5 | 3.0 | |
| endo endo | |||||
| 3 | –2576.4 | 10.8 | –2589.4 | 8.1 | |
| exo exo | |||||
| 4 | –2575.1 | 12.1 | –2586.5 | 11.1 | |
| endo exo |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cl | F | |||
|---|---|---|---|---|
| r(a1)/Å | r(a2)/Å | r(a1)/Å | r(a2)/Å | |
| 1 | 2,359 | 2,378 | 2,394 | 2,397 |
| 2 | 2,330 | 2,360 | 2,374 | 2,362 |
| 3 | 2,611 | 2,153 | 2,486 | 2,294 |
| 4 | 2,840 | 2,055 | 2,881 | 2,080 |
| Cl | F | |||||
|---|---|---|---|---|---|---|
| ạ(ClCCCl) | β(HCCH) | γ(HCCH) | ạ(CFCCCF) | β(HCCH) | γ(HCCH) | |
| 1TS | –175.10 | 149.89 | 177.26 | –170.33 | 152.32 | –177.51 |
| 2TS | 146.48 | 149.12 | –176.55 | –173.56 | 152.55 | –176.52 |
| 3TS | –172.56 | 132.81 | 178.30 | –167.21 | 146.31 | 178.02 |
| 4TS | –173.76 | 126.29 | –174.89 | –168.71 | 159.23 | –173.23 |
| Cl B3LYP/6-31++G | Cl B3LYP/6-31+G* | F B3LYP/6-31++G | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Structure | ΔEf/ kcal/mol | ΔE / kcal/mol | ΔE# / kcal/mol | ΔEf/ kcal/mol | ΔE / kcal/mol | ΔE# / kcal/mol | ΔEf/ kcal/mol | ΔE / kcal/mol | ΔE# / kcal/mol | |
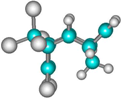
| 1 exo endo aldrin |  | –30.0 –26.1 | 0.0 0.0 | 23.9 24.7 | –26.9 –22.8 | 0.0 0.0 | 25.9 26.8 | –45.4 –41.2 | 0.0 0.0 | 13.0 14.0 |
| 2 endo endo |  | –28.1 –24.2 | 1.9 1.9 | 27.2 27.9 | –24.9 –20.9 | 1.9 1.9 | 29.2 30.0 | –45.0 –40.8 | 0.9 0.7 | 13.9 14.7 |
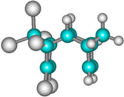
| 3 exo exo |  | –14.6 –10.9 | 15.4 15.2 | 36.5 36.9 | –12.2 –8.3 | 14.7 14.4 | 37.7 38.5 | –37.0 –32.8 | 8.9 8.8 | 21.9 22.8 |
| 4 endo ex |  | –12.6 –8.9 | 17.4 17.2 | 41.17 41.4 | –10.5 –6.6 | 16.4 16.2 | 42.7 43.4 | –35.0 –31.0 | 13.5 12.8 | 26.5 26.8 |


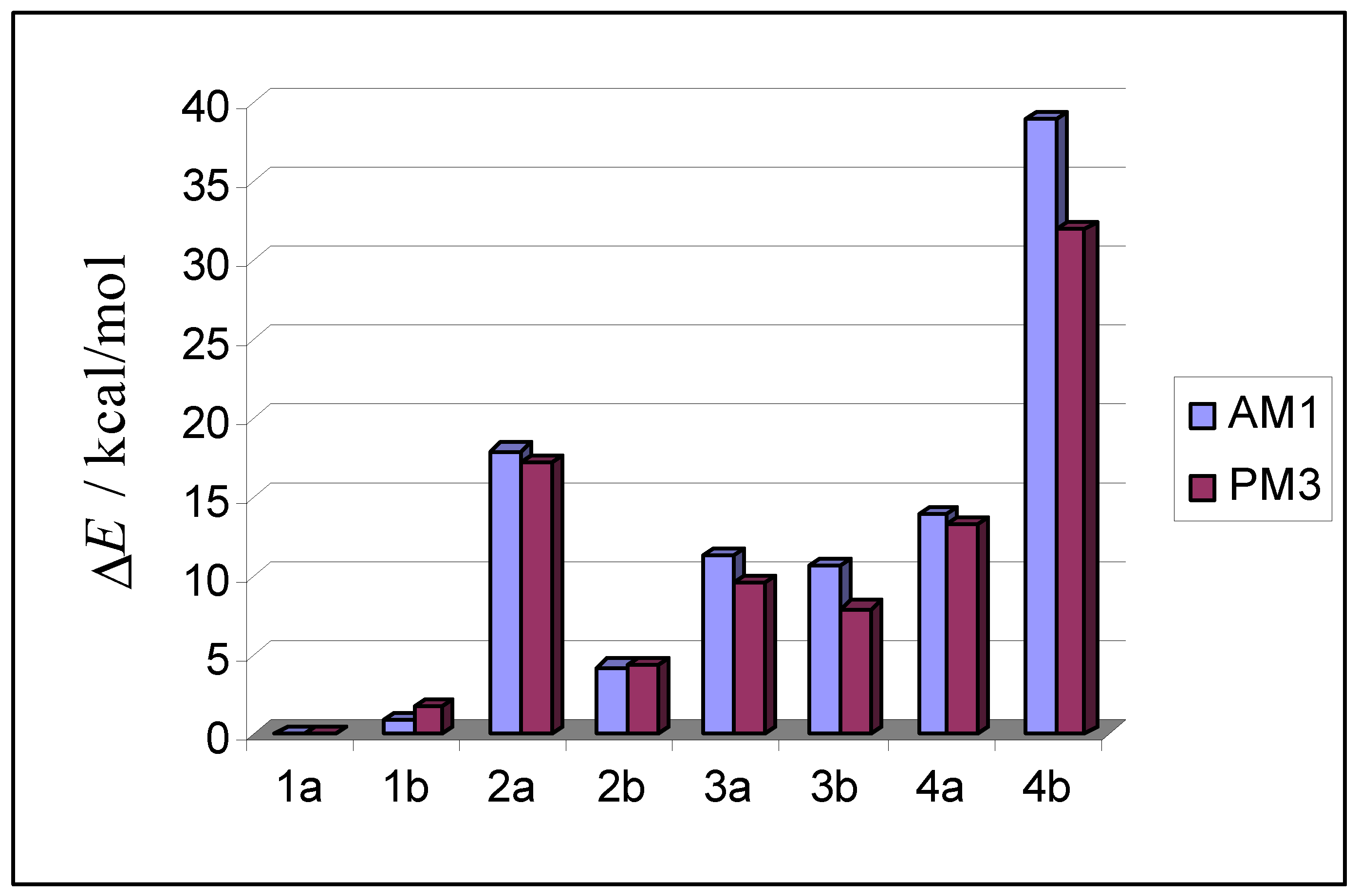
| AM1 structure | E(AM1) / (kcal/mol) | ΔE(AM1)/ (kcal/mol) | E(PM3) / (kcal/mol) | ΔE(PM3)/ (kcal/mol) | |
|---|---|---|---|---|---|
| 1a |  | –2667.6 | 0.0 | –2677.7 | 0.0 |
| 1b |  | –2666.7 | 0.9 | –2675.8 | 1.8 |
| 2a | 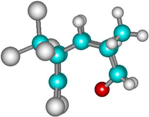 | –2649.7 | 17.9 | –2660.5 | 17.1 |
| 2b | 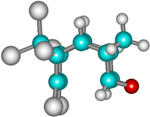 | –2663.3 | 4.3 | –2673.2 | 4.4 |
| 3a |  | –2656.3 | 11.3 | –2668.1 | 9.6 |
| 3b |  | –2656.9 | 10.7 | –2669.7 | 7.9 |
| 4a |  | –2653.6 | 14.0 | –2664.3 | 13.3 |
| 4b | 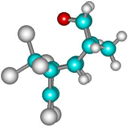 | –2628.8 | 38.8 | –2645.7 | 32.0 |
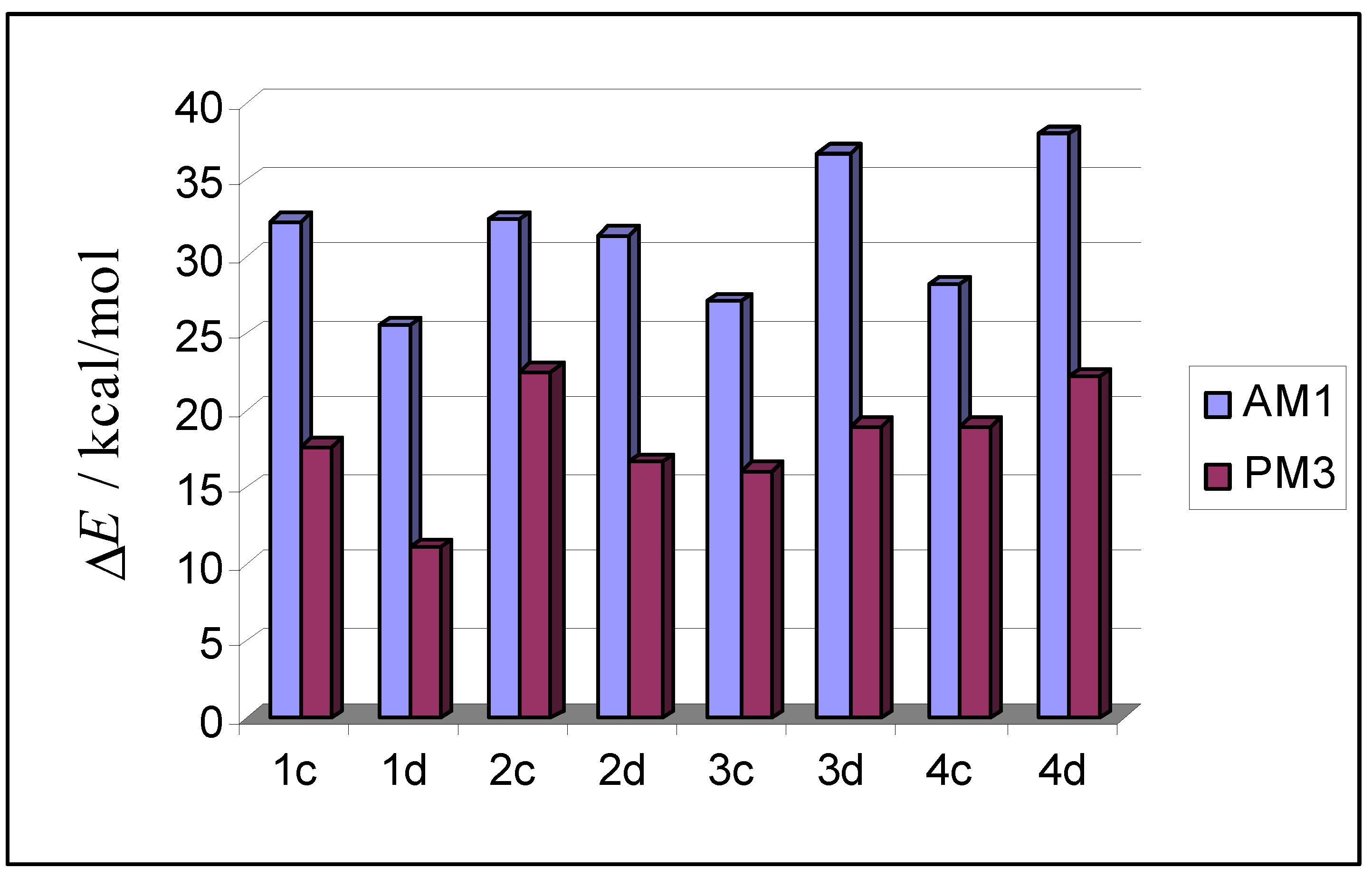
| AM1 structure | E(AM1) / (kcal/mol) | ΔE(AM1)/ (kcal/mol) | E(PM3) / (kcal/mol) | ΔE(PM3)/ (kcal/mol) | |
|---|---|---|---|---|---|
| 1c | 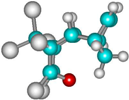 | –2635.3 | 32.3 | –2660.0 | 17.7 |
| 1d | 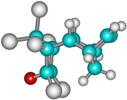 | –2642.1 | 25.5 | –2666.6 | 11.1 |
| 2c | 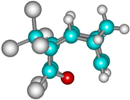 | –2635.1 | 32.5 | –2655.1 | 22.6 |
| 2d | 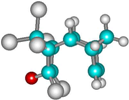 | –2636.2 | 31.4 | –2661.1 | 16.6 |
| 3c | 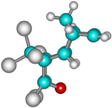 | –2640.5 | 27.1 | –2661.5 | 16.1 |
| 3d |  | –2630.8 | 36.8 | –2658.6 | 19.0 |
| 4c |  | –2639.4 | 28.2 | –2658.6 | 19.0 |
| 4d |  | –2629.6 | 38.0 | –2655.5 | 22.2 |


Computational Methods
Acknowledgements
References
- a)Carruthers, W. Cycloaddition Reactions in Organic Synthesis; Pergamon Press: New York, 1990. [Google Scholar]Brocksom, T. J.; Nakamura, J.; Ferreira, M. L.; Brocksom, U. The Diels-Alder Reaction: an Update. Journal of the Brazilizan Chemical Society 2001, 12(5), 597–622. [Google Scholar]
- Zdravkovski, Z. Theoretical Study of the Diels-Alder Reaction of Benzene with Fluorinated Dienophiles. Journal of Molecular Structture (Theochem) 2004, 684, 99–102. [Google Scholar]Jursic, B. S.; Zdravkovski, Z. Reaction of Imidazoles with Ethylene and Singlet Oxygen. An Ab Initio Theoretical Study. Journal of Organic Chemistry 1995, 60, 2865–2869. [Google Scholar]Jursic, B. S.; Zdravkovski, Z.; Whittenburg, S. L. Ab Initio Study of the Low Reactivity of Thiophene in Diels-Alder Reactions with Carbon Dienophiles. Journal of Physical Organic Chemistry 1995, 8, 753–760. [Google Scholar]Jursic, B. S.; Zdravkovski, Z.; Whittenburg, S. L. Theoretical Study of the Diels-Alder Reaction between S-Methylthiophenium Ion and Ethane. Journal of the Chemical Society, Perkin Transactions 2 1995, 455–459. [Google Scholar]Jursic, B. S.; Zdravkovski, Z. Ab Initio Calculations of Diels-Alder Transition Structures for Heterodienophile Additions to Cyclopentadiene. Journal of Physical Organic Chemistry 1994, 7, 641–645. [Google Scholar]Jursic, B. S.; Zdravkovski, Z. Ab Initio Study of Heterodienophile Addition to Oxazole. Journal of the Chemical Society, Perkin Transactions 2 1994, 1877–1881. [Google Scholar]Jursic, B. S.; Zdravkovski, Z. Theoretical Study of Ethylene and Vinyl Alcohol Addition to 1,4-Dioxa-1,3-butadiene. Journal of Organic Chemistry 1994, 59, 7732–7736. [Google Scholar]
- Marchand, A. P.; Ganguly, B.; Malagón, C. I.; Lai, H.; Watson, W. Experimental and Theoretical Studies of Diels-Alder Dimerization of 1,2,3,4,5-Pentachloropentadiene and of Diels-Alder Cycloaddition of Polychlorinated Cyclopentadienes to Norbornadiene. Tetrahedron 2003, 59, 1763–1771. [Google Scholar] [CrossRef]
- Mazzocchi, P. H.; Stahly, B.; Dodd, J.; Rondan, N. G.; Domelsmith, L. N.; Rozeboom, M. D.; Carmella, P.; Houk, K. N. π-Facial Stereoselectivity: Rates and Stereoselectivities of Cycloadditions of Hexachlorocyclopentadiene to 7-Substituted Norbornadienes, and Photoelectron Spectral and Molecular Orbital Computational Investigations of Norbornadienes. Journal of the Americal Chemical Society 1980, 102, 6482–6490. [Google Scholar]Caramella, P.; Rondan, N. G.; Paddon-Row, M. N.; Houk, K. N. Origin of π-Facial Stereoselectivity in Additions to π-Bonds: Generality of the Anti-Periplanar Effect. Journal of the Americal Chemical Society 1981, 103, 2438–2440. [Google Scholar]Marchand, A. P.; Chong, H.S.; Ganguly, B.; Coxon, J. M. π-Facial Selectivity in Diels-Alder Cycloadditions. Croatica Chemica Acta 2000, 73, 1027–1038. [Google Scholar]
- Rondan, N. G.; Paddon-Row, M. N.; Caramella, P.; Houk, K. N. Nonplanar Alkenes and Carbonyls: A Molecular Distortion Which Parallels Addition Stereoselectivity. Journal of the Americal Chemical Society 1981, 103, 2436–2438. [Google Scholar] [CrossRef]
- Gaussian 98; Revision A.11.2; Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Zakrzewski, V. G.; Montgomery, J. A., Jr.; Stratmann, R. E.; Burant, J. C.; Dapprich, S.; Millam, J. M.; Daniels, A. D.; Kudin, K. N.; Strain, M. C.; Farkas, O.; Tomasi, J.; Barone, V.; Cossi, M.; Cammi, R.; Mennucci, B.; Pomelli, C.; Adamo, C.; Clifford, S.; Ochterski, J.; Petersson, G. A.; Ayala, P. Y.; Cui, Q.; Morokuma, K.; Rega, N.; Salvador, P.; Dannenberg, J. J.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Cioslowski, J.; Ortiz, J. V.; Baboul, A. G.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Gomperts, R.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Andres, J. L.; Gonzalez, C.; Head-Gordon, M.; Replogle, E. S.; Pople, J. A. Gaussian, Inc.: Pittsburgh PA, 2001. [Google Scholar]
- Gaussian 03; Revision B.03; Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery, J. A., Jr.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A. Gaussian, Inc.: Wallingford CT, 2003. [Google Scholar]
- Gauss View 03; Gaussian, Inc.: Pittsburgh PA, 2003.
- Stewart, J. J. P. Optimization of Parameters for Semi-Empirical Methods I-Method. Journal of Computational Chemistry 1989, 10, 209–220. [Google Scholar] [CrossRef]
- Becke, A. D. A New Mixing of Hartree-Fock and Local Density-functional Theories. Journal of Chemical Physics 1993, 98, 1372–1377. [Google Scholar]Lee, C.; Yang, W.; Parr, R. G. Development of the Colle-Salvetti Correlation Energy Formula into a Functional of the Electron Density. Physical Review B 1988, 37, 785–789. [Google Scholar]
- Branchadell, V. Density Functional Study of Diels-Alder Reactions Between Cyclopentadiene and Substituted Derivatives of Ethylene. International Journal of Quantum Chemistry 1997, 61(3), 381–388. [Google Scholar] [CrossRef]
© 2006 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Nestorovska-Krsteska, A.; Zdravkovski, Z. Theoretical Study of the Diastereofacial Isomers of Aldrin and Dieldrin. Int. J. Mol. Sci. 2006, 7, 35-46. https://doi.org/10.3390/i7020035
Nestorovska-Krsteska A, Zdravkovski Z. Theoretical Study of the Diastereofacial Isomers of Aldrin and Dieldrin. International Journal of Molecular Sciences. 2006; 7(2):35-46. https://doi.org/10.3390/i7020035
Chicago/Turabian StyleNestorovska-Krsteska, Aleksandra, and Zoran Zdravkovski. 2006. "Theoretical Study of the Diastereofacial Isomers of Aldrin and Dieldrin" International Journal of Molecular Sciences 7, no. 2: 35-46. https://doi.org/10.3390/i7020035
APA StyleNestorovska-Krsteska, A., & Zdravkovski, Z. (2006). Theoretical Study of the Diastereofacial Isomers of Aldrin and Dieldrin. International Journal of Molecular Sciences, 7(2), 35-46. https://doi.org/10.3390/i7020035